

1 Introduction
Silyl-substituted cyclopentadienyl anions play an important role in organometallic chemistry, in particular in Ziegler-Natta type olefin polymerization catalysts where the presence of the electron-withdrawing silyl substituent improves their catalytic activity [1,2]. An additional benefit is that the silyl group may readily be adapted to allow fixation of the catalyst on solid supports [3–5]. An alternative approach, which imparts similar electron withdrawing properties on cyclopentadienyl units, is the substitution of CH vertices in the ring for isolobal P centres, a strategy that is known to promote δ-backbonding from the metal to the ring [6–8]. However, the combination of these concepts, i.e. silyl-substituted phospholide anions is relatively under explored. Niecke and co-workers isolated the bis(trimethylsilyl)triphospholide anion as its lithium salt whilst investigating the reactivity of the 2,3,4-triphosphapentadienide anion [9]; unfortunately, one of the steps involved oxidation of the 1,2,4-triphospholenide precursor using atmospheric oxygen, which has limited a more extensive survey of its reactivity.
Regarding the di- and triphospholide anions [10–16], which are the subject of this report, the most commonly accessed species contain tBu substituents, a feature that is inbuilt as their synthesis often derives from the phosphaalkyne, tBuCP. Indeed, a considerable number of reactions of tBuCP have been reported to yield phospholide anions [17–19]. One of the most commonly used syntheses is from the reaction of tBuCP and alkali metals giving a mixture of the 1,3,4-triphospholide anion (42% yield) and the 1,3-diphospholide anion (37% yield) [17]. Recently, we have developed new strategies, which allow selective access to the triphospholide anion and make better economic use of the valuable tBuCP [20,21]. Probing the chemistry of diphospholide and triphospholide anions is greatly assisted by their distinctive 31P NMR spectroscopy signatures, e.g. the bis(trimethylsilyl)triphospholide anion give low-field shifts for all 31P NMR signals (δ = 313(t), 328(d), 2J(PP) = 31.8 Hz) [9].
We believed that the title compound, Me3SiCP, may be a convenient starting point in our goal towards silyl-substituted phospholide anions, and we were encouraged to observe that there exist several literature routes to Me3SiCP. For example, Appel et al. reported that elimination of Me3SiCl from (Me3Si)2C = PCl occurs at 750 °C giving a quantitative yield of the silyl functionalised phosphaalkyne [22]; similarly, Denis and co-workers found that passing Me3SiCCl2PH2 over K2CO3 at 350 °C effects the base induced elimination of two equivalents of HCl giving a good yield of Me3SiCP [23,24]. However, the harsh conditions that these preparations require are not routinely accessible to all groups, so we sought a more convenient bench-top synthesis using conventional Schlenk-line techniques. In this report, we describe this synthesis, the subsequent conversion of the product to di- and triphospholide anions and the binding of the anions to an iron centre.
2 Results and discussion
2.1 Preparation of Me3SiCP
Me3SiCP was prepared by an adaptation of the route employed by Grützmacher and co-workers towards Ph3SiCP [25]. The dichlorophosphine, Me3SiCH2PCl2 (1), serves as a convenient precursor to Me3SiCP (Scheme 1). It was prepared in excellent yield and on multi-gram scales by reaction of chloromethyltrimethylsilane with magnesium to form the corresponding Grignard reagent followed by reaction with PCl3 at −78 °C. Bubbling dry HCl gas through the mixture was necessary to facilitate precipitation of the magnesium salts and aid product separation. Filtration of the salt residues and removal of the solvent yielded pure 1. Reaction of 1 with two equivalents of DABCO (1,4-diazabicyclo[2.2.2]octane) and silver triflate in toluene (room temperature, 1 h) in the absence of light, followed by filtration and distillation gives colourless solutions with Me3SiCP 2 as the only phosphorus-containing product (Scheme 1). The addition of silver triflate is vital in order to cause the immediate precipitation of any chloride salts as AgCl so as to eliminate the possibility of chloride acting as a nucleophile and attacking the SiMe3 group. It is important to note that replacing toluene with THF, Et2O, or indeed many other common non-aromatic solvents, gives rise to an entirely different reaction, the products of which have not been identified. Yields are modest (20–40%), but this is compensated for in part by relatively fast reaction times and commonly available reagents.
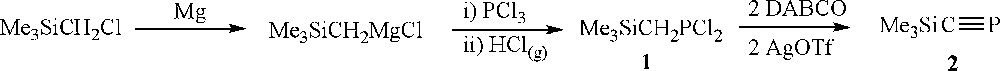
Unfortunately, despite repeated attempts, it was not possible to separate Me3SiCP from the toluene in which it was prepared; similar observations were made if aromatic solvents of different volatilities (e.g., mesitylene, benzene) were employed. In order to facilitate the systematic evaluation of the properties of Me3SiCP, a means of estimating the concentration of the solution was needed. The concentrations of solutions produced were determined by adding a known amount of PPh3 in toluene solution and integrating it against the signal for Me3SiCP (at 99.4 ppm) in the 31P NMR spectra. In order to obtain reliable results it was necessary to take into account a number of factors; the long relaxation time of 31P nuclei and the variable relaxation time of 31P nuclei, as well as NOEs from nearby protons, which can also impact on the integrals in 31P NMR spectra. [Cr(acac)3] was used as a relaxation agent in order to ensure that the 31P nuclei in PPh3 and in Me3SiCP relax completely before the next radio frequency pulse was broadcast when collecting data. The time between pulses was also increased slightly in order to facilitate this as well. The proton decoupling protocol was also amended in order to eradicate NOEs that could bias the results. All of this was used to achieve an estimation of the concentration of Me3SiCP in toluene solution to approximately ± 10%, which enabled the matching of stoichiometries in all subsequent reactions. As empirical evidence, in 31P NMR spectra of reactions where Me3SiCP reacted cleanly with 31P-containing reagents, no excess of either reagent could be seen by NMR spectroscopy.
2.2 Stability of Me3SiCP
It has been reported that 2 is unstable at room temperature decomposing with a half-life of one day for THF solutions of between 2 and 5% at 20 °C [24]. However, it is unclear whether this is an inherent property of the species or a by-product of the harsh reaction conditions employed in its synthesis. In order to probe the utility of the above procedure, a better qualitative understanding of the stability of 2 prepared in aromatic solvent was desirable. No evidence was observed that solutions of 2 decompose at −78 °C, and furthermore these solutions do not appear to decompose (31P NMR) at room temperature if stored under an inert atmosphere even over several days. However, solutions of 2 were observed to decompose if exposed to atmospheric oxygen and decomposition was also observed when a toluene solution of 2 was heated to 100 °C for 16 h, although short exposure to such temperatures did not adversely affect the sample.
2.3 Synthesis of trimethylsilyl-substituted di- and triphospholides
2.3.1 Reactions of Me3SiCP with group 1 alkoxides, alkyls and phosphides
Reaction of 2 with an equimolar amount of either LiOMe or NaOMe/PMDETA (PMDETA = N,N,N′,N″,N″-pentamethyldiethylenetriamine) produces highly coloured solutions which contain a ca. 1:2 mixture of alkali metal salts of the diphospholide 3 and triphospholide 4 anions, respectively (Scheme 2). The 31P NMR spectrum for the lithium salts show a singlet resonance for the diphospholide anion at δ = 270 ppm, and two peaks for the triphospholide anion (δ = 327(d), 316(t) ppm, 2J(PP) = 30.2 Hz), the latter signals being very similar to those in the existing literature report of the lithium salt of [C2P3(SiMe3)2] anion (δ = 328(d), 313(t) ppm, 2J(PP) = 31.8 Hz) [9], with the minor discrepancies presumably being accounted for by the different solvents employed. The sodium salts of solutions of 3 and 4 display similar spectra (3Na(PMDETA)2 δ = 272(s) ppm; 4Na(PMDETA)2 δ = 326(t), 315(d) ppm, 2J(PP) = 34.4 Hz).
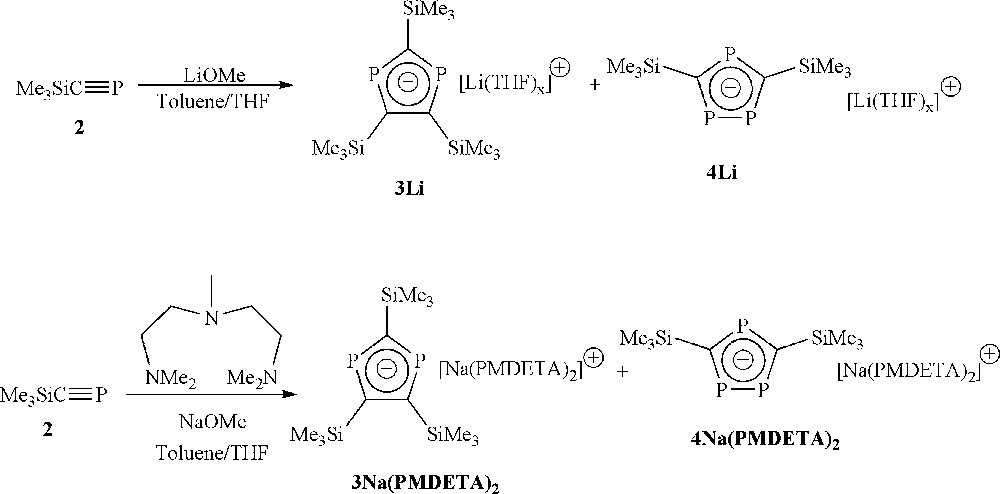
Formation of di- and triphospholides by reaction of 2 with LiOMe or NaOMe.
With this promising lead, we extended the range of bases employed by examining reactions of 2 with some alkyllithium reagents. In this respect, we noted that reactions of tBuCP with MeLi have been reported to give wholly different reactivity, yielding the simple addition product forming a lithiated phosphaalkene [26]. In contrast, reaction of MeLi with 2 in toluene led selectively to the formation of the lithium diphospholide, 3Li (Scheme 3), the reaction being fairly independent of stoichiometry as using anywhere between half to one equivalent of MeLi generates 3Li with only very minor by-products (by 31P NMR spectroscopy). The lithium phosphide that is presumed to be the by-product of the reaction was not observed experimentally, but we note literature precedent of similar reactions in the elimination of phosphorus species [27–29]. Addition of 12-crown-4 and storage of a concentrated THF solution layered with n-hexane gave crystals of 3Li(12-crown-4)2 that were suitable for an X-ray diffraction study. Identical reactivity was also observed using nBuLi, allowing the synthesis of toluene soluble, base-free 3Li, although, in our hands, the only successful crystallisation to date was achieved using the MeLi/12-crown-4 combination. The molecular structure of 3Li(12-crown-4)2 (Fig. 1, Table 1) showed a planar diphospholide ring ion-separated from the [Li(12-crown-4)2]+ cation.
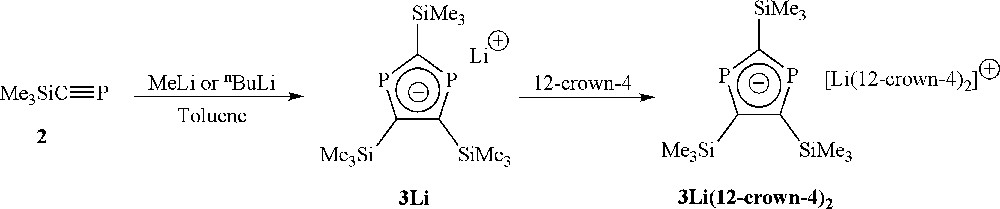
Selective formation of the diphospholide anion by reaction of 2 with alkyllithium reagents.
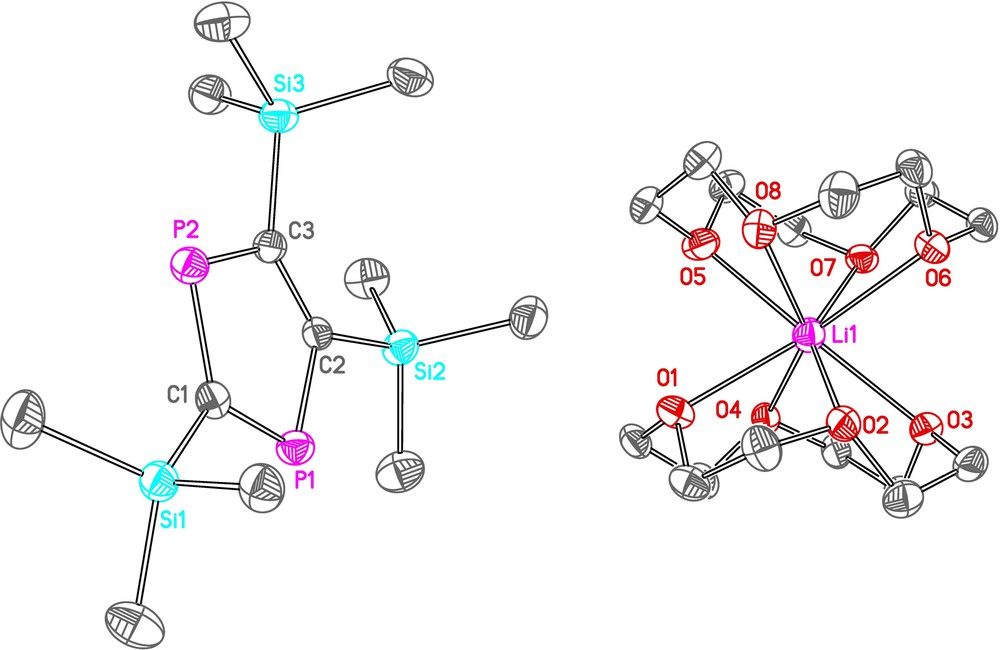
Thermal ellipsoid plot (at 50% probability level) of 3Li(12-crown-4)2. All hydrogen atoms have been omitted for clarity.
Selected bond lengths (Å) and angles (°) for 3Li(12-crown-4)2.
| P1C1 | 1.739(4) | C2C3 | 1.421(5) |
| P1C2 | 1.774(4) | C1Si1 | 1.849(4) |
| P2C1 | 1.743(4) | C2Si2 | 1.870(4) |
| P2C3 | 1.767(4) | C3Si3 | 1.864(4) |
| C1P1C2 | 97.38(19) | C3C2Si2 | 130.9(3) |
| C1P2C3 | 97.79(19) | P1C2Si2 | 113.6(2) |
| P1C1P2 | 114.4(2) | C2C3P2 | 114.9(3) |
| P1C1Si1 | 123.7(2) | C2C3Si3 | 129.9(3) |
| P2C1Si1 | 121.5(2) | P2C3Si3 | 115.1(2) |
| C3C2P1 | 115.4(3) |
As expected, the PC and CC bond lengths in (3) are suggestive of aromatic delocalisation and the bond dimensions are very similar to those seen in the relatively rare examples of tert-butyl substituted diphospholide anions [27,29].
2.3.2 Reaction of Me3SiCP with group 1 and group 2 metals, and ytterbium
As noted above, one of the most commonly used syntheses of tert-butyl substituted di- and triphospholides is from the direct reaction of tBuCP with alkali metals [17,19]. We therefore explored whether a similar method could be employed for the silyl substituted analogues. Overnight stirring (room temperature) of toluene/THF solutions of 2 and any of the three lightest alkali metals (Li, Na and K) gave a mixture of the di- and triphospholide anions (Scheme 4) as the only products in a ratio of approximately 1:1 (31P NMR spectroscopy). Attempts to separate and purify these mixtures of phospholides have so far proved unsuccessful. The 31P NMR spectra of the Li and Na salts have been previously mentioned; unsurprisingly the potassium salts show very similar spectra ((3K δ = 274(s) ppm); 4K δ = 326(d), 318(t) ppm, 2J(PP) = 33.5 Hz).

M = Li, Na, K. Formation of di- and triphospholides by reaction of 2 with alkali metals.
In contrast, alkaline earth metal salts of phospholide anions are much less well known [21,30], and the synthesis of phospholide anions from the direct reaction of a phosphaalkyne with an alkaline earth metal has not, to our knowledge, been reported in the literature. With this in mind, we attempted the direct reduction of Me3SiCP with calcium (Scheme 5).
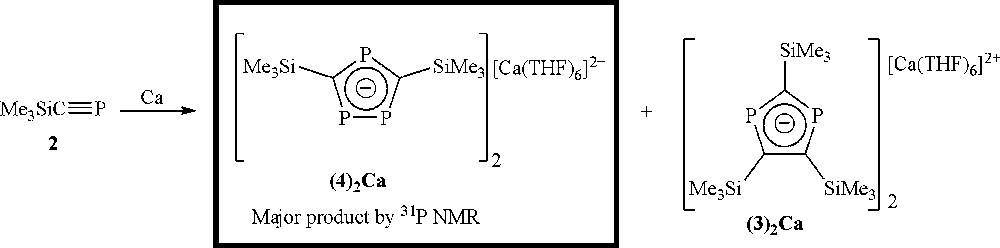
Formation of the triphospholide anion as the major product using calcium metal.
Reaction of 2 in a mixture of THF and toluene with calcium metal, which had been activated with 1,2-dibromoethane, was slow and took 3 days before the starting material had been consumed to give a dark red solution, which 31P NMR spectroscopy revealed to contain peaks corresponding to the triphospholide anion (δ = 325(d) and 315(t) ppm, 2J(PP) = 29.8 Hz) as the major product (94%), with only a very small signal for the diphospholide anion (δ = 270 ppm). The reason for the preference for the triphospholide anion is not readily apparent. Crystallisation from a THF/n-hexane layer gave a complicated ion-separated structure with four crystallographically independent [Ca(THF)6]2+ cations and eight crystallographically independent triphospholide anions (Fig. 2). This large number of atoms in the asymmetric unit (564) and the unit cell (2256) meant that, together with disorder present in some of the THF molecules and SiMe3 groups, the dataset is not of the highest quality; bond lengths and angles for a representative anion are given in Table 2.
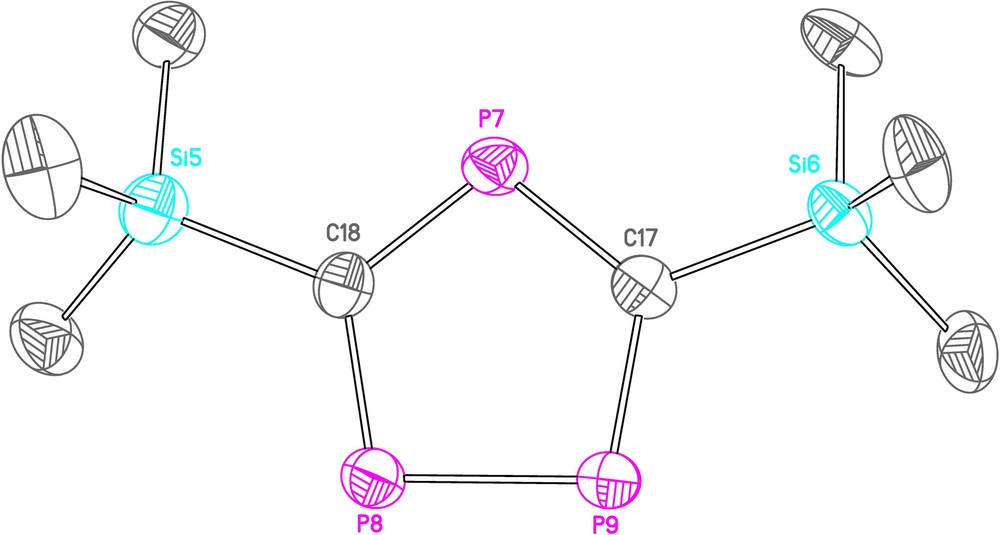
Thermal ellipsoid plot (at 50% probability level) of one triphospholide anion from the structure of (4)2Ca. Hydrogen atoms have been omitted for clarity.
Selected bond lengths (Å) and angles (°) for one triphospholide anion from the structure of (4)2Ca.
| P7C17 | 1.735(7) | C17P9 | 1.749(7) |
| P7C18 | 1.734(7) | P8P9 | 2.097(3) |
| C18P8 | 1.751(7) | ||
| C17P7C18 | 102.0(3) | C17P9P8 | 100.0(3) |
| P8C17P7 | 119.2(4) | P7C17P9 | 119.0(4) |
| P9P8C17 | 99.7(3) |
All of the anions are planar with an average PP bond length of 2.098 Å and contain CP bond lengths ranging from 1.711(10) to 1.761(9) Å. These dimensions and the associated bond angles are very similar to those seen in the structure of [Mg2Cl3(THF)6][C2P3(tBu)2], which has a PP bond length of 2.091(1) Å and PC bond lengths of 1.731(4) to 1.748(3) Å [21]. The [Ca(THF)6]2+ dication is unexceptional, although crystallographically characterised examples of it are rare [31].
In attempting to extend the scope of these reactions beyond the s-block metals, we explored the analogous reaction between 2 and ytterbium metal (Scheme 6). As with calcium, this proceeded very slowly, taking several days to consume all of the starting material and yielding a complex mixture of products including the di- and triphospholide anions, as well as a number of unidentified products.

Formation of multiple products by reaction of Me3SiCP with Yb metal.
2.3.3 Reactivity of the di- and triphospholides with iron salts
Di- and triphospholide anions bearing tert-butyl substituents have been coordinated to a range of metals, and naturally it was of interest to establish that the trimethylsilyl substituted analogues that we have prepared by the methods outlined above may be coordinated to a transition element. The target metal we chose was iron.
Reaction of the lithium di- and triphospholides (formed from the reaction of lithium metal) and 2 with FeCl2 (Scheme 7) was only successful in dme (not THF) and without the addition of 12-crown-4; the reason for this lack of reactivity in THF, or with 12-crown-4 added, is not apparent. The brown reaction mixture was purified either by column chromatography on silica, eluting a green band with n-hexane, or by sublimation at 120 °C and 1 × 10−2 Torr yielding a green product. Crystallisation has so far not been achieved possibly due to the mixture of phosphaferrocenes produced.
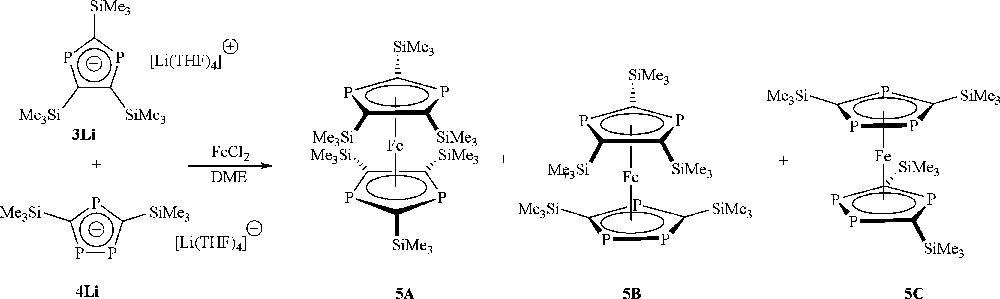
Formation of Me3Si functionalised tetra- 5A, penta- 5B, hexaphosphaferrocene 5C.
EI mass spectrometry revealed the presence of tetra- (m/z 690.1), penta- (m/z 636.0) and hexaphosphaferrocene (m/z 582.0). Interestingly, there are in addition two other products with masses of 466.0 and 520.0, which are consistent with the loss of “Me3SiCP” units from both hexa- and pentaphosphaferrocene. There are many intriguing possibilities as to how such a formulation may be constructed, which provides an interesting stimulus for further research. Whether these are fragmentation products from the mass spectrometry experiment or are minor products arising from the synthesis is unclear.
31P NMR spectroscopy at room temperature of the mixture of phosphaferrocenes revealed surprisingly few signals, displaying a triplet (δ = 114.9 pmm) and doublet (δ = 106.4 pmm) with a common coupling constant of 30.0 Hz, consistent with a triphospholide unit, and a singlet (δ = 102.3 pmm) consistent with a diphospholide unit, as well as two additional peaks (vide infra). The very different chemical shifts of both groups compared to those observed in the s-block metal salts above suggest that the phospholide groups are bound to iron. It is likely that these signals arise from the pentaphosphaferrocene, 5B, which contains both a di- and triphospholide ligand as the signals are sharp and the structure would be expected to be eclipsed giving a conformationally locked system.
The two additional resonances in the room temperature 31P NMR spectrum, a complex multiplet centred at δ = 128 ppm and an extremely broad peak (barely visible above the baseline) centred at ca. δ = 89 ppm, which do not couple to the other signals visible in the room temperature spectrum (deduced from 31P31P NMR correlation spectroscopy), remained unresolved. Variable temperature 31P NMR experiments were carried out, and upon cooling to −90 °C, five further signals became visible. All of these additional signals are significantly broad and show complicated, not fully resolved coupling patterns and thus far we have been unable to assign these signals definitively, but taken with mass spectroscopy results, there is strong evidence that the trimethylsilyl substituted phospholides bind to transition metal centres. We hope to provide definitive proof and detailed structural information regarding such transition metal phospholide complexes in the near future.
3 Conclusions
In summary, it has been shown that Me3SiCP may be prepared using conventional Schlenk-line techniques under ambient conditions by dehydrochlorination of Me3SiCH2PCl2 in aromatic solvents using DABCO in the presence of AgOTf. Subsequent reaction of Me3SiCP with a selection of s-block metals, s-block metal salts and ytterbium affords access to silyl substituted di- and triphospholide anions. This mixture of anions was subsequently reacted with FeCl2 to yield a mixture of tetra-, penta- and hexaphosphaferrocenes.
4 Experimental
4.1 General experimental
Reactions were performed under either a N2 or Ar atmosphere using standard Schlenk and glovebox techniques. Solvents used were distilled HPLC grade and further dried and degassed using a commercially available solvent purification system (Anhydrous Engineering). All chemicals used were purchased from commercial sources and used as supplied. Solution NMR spectroscopy samples were prepared using dry and degassed deuterated solvent sealed in air-tight NMR tubes (Wilmad WG-5 M economy) and were run on either a Jeol Eclipse 300 MHz or Varian 500 MHz Multinuclear FT spectrometer. 1H NMR spectroscopy samples were referenced to the internal solvent peaks. Solution 31P NMR spectra were referenced to an external sample of 85% H3PO4. Solution 29Si NMR spectra were referenced to an external sample of SiMe4.
4.1.1 Synthesis of Me3SiCH2PCl2 1
Me3SiCH2Cl (17.8 cm3, 0.129 mmol) in Et2O (70 cm3) was added to activated magnesium turnings (ca. 7 g) suspended in Et2O (80 cm3). After ca. 1 h, the dark coloured solution was filtered (porosity 3 sinter and Celite) giving a pale yellow solution, which was added slowly to a −78 °C solution of PCl3 (11.2 cm3, 0.129 mmol) in Et2O (100 cm3) causing the immediate formation of a white precipitate. The mixture was allowed to warm to room temperature and then stirred for 1 h. Dry HCl gas was bubbled through the reaction mixture for 30 min. The solution was filtered (Celite) and washed through with a further portion of Et2O (40 cm3) before all the Et2O was removed under reduced pressure yielding pure 1 as a pale air- and moisture-sensitive yellow liquid (14.33 g, 14 cm3, 76 mmol, 59%). 1H NMR (300 MHz, 25 °C, CDCl3): δ(ppm) = 2.06 (d 2J(1H31P) = 15.6 Hz, 2 H, CH2), 0.22 (s, 9 H, Me3Si). 31P{1H} NMR (121.4 MHz, 25 °C, CDCl3): δ(ppm) = 205.9. 31P NMR (121.4 MHz, 25 °C, CDCl3): δ(ppm) = 205.9 (t, 2J(1H31P) = 15.8 Hz). 13C NMR (75.4 MHz, 25 °C, CDCl3): δ(ppm) = 35.31 (d, 1J(13C31P) = 60.9 Hz, Cl2PCH2), −0.08 (d, 3J(1C31P) = 5.0 Hz, Cl2PCH2Si(CH3)3). 29Si NMR (59.6 MHz, 25 °C, CDCl3): δ(ppm) = 0.81 (d, 2J(29Si31P) = 16 Hz).
4.1.2 Synthesis of Me3SiCP 2
Me3SiCH2PCl2 (1, 0.714 g, 0.7 cm3, 3.8 mmol, one equivalent) was dissolved in toluene (30 cm3) and AgOTf (2.134 g, 8.3 mmol, 2.2 equivalents) was added at room temperature with the exclusion of light. The mixture was stirred for 5 min and then DABCO (0.932 g, 8.3 mmol 2.2 equivalents) was added and the mixture stirred for another 1 h in darkness. The solution was filtered through Celite giving a red solution, which was washed with toluene (10 cm3). The solution was distilled (static vacuum, ca. 5 × 10−1 Torr) giving a colourless solution of 2, which could be stored indefinitely at −78 °C.
In order to estimate the concentration of 2 in the toluene solution, a 0.5 cm3 portion of this solution was placed in an NMR tube along with 0.2 cm3 of a PPh3 toluene solution (containing 0.0133 mmol PPh3) along with ca. 30 mg [Cr(acac)3]. The mixture was sonicated (ca. 10 min) in order to produce a homogenous solution. Integrating the peaks obtained in the 31P NMR spectrum obtained using an experiment, which negates NOEs gave the concentration of the solution, which is usually between 0.023 and 0.050 M, with an average yield of 29% over multiple experiments.
The following NMR experiments were performed on toluene solutions of 2 with 0.2 cm3 CDCl3 added for locking purposes. 1H and 13C NMR spectra show large signals for toluene. 1H NMR (300 MHz, 25 °C, CDCl3): δ(ppm) = 0.09 (s). 31P{1H} NMR (121.4 MHz, 25 °C, CDCl3): δ(ppm) = 99.4. 13C NMR (75.4 MHz, 25 °C, CDCl3): δ(ppm) = −0.35 (d, 3J(1C31P) = 3.1 Hz, (CH3)3SiCP). Note that PC was not observed. 29Si NMR (59.6 MHz, 25 °C, CDCl3): δ (ppm)−16.1 (d, 2J(29Si31P) = 7.6 Hz).
4.1.3 Syntheses of tris(trimethylsilyl)diphospholide 3 and bis(trimethylsilyl)triphospholide 4
4.1.3.1 From reaction of Me3SiCP with LiOMe
Me3SiCP (0.60 mmol in 10 cm3 toluene) was added (room temperature) to a solution of LiOMe (23 mg, 0.60 mmol) in THF (10 cm3) and was stirred overnight. A dark orange solution was observed and 31P NMR spectroscopy revealed the formation of di- and triphospholide species, along with two other signals. 31P{1H} NMR (121.4 MHz, 25 °C, THF): δ(ppm) = 327.0 (d, 2J(31P31P) = 30.2 Hz, triphospholide), 315.6 (t, 2J(31P31P) = 30.2 Hz, triphospholide), 269.8 (s, diphospholide).
4.1.3.2 From reaction of Me3SiCP with NaOMe
Me3SiCP (0.52 mmol in 10 cm3 toluene) was added (room temperature) to a solution of NaOMe (28 mg, 0.52 mmol) and PMDETA (0.11 cm3, 0.52 mmol) in THF (10 cm3) and stirred overnight. A red-orange solution was observed and 31P NMR spectroscopy revealed the formation of di- and triphospholide species. 31P{1H} NMR (121.4 MHz, 25 °C, THF): δ(ppm) = 326.3 (t, 2J(31P31P) = 34.4 Hz, triphospholide), 315.2 (d, 2J(31P31P) = 34.4 Hz, triphospholide), 271.8 (s, diphospholide).
4.1.3.3 Selective synthesis of (3) by reaction of Me3SiCP with alkyllithiums
The respective alkyllithium (1 equivalent MeLi, 0.5 equivalent MeLi or 2/3 equiv. nBuLi) was added at room temperature to Me3SiCP (1 equivalent in toluene) and the clear orange solution was stirred for 30 min. 31P NMR spectroscopy revealed selective formation of the diphospholide anion. The reaction using one equivalent of MeLi was crystallised by addition of 12-crown-4 (one equivalent) and layering of a THF solution with n-hexane producing a small crop of colourless crystals of 3Li(12-crown-4)2. 31P{1H} NMR (121.4 MHz, 25 °C, Toluene): δ(ppm) = 269.7 (s, diphospholide). All attempts at obtaining more extensive characterization data on 3Li(12-crown-4)2 were unsuccessful, being hampered by the low crystalline yield and high air-and moisture-sensitivity of the sample.
4.1.3.4 From reaction of Me3SiCP with potassium
A freshly prepared potassium mirror was reacted at room temperature with a solution of Me3SiCP (0.435 mmol in 14.5 cm3 toluene) in THF (15 cm3); this rapidly turned dark red in colour and stirring was continued overnight. 31P NMR spectroscopy revealed formation of di- and triphospholides. 31P{1H} NMR (121.4 MHz, 25 °C, THF): δ(ppm) = 325.7 (d, 2J(31P31P) = 33.5 Hz, triphospholide), 318.1 (t, 2J(31P31P) = 33.5 Hz, triphospholide), 273.7 (s, diphospholide). Mass spectrometry (ESI, negative ion): m/z 263 ([P3C2(SiMe3)2]− with correct isotopic distribution).
4.1.3.5 From reaction of Me3SiCP with sodium
An excess of sodium was reacted (room temperature) in THF (15 cm3) with Me3SiCP (0.75 mmol in 15 cm3 toluene); the solution quickly turned dark orange and was stirred overnight. 31P NMR spectroscopy revealed formation of di- and triphospholides. 31P{1H} NMR (121.4 MHz, 25 °C, THF/toluene): δ(ppm) = 322.8 (t, 2J(31P31P) = 33.5 Hz, triphospholide), 320.2 (d, 2J(31P31P) = 33.5 Hz, triphospholide), 271.2 (s, diphospholide).
4.1.3.6 Reaction of Me3SiCP with lithium
Under argon, an excess of lithium was added (room temperature) to Me3SiCP (0.57 mmol) in a mixture of toluene (15 cm3) and THF (10 cm3). The reaction mixture turned a dark red/brown colour over 30 min and 31P NMR spectroscopy revealed formation of di- and triphospholides. Further reactions (see below) with the solution were carried out after filtering (Celite) in order to remove any excess lithium. 31P{1H} NMR (121.4 MHz, 25 °C, THF): δ(ppm) = 322.7 (d, 2J(31P31P) = 29.9 Hz, triphospholide), 316.9 (t, 2J(31P31P) = 29.9 Hz, triphospholide), 270.1 (s, diphospholide).
4.1.3.7 Reaction of Me3SiCP with calcium
Calcium metal (in excess) was flattened in a press to increase its surface area, and added to THF (10 cm3) followed by 1,2-dibromoethane (one drop) in order to activate the surface of the calcium, and was stirred for 15 min. Me3SiCP (0.39 mmol in 10 cm3 of toluene) was then added at room temperature and the reaction sonicated for ca. 10 min. Upon stirring for 3 days, a red solution was observed. The solvent was removed in vacuo and the residue extracted into THF (2 cm3); 31P NMR spectroscopy indicated formation of the triphospholide anion as the major product. Layering this solution with n-hexane followed by storage at room temperature for 2 weeks gave a good crop of yellow crystals, which were identified (X-ray crystallography) as the ion-separated calcium salt of the triphospholide anion. 31P{1H} NMR (121.4 MHz, 25 °C, THF): δ(ppm) = 325.0 (d, 2J(31P31P) = 29.8 Hz, triphospholide), 315.3 (t, 2J(31P31P) = 29.8 Hz, triphospholide), 269.7 (s, diphospholide).
4.1.3.8 Reaction of Me3SiCP with ytterbium
Ytterbium metal (in excess) was cut into small pieces in a glove box and added to THF (30 cm3) followed by 1,2-dibromoethane (one drop) in order to activate the surface of the metal, and stirred for 15 min. Me3SiCP (1.2 mmol in 30 cm3 of toluene) was then added and the reaction sonicated for 15 min producing a pale yellow solution. Upon stirring for 5 days a red/brown cloudy solution was observed. The solvent was removed in vacuo and the solid extracted into THF (30 cm3), filtered through Celite and concentrated to ca. 3 cm3 in order to carry out 31P NMR spectroscopy, which indicated the formation of several products including di- and triphospholides. 31P{1H} NMR (121.4 MHz, 25 °C, THF): δ(ppm) = 463.0 (s), 321.5 (m, triphospholide), 318.8 (d, 2J(31P31P) = 32 Hz, triphospholide), 271.3 (s, diphospholide), 80.8 (d, J(31P31P) = 178.6 Hz Hz), 63.5 (d, J(31P31P) = 178.6 Hz), −18.2 (s).
4.1.4 Synthesis of phosphaferrocenes 5A, 5B and 5C
A mixture of di- and triphospholides was prepared as outlined in Section 4.1.3.6 above. The solution was filtered through Celite and the solvent removed in vacuo yielding a red oil. Dimethoxyethane (10 cm3) was added to redissolve the phospholides and the solution was then added to a suspension of FeCl2 (103 mg, 0.81 mmol) in dme (5 cm3) and the red mixture was stirred overnight. All of the solvent was removed from the brown solution, which was then extracted into n-hexane (30 cm3) and filtered (Celite) yielding a brown solution from which all the solvent was removed yielding a brown oil. This was purified by one of two methods: using high vacuum sublimation (120 °C at 1 × 10−2 Torr) to give a green sublimate; or by column chromatography (on silica using n-hexane as the eluent) taking the first green fraction (33 mg). 31P{1H} NMR (161.8 MHz, 25 °C, CDCl3): δ(ppm) = 128.0 (m), 114.9 (t, 2J(31P31P) = 30.0 Hz), 106.4 (d, 2J(31P31P) = 30.0 Hz), 102.3 (s), 89 (very broad signal). 31P31P NMR correlation spectroscopy (162.0 MHz, 25 °C, CDCl3): δ(ppm) = 128.0 (m, not coupling to any visible signal), 114.9 (t, coupled to d at 106.4 ppm), 106.4 (d, coupled to t at 114.9 ppm), 102.3 (s, not coupling to any other signal). 1H NMR (300 MHz, 25 °C, C6D6): δ(ppm). Many overlapping SiMe3 signals.
4.1.5 X-ray crystallography
Crystals of 3Li(12-crown-4)2 and (4)2Ca suitable for X-ray diffraction analysis were grown from concentrated THF solutions of the respective compound layered with n-hexane. They were mounted in an inert oil and then transferred to the cold gas stream of the diffractometer. Experiments were performed on 3Li(12-crown-4)2 using a Bruker-Nonius KappaCCD Area Detector; the corresponding measurements for (4)2Ca were made on a Bruker-Nonius Kappa APEX2 Area Detector. Both diffractometers employed Mo-Kα radiation (λ = 0.71073 Å). Intensities were integrated from several series of exposures and absorption corrections were applied based on multiple- and symmetry-equivalent measurements [32]. The structures were solved by direct methods and refined by least squares on weighted F2 values for all reflections [33]. All non-hydrogen atoms were assigned anisotropic displacement parameters and refined without positional constraints. Hydrogen atoms were constrained to ideal geometries and refined with fixed isotropic displacement parameters. Refinement proceeded smoothly to give the residuals shown in Table 3. Complex neutral-atom scattering factors were used.
Crystallographic data for 3Li(12-crown-4)2 and (4)2Ca.
| Compound | 3Li(12-crown-4)2 | (4)2Ca |
| Colour, habit | Colourless Rod | Yellow block |
| Size (mm) | 0.20 × 0.06 × 0.06 | 0.28 × 0.27 × 0.25 |
| Empirical Formula | C28H59LiO8P2Si3 | C40H84CaO6P6Si4 |
| Fw | 676.9 | 999.33 |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | Pbca | P212121 |
| a (Å) | 11.6934(2) | 21.1186(13) |
| b (Å) | 22.3077(6) | 23.2144(14) |
| c (Å) | 28.6305(8) | 46.396(3) |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 90 | 90 |
| V (Å3) | 7468.3(3) | 22746(3) |
| Z | 8 | 16 |
| μ (mm−1) | 0.254 | 0.401 |
| T (K) | 120 | 100 |
| Reflections: | ||
| total/independent/Rint | 32946/4553/0.1160 | 213733/41617/0.0453 |
| Final R1 and R2 | 0.0533, 0.1006 | 0.0698, 0.2178 |
| Largest peak, hole | 0.249, −0.253 | 1.520, −1.020 |
| ρ calc (gcm−3) | 1.204 | 1.167 |
| Flack parameter | Not applicable | 0.01(5) |
Crystals of 3Li(12-crown-4)2 were weakly diffracting at high angle due to their small size and hence the data were cut off at 0.95 Å resolution. The structure of (4)2Ca contained disorder in a number of THF molecules and SiMe3 groups, which was modelled with a number of restraints.
CCDC 764240 (3Li(12-crown-4)2) and 764241 ((4)2Ca) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgments
We acknowledge the University of Bristol for funding and Dr Louise Male and the EPSRC UK National Crystallography Service at the School of Chemistry, University of Southampton, for collection of the data set of 3Li(12-crown-4)2. We thank the European Union for funding PhoSciNet through COST Action CM0802.