

1 Introduction
C-glycosides, both synthetic and naturally-occurring, are an important category of bioactive compounds of extensive therapeutic potential [1]. These compounds can also be used as tools to study the role of the carbohydrate moiety in biological processes [2–4] and to design potent drugs. In conventional drug design, carbohydrates are largely avoided due to the easy hydrolysis of the O-glycoside bond in human metabolism. Replacing the anomeric oxygen atom with a methylene group leads to C-glycoside analogues with higher chemical and enzymatic stability,along with lower polarity and a greater flexibility. C-glycosides can also serve as elaborated chiral synthons and are useful building blocks for the synthesis of many natural products. Consequently, a great deal of effort has been directed towards the stereoselective synthesis of various C-glycosides from simple carbohydrate precursors. For a non-exhaustive list of reviews on C-glycoside synthesis, see references [5–15].
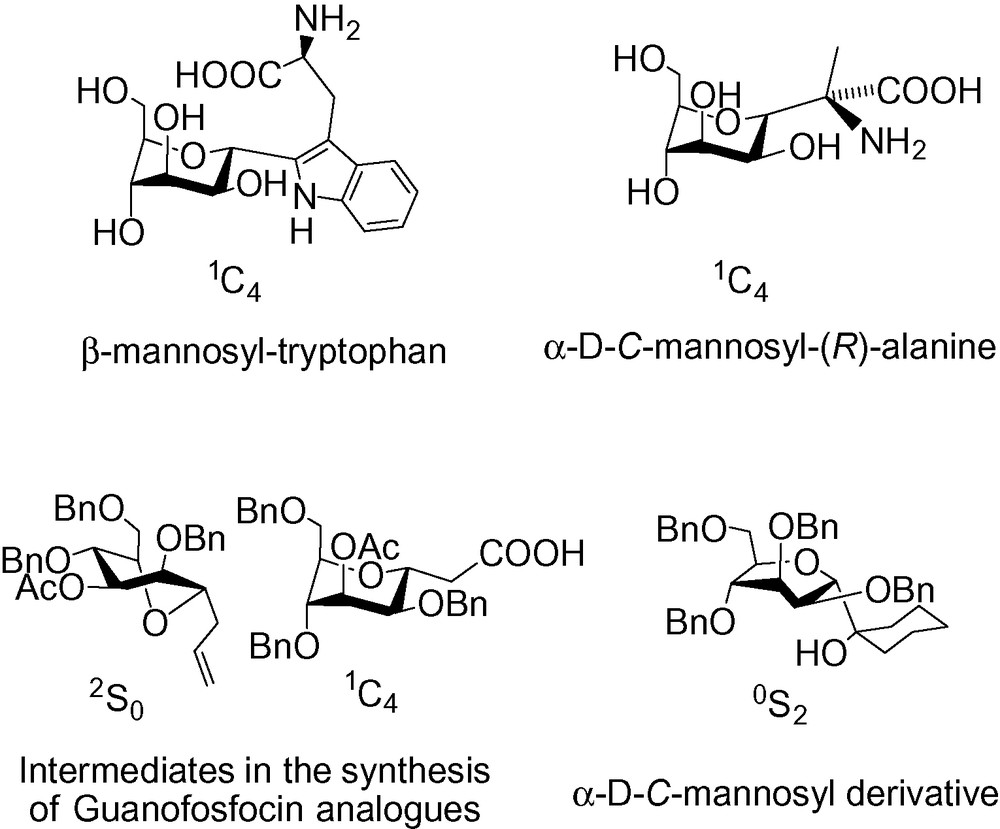
Carbohydrates play important roles in many biological processes including cell recognition, cell migration, inflammation, and bacterial and viral infections [16–18]. d-mannose is a carbohydrate commonly present as a recognition element in biological processes as part of the core trisaccharide of the asparagine-linked oligosaccharides [19,20]. As a C-glycoside, it is present in natural products such as d-mannosyl tryptophan [21–29] or chafuroside [30]. It has also been incorporated in synthetic entities such as modified glycosyl amino-acids [31–35], or used in the course of total syntheses, that is a guanofosfocin analogue,for chitine synthase inhibition [36], (−)-daucic acid [37,38], zooxanthellatoxins [39] and the IJK ring framework of brevetoxin B [40]. Various d-C-mannopyranoside derivatives have been synthesized in order to evaluate their biological activity as lectin inhibitors relative to their O-glycoside counterpart [41–57]. C-mannosides have also been prepared and tested as potential glycosidase and glycosyltransferase inhibitors [58–64], as anti-inflammatory derivatives [65], anti-tumor agents [30,66], and anti-bacterial agents [67,68].
The synthesis of d-mannosyl tryptophan was recently reviewed [69], but, to the best of our knowledge, no literature review exists that is dedicated solely to the synthesis of d-C-mannopyranosides. It is for this reason that we wish to present this article as a reference for d-C-mannopyranosides synthesis.
2 d-C-mannopyranoside conformations
Before discussing the synthetic strategies that have been applied to the preparation of d-C-mannopyranosides, it is important to understand their possible conformations, as this is one of the key factors in explaining reactivity.
2.1 C-pyranosides vs O-pyranosides
In general, when the anomeric oxygen is replaced by a methylene or a substituted methylene unit, conformational changes occur. C-glycosides are known to be more flexible, showing a distribution of different conformers possibly due to the absence of the exo-anomeric effect [70,71]. Chemical changes at the pseudoanomeric center have important effects on the behavior of C-glycosides, but it seems that the geometry corresponding to an exo-anomeric “like” orientation is favored [72].
2.2 α-d-C-mannopyranosides vs β-d-C-mannopyranosides
It is clear that β-C-mannopyranosides are more stable than their α-C-counterparts [73]. In C-glycosides, there is no longer an anomeric effect and the C-1 substituent is more stable in an equatorial position, as illustrated in the 4C1 conformation (Fig. 1).
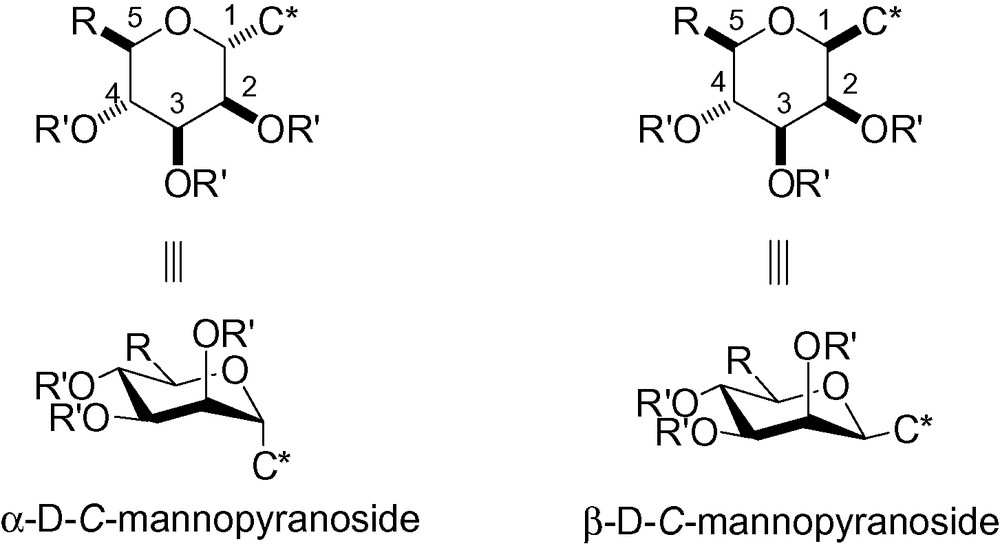
Stability of C-mannopyranosides.
This is a general rule for all C-glycosides (i.e., d-C-gluco- and d-C-galactosides). The particularity of the d-manno series lies in the possible 1,2-cis interaction that may occur between the groups in the positions 1 and 2 depending on their relative sizes (Fig. 2).
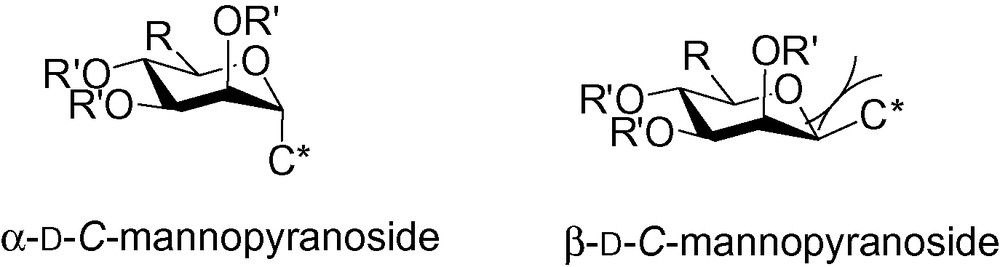
Relative sizes of the C-mannopyranosides.
To the best of our knowledge, all β-C-mannopyranosides adopt a 4C1 conformation, meaning that this 1,2-cis interaction is not strong enough to force the system to move to another conformation. This is not surprising when considering the Newman projection between C-1 and C-2 because, in reality, only a negligible C*/O interaction occurs in β-C-mannopyranosides (Fig. 3).
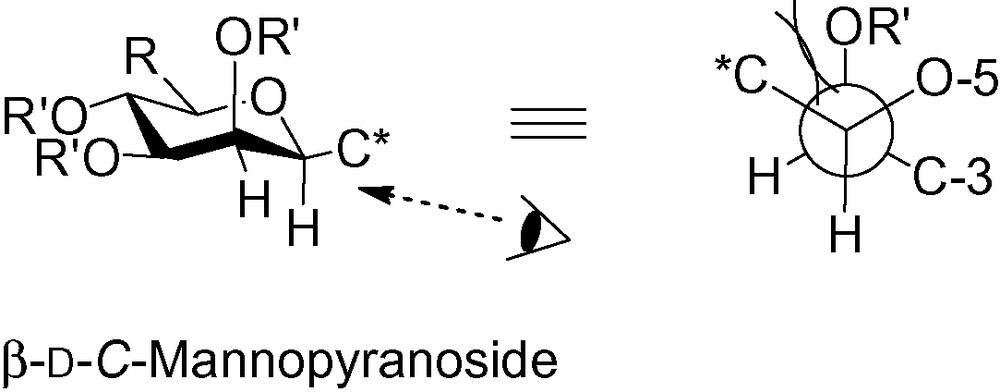
The Newman projection of the β-C-mannopyranoside.
The α-face of the C-mannopyranosides is clearly more accessible, because the axial group in position 2 blocks the upper β-face. With a C* anomeric group on the α-face, there is also no 1,2-cis interaction. The problem is that the C-glycoside now possesses two axial groups. The resulting Newman projections between C-1 and C-2, as well as between C-1 and O, show the C/C interactions which occur in an α-C-mannopyranoside (Fig. 4).
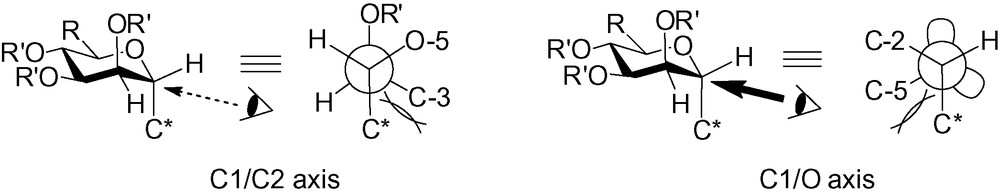
The Newman projection of the α-C-mannopyranoside.
Furthermore, because the ring CO bond lengths are smaller than the CC ones (1.43 Å versus 1.54 Å on average), the interaction between C* and C-5 can be significant. When this interaction is sufficiently large, the α-oriented group in the anomeric position will try to avoid these non-favorable interactions and adopt another conformation, even if this places other groups in axial positions (Fig. 5) [30,36,74–76].
The interaction between C* and C-5.
To better understand and control the formation of d-C-mannopyranosides, knowing the conformation of the possible transition states (oxonium or radical) leading to α and β addition is essential. d-mannosyl oxonium species are clearly planar, and ESR studies have indicated that d-mannosyl radicals preferentially adopt a 4C1 chair conformation [77,78] (Fig. 6).
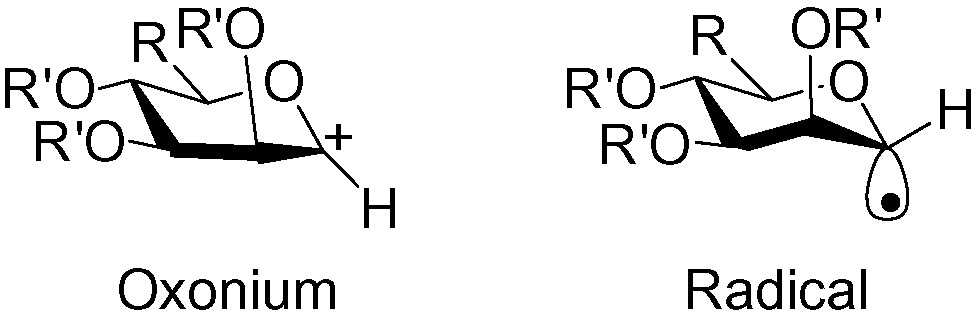
The 4C1 chair conformation of the d-mannosyl radicals.
In the case of the radical species, a maximum overlap exists between its SOMO and the LUMO of the neighboring periplanar carbonoxygen bond. The presence of an electron-withdrawing substituent (e.g. acetoxy) in this position increases the stabilizing effect of the SOMO–LUMO interactions, lowering the LUMO energy. This effect differs from the known anchimeric assistance but leads to the same results in terms of selectivity.
In summary, the stereoselective formation of α-d-C-mannopyranosides (e.g., kinetic addition to an oxonium or a radical) is relatively straightforward but gives thermodynamically unfavored compounds. In contrast, the stereoselective formation of β-d-C-mannopyranosides (e.g. thermodynamic control, axial addition of a hydride, kinetic selection by substitution of the α-anomer in rapid equilibrium) is more complicated and involves special strategies in order to circumvent steric issues between the substituents in positions 1 and 2.
In many of the cited examples, it is not always clear whether a reaction is stereospecific or stereoselective. Selectivity can be a combination of stereoselectivity as well as selectivity between competing mechanisms (SN1 vs SN2, isomerization of organometallic intermediates, etc.), or reversible (thermodynamic control) and irreversible (kinetic control) mechanisms. As a result, we will use the general term “selectivity” to reflect the observed α/β ratio. Unfortunately, in several cases described in the literature, the α/β ratios are missing, and these cases are referred by the term “not given” in schemes and/or tables.
3 General presentation and strategy
This section will be divided into six sub-sections, each one dealing with a different retrosynthetic possibility. The discussion will be focused on the key transformation which forms the targeted d-C-mannopyranoside. Fig. 7 summarizes the disconnection steps (A to E) that will be discussed.
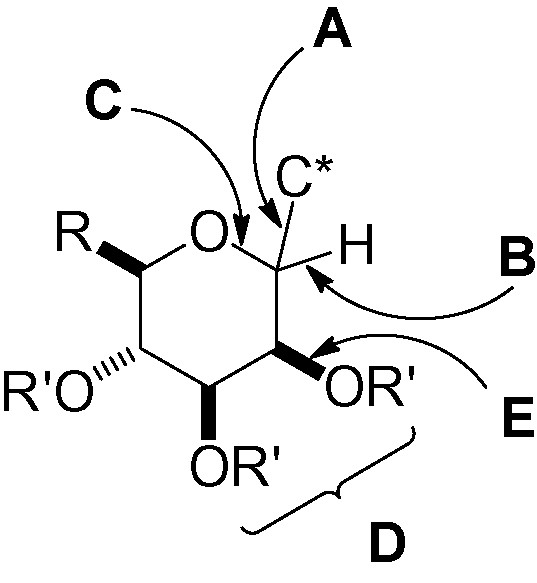
The disconnection steps.
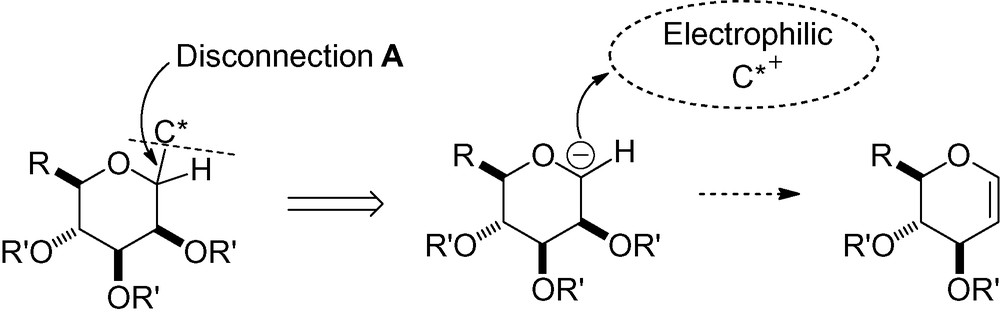
3.1 Disconnection A
3.1.1 Coupling between an electrophilic anomeric carbon and a nucleophilic carbon
This approach is obviously one of the most popular, and was studied very early in the field of C-glycoside synthesis. The general concept is shown in Scheme 1.
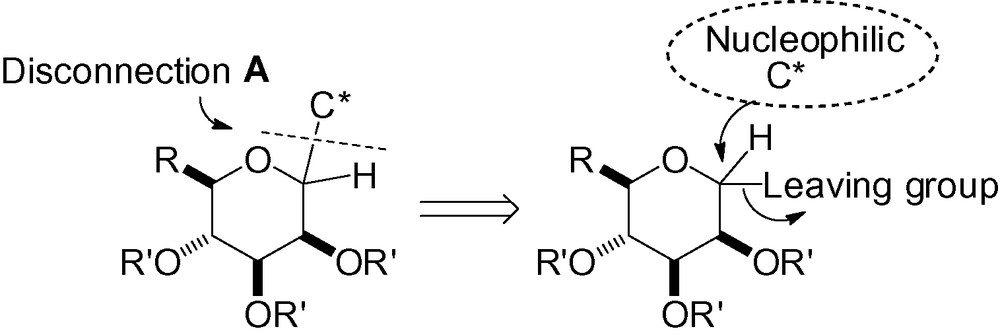
Several types of leaving groups have been studied, each chosen according to the nucleophilic C* group used. The electrophilic anomeric carbon can react through several different pathways following an SN1 or SN2-type process, a transition metal catalyzed mechanism, or via a Friedel-Crafts or allylsilane-type process.
3.1.1.1 Access to alkyl or aryl side chains
The addition of simple alkyl magnesium derivatives to glycosyl bromides has been reported but with only moderate selectivities (Table 1).
Alkyl magnesium bromide additions to glycosyl bromides. .
| Entry | R’ | R | Solvent | T | Yield (%) | α/β | ref |
| 1 | Me | (CH2)2CHCH2 | – | – | nd | nd | [79] |
| 2 | Ac | Ph | Et2O | 0 °C → r.t. | 73 | 1/1 | [80] |
| 3 | Ac | CHCH2 | THF | Reflux | 45 | 29/71 | [81,82] |
Halogenated anomeric sugars can also be substituted with zinc derivatives in the presence of nickel complexes in an aprotic polar solvent (Negishi cross-coupling). This reaction selectively gives the α-C-glycosides in moderate to good yields (Table 2) and reflects nickel insertion into the carbonhalogen bond with retention of configuration [83,84].
C-alkylation with functionalized zinc reagents. .
| Entry | R’ | X | Reaction Conditions | Zinc derivative | Yield (%) | α/β |
| 1 | Bn | Cl | 76 | α | ||
| 2 | Bn | Cl | 40 | α | ||
| 3 | Bn | Cl | NiCl2 10 mol% PyBOX 15 mol% DMI | 43 | α | |
| 4 | Bn | Cl | 65 | α | ||
| 5 | Bn | Cl | 61 | α | ||
| 6 | Bn | Cl | Ni(COD)2 10 mol% tBuTerpy 15 mol% DMF | IZnPh. LiCl | 28 | > 20/1 |
| 7 | Ac | Br | NiCl2 10 mol% PyBOX 15 mol% DMI | 75 | 8/1 | |
| 9 | Ac | Br | 70 | 8/1 |
By modifying the catalyst and the ligands, the same authors optimized the reaction conditions necessary to achieve a high yield of the α-C-phenyl mannoside 1 (Table 3) [83].
Optimal conditions for C-Ph-mannoside formation. .
| Entry | Conditions | Yield (%) | α/β |
| 1 | NiCl2.glyme PyBox, DMI | 40 | α |
| 2 | Ni(COD)2, PyBox, DMA | 30 | α |
| 3 | Ni(COD)2, Terpy, DMA | 56 | 6.6/1 |
| 4 | Ni(COD)2, tBuTerpy, DMA | 65 | 1.6/1 |
| 5 | Ni(COD)2, PyBox, DMF | 80 | 20/1 |
| 6 | Ni(COD)2, Terpy, DMF | 79 | 10/1 |
| 7 | Ni(COD)2, tBuTerpy, DMF | 76 | 2.9/1 |
Hurd and Holysz also reported phenyl addition to glycosyl bromide 1 in the presence of a cadmium derivative (Ph2Cd) [85]. The reaction conditions were relatively harsh in refluxing toluene with a low yield of the α-C-mannoside (29%).
Dialkylzinc derivatives have also been used for the formation of C-mannosides albeit in variable yield and moderate selectivity (Table 4) [86]. The authors postulated a double role of the zinc reagent with thioglycosides in particular because of the affinity of the zinc for the sulfur atom, as well as the nucleophilic property of the alkyl chain.
Dialkylzinc additions to thioglycosides. .
| R | T (°C) | Yield (%) | α/β |
| Me | 40 | 80 | 5.3/1 |
| Ph | 50 | 40 | 4.3/1 |
Kametani and co-workers reported the reaction of thioglycosides with azoderivatives in the presence of rhodium (II) acetate. When this reaction was applied to a thiomannoside derivative, the corresponding β-C-mannopyranoside structure was surprisingly obtained (Scheme 2) [87].
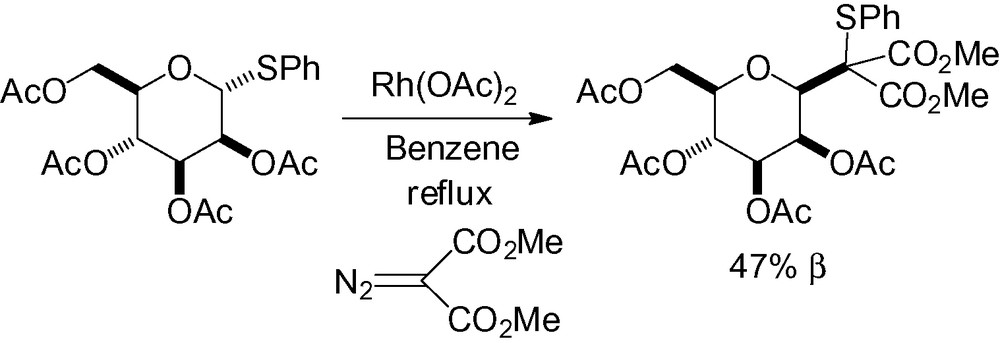
Nucleophilic opening of the Brigl-type epoxide 2 with a lithium reagent is another interesting way to introduce a side chain in the “anomeric” position (Table 5). This type of reaction has been successfully used for the synthesis of C-mannosyltryptophans [69,88–90]. Selective opening on the α-face is observed but is greatly dependent on the nitrogen protecting group.
Nucleophilic mannosyl epoxide opening. .
| Entry | R | X | Yield (%) | α/β |
| 1 | H | SO2Ph | 39 | 69/31 |
| 2 | H | Boc | 49 | 34/66 |
| 3 | Me | SO2Ph | 39 | 87/13 |
| 4 | Me | Boc | 56 | 17/83 |
| 5 | Et | SO2Ph | 38 | > 95/5 |
| 6 | CH2OTBS | Boc | 17 | 77/23 |
| 7 | CH2OTBS | SO2Ph | 50 | > 95/5 |
| 8 | CH2CH2OTBS | Boc | 50 | > 95/5 |
| 9 | SO2Ph | 63 | 95/5 | |
| 10 | SO2Ph | 18 | 55/45 | |
| 11 | SO2Ph | 18 | 56/44 |
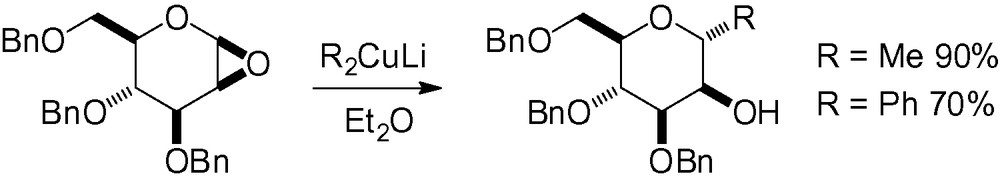
Cuprate derivatives have also proven to be highly selective (Scheme 3) [91,92].
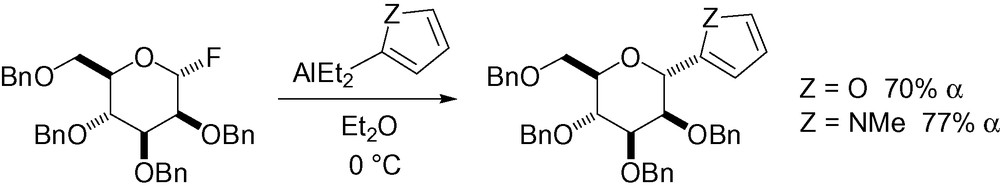
Proceeding through an oxonium intermediate, fluorinated carbohydrates can be activated in the presence of aluminum derivatives (Scheme 4) [93].
3.1.1.2 Access to C-mannosyl cyanide derivatives
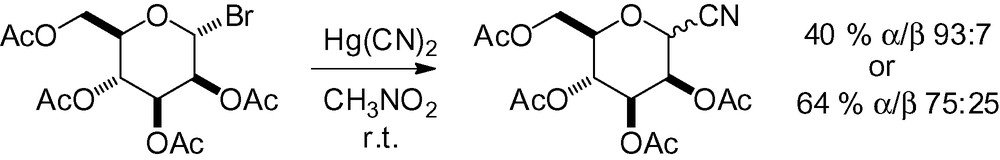
Another useful group that can be introduced in the anomeric position and further elaborated to a functionalized C-mannoside is the cyano group. A cyanide nucleophile can be introduced with various procedures. Early studies used Hg(CN)2 in nitromethane at room temperature with only moderate selectivity (Scheme 5) [94,95]. Low yields were observed due to the formation of a large quantity of the cyano “orthoester” (40%). This problem was resolved in a later publication by treatment of the crude reaction mixture with BF3·Et2O to give 67% yield (α/β 93:7) of the cyano derivative [96].
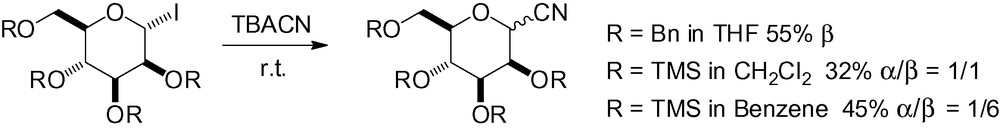
Glycosyl iodides have also been used in the preparation of mannosyl cyanides in the presence of tetrabutylammonium cyanide (Scheme 6). Selectivity was variable, and the choice of solvent important as the more apolar benzene clearly gave better results than methylene chloride [97,98].
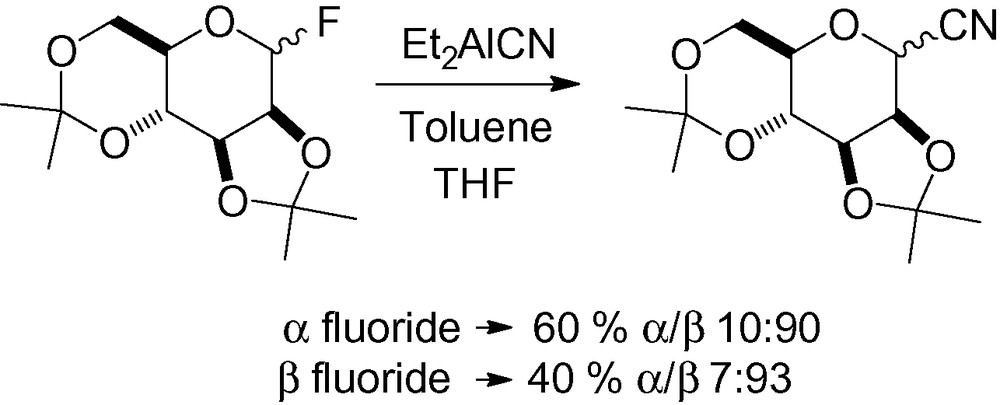
In isopropylidene protected carbohydrates, cyanation is possible with a poor-leaving group, like a fluorine derivative. The β-C-glycoside is the major reaction product starting from either α- or β-anomeric glycosyl fluorides, and is activated by the electrophilic character of the aluminum derivative (Scheme 7) [99].
α-d-mannose pentacetate, used as a mixture of anomeric diastereoisomers or in pure form, can be used as a carbohydrate donor to give the corresponding cyano C-glycosides [96,99]. This methodology has been extensively used in the synthesis of lectin inhibitors [52,57] and in studies on the synthesis of (−)-daucic acid [37,38] and the ring K of brevetoxin B [40].
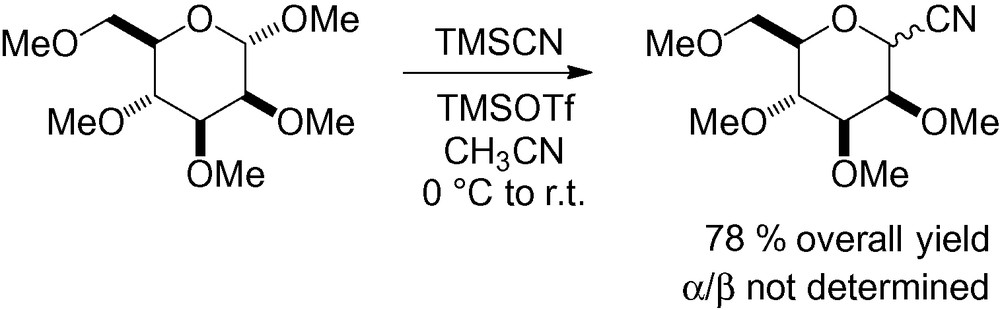
Permethylated α-d-mannopyranoside has also been used to prepare the corresponding cyano sugar (Scheme 8) [100]. The harsh conditions required for deprotection of the methyl ethers limit the usefulness of this reaction. In some cases, deprotection is unnecessary, as the methylated sugars have similar biological activities as the unprotected ones. The pentasaccharide idraparinux, for example, is more active then the hydroxyl derivative in the treatment and secondary prevention of thrombosis [101].
With a sulfide or phosphate in the anomeric position, Lewis acidic activation generating an oxonium moiety is required for introduction of the cyano group (Table 6) [102–104].
Cyano sugars from thioglycosides or anomeric phosphates. .
| R | Lewis acid | Yield (%) | α/β |
| SEt | MeOTf | 68 | 3/1 |
| BF3·Et2O | 78 | 2.2/1 | |
| TMSOTf | 81 | 1.5/1 |
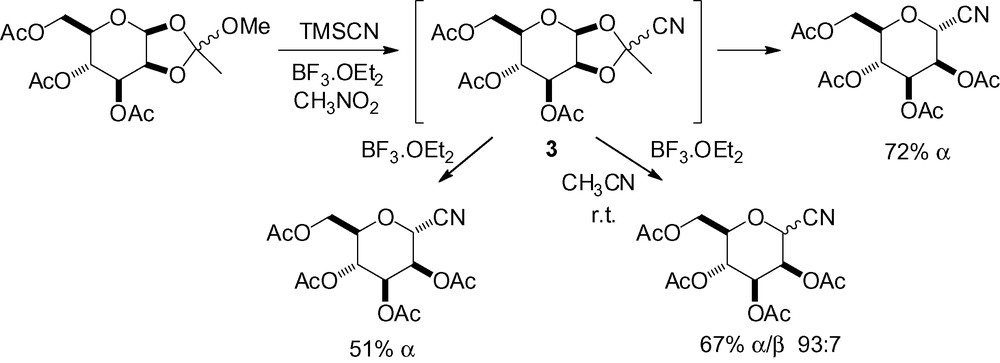
Orthoester species involving positions 1 and 2 have also been used for anomeric cyanation. The cyanoorthoester 3 was first proposed as an intermediate in the activation of the starting substrate. Subsequent isolation and reaction of 3 in the presence of Lewis acids gave the same product, however with lower yields and selectivity (Scheme 9) [62,96,105].
3.1.1.3 Access to alkynyl side chains
Nucleophilic substitution of anomeric acetates with an alkynyl group is a convenient method for a controlled two carbon homologation. This reaction can be used starting from the pure α anomer with fair to excellent yields and selectivity as well as from an anomeric mixture (Scheme 10), although, in this case, selectivity is often worse. Isobe and co-workers observed a drop in yield and selectivity with a benzyl protecting group in position 6 which they attributed to an arming/disarming effect [106,107]. This result was not confirmed by Dondoni et al. as good yield and selectivity were obtained with a benzyl protecting group in their work [108].
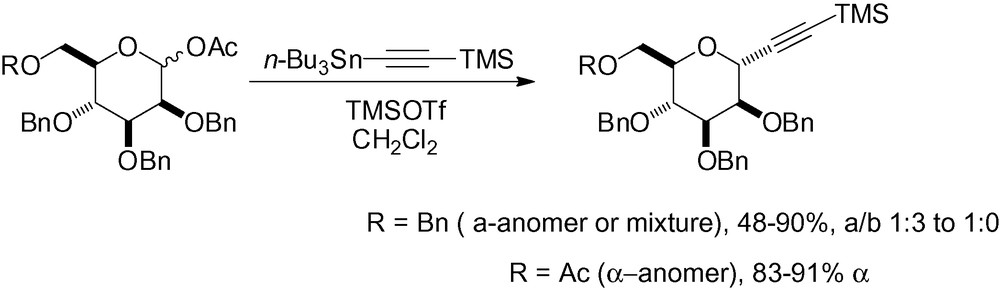
In the following examples, the palladium catalyzed addition of a functionalized alkynyl chain to a glycosyl bromide gave good α-selectivity (Scheme 11) [109].
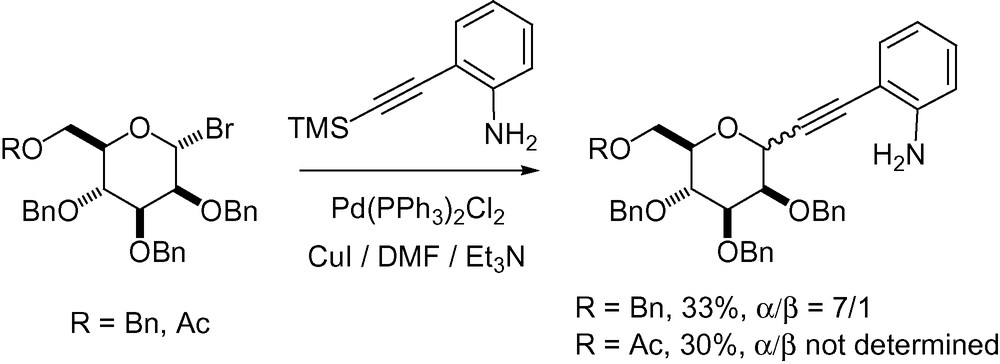
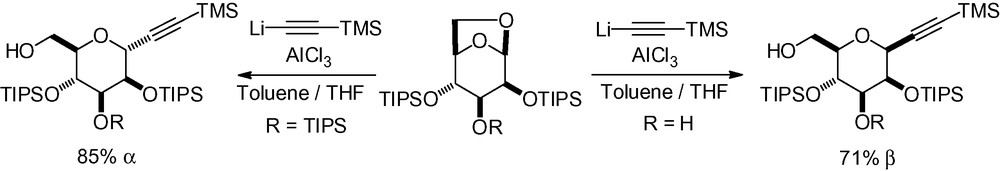
Indium-mediated alkynylation has also been reported starting from the protected mannose derivative 4 (Scheme 12) [110]. The α-anomer was obtained in good yield as the major reaction product.
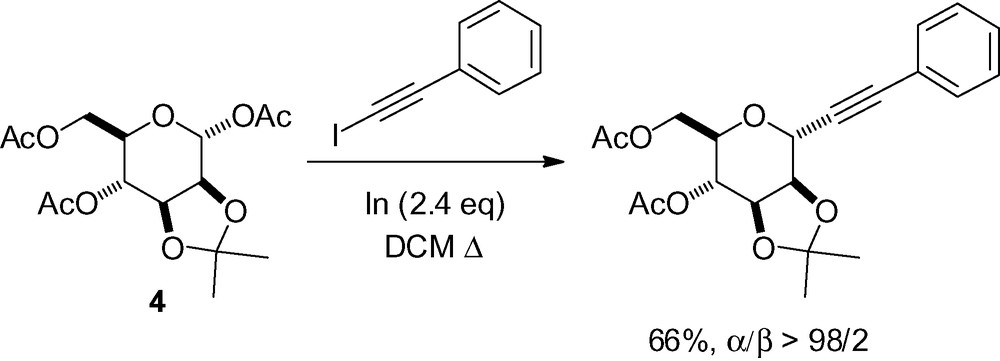
In the total synthesis of the protein phosphatase inhibitor okadaic acid, Ley et al. described a complex alkynylation reaction with the mannose derivative 5 (Scheme 13) [111]. This compound was first protected with a sulfide in the anomeric position which was later oxidized to the corresponding sulfone as an activating group. Trans-metallation of the deprotonated alkyne with dimethylaluminum chloride and reaction with 5 gave the desired C-mannoside in good yield and complete α-selectivity.
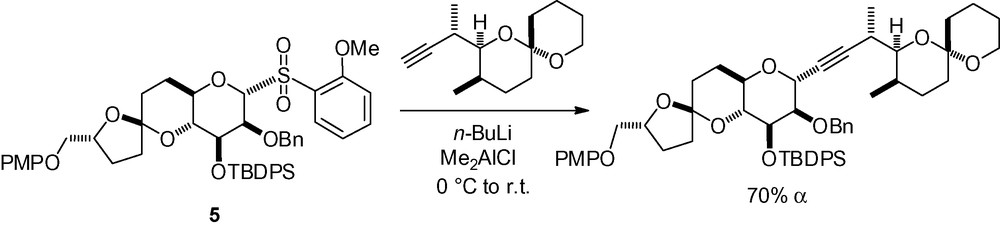
Alkynylation of a protected 1,6-anhydro-β-d-mannopyranose derivative was studied, and gave opposite selectivities depending on the protecting group pattern (Scheme 14) [112,113].
The role of the aluminum in these reactions is very important. In both cases, it acts as a Lewis acid and helps to cleave the oxygen bridge, and an oxocarbenium intermediate is formed. The presence of a free hydroxyl group in position 3 chelates the aluminum, thus promoting an intramolecular delivery of the alkynyl moiety to the upper face, giving the β-C-mannopyranoside (Fig. 8).
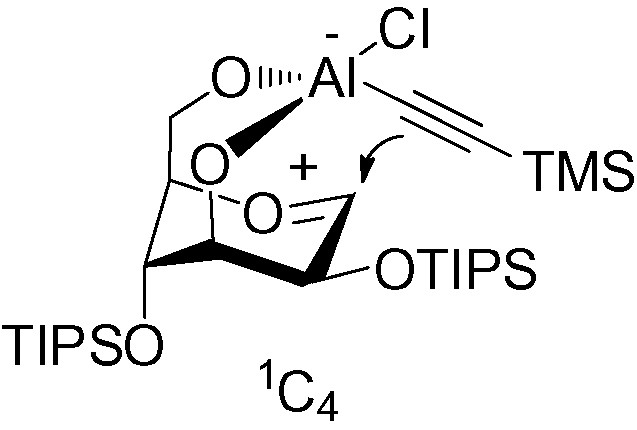
β-C-mannopyranoside.
With a protected hydroxyl group in position 3, chelation is no longer possible and the acetylene is delivered to the less hindered α-face.
3.1.1.4 Friedel-Crafts pathway
If the anomeric position has an appropriate leaving group and is further activated with Lewis acid to form an oxocarbenium cation, it is possible to envisage a Friedel-Crafts reaction with diverse electron-rich aromatic moieties. The introduction of indole species has been described with excellent α-selectivities using indium (III) chloride as an activator (Scheme 15) [114].
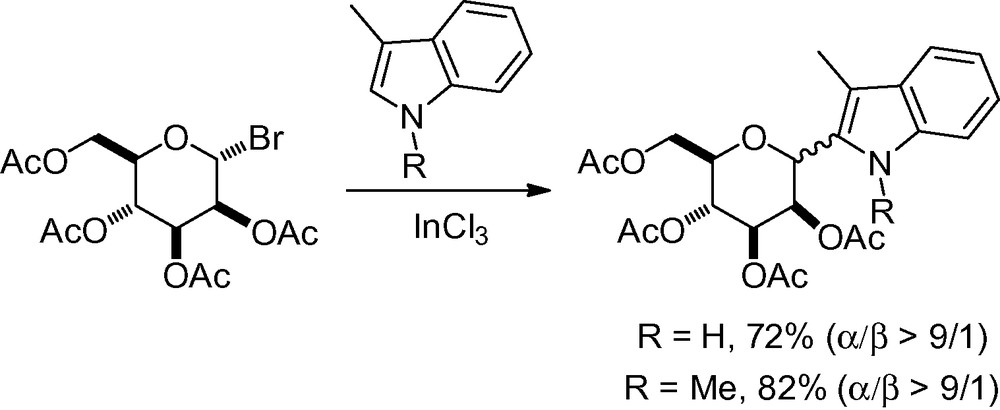
Two “Friedel-Crafts” examples have been studied with compound 6 (Table 7). Using more traditional glycosylation methods, the desired C-aryl-mannoside was obtained as an anomeric mixture in fair yield [115,116]. When the same substrate was coupled to an aryl moiety in the presence of an ionic liquid and HBF4, the reaction gave a good yield of the α-anomer [117].
Friedal-Crafts synthesis of C-arylmannosides. .
| H-Ar | Reaction conditions | Yield (%) | α/β |
| BF3·OEt2, 4Å MS CH2Cl2, −78 to 0 °C | 39 | 1/2.5 | |
| HBF4, C6mim[BF4] 60 °C | 82 | α |
Friedel-Crafts reactions have also been used with trichloroacetimidates in the presence of a Lewis acid, giving the α-anomers in good yield and selectivity (Scheme 16) [118,119].
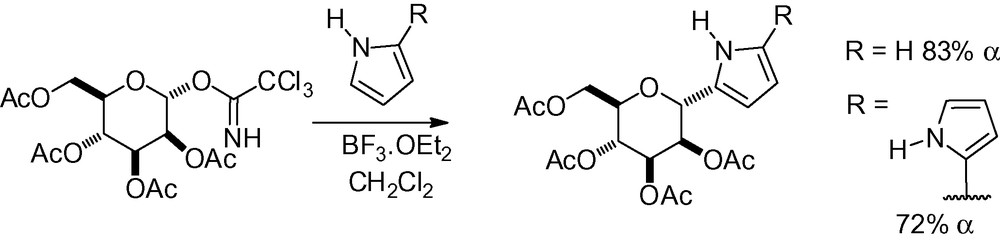
Trifluoroacetimidates have also been studied with different aryl acceptors (Table 8) [120]. The anomeric configuration of the products was assumed to be α in comparison to the known 1-(2,3,4,6-tetra-O-benzyl-α-d-mannopyranosyl)-naphthen-2-ol [121].
Trifluoroacetimidates for the synthesis of C-arylmannosides. .
| Ar | Yield (%) |
| 48 | |
| 76 | |
| 96 | |
| 74 | |
| Not purified | |
| Not purified |
2′-carboxybenzyl glycosides have proven to be useful glycosyl donors for the synthesis of C-arylmannosides as demonstrated in the following example (Table 9) [122].
Friedel-Crafts reaction with 2′-carboxybenzyl glycosides. .
| Ar | Yield (%) |
| 74 | |
| 69 | |
| 71 | |
| 52 |
Palmacci et al., Palmacci and Seeberger and Plante et al. have extensively studied the reaction of glycosyl phosphates and their application in intermolecular Fries-like O to C rearrangements. Upon coupling with phenolic acceptors, the initially formed O-glycoside rearranges to the C-glycoside with net retention of configuration at the anomeric center in the presence of a Lewis acid (Table 10) [5,121,123]. These reactions were stereo- and regioselective, yielding only the α-anomer.
Reaction of glycosyl phosphates. .
| Ar | Yield (%) |
| 79 | |
| 85 | |
| 82 |
Activation of a pentenyl ether in the presence of iodonium dicollidine perchlorate (IDCP), well known for O-glycosylation, followed by an intramolecular Friedel-Crafts attack gave the following tricyclic framework (Scheme 17) [124].
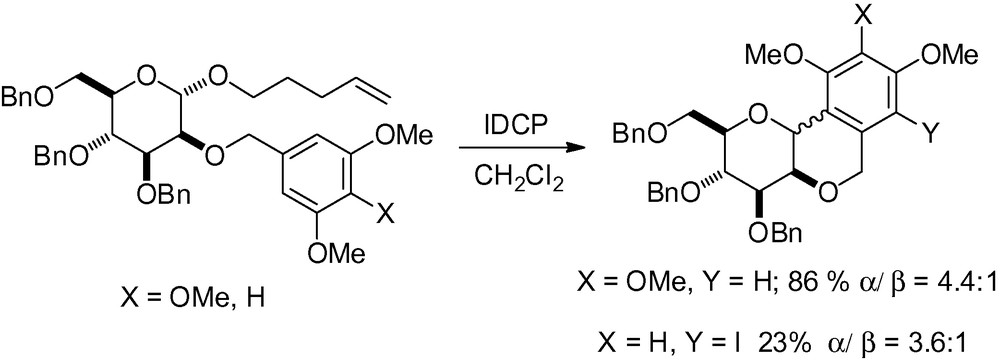
Surprisingly, the 1,2-trans adduct was the major reaction product even though steric and entropic factors should have favored the 1,2-cis derivative. Treatment of the 1,2-trans compound with BF3·OEt2 in methylene chloride gave good yields of the 1,2-cis derivative in both cases proving that the 1,2-trans derivative was the kinetic reaction product and the 1,2-cis the thermodynamically more favored one. Activation with BF3·OEt2 alone or in the presence of acid did little to improve the reaction yield.
When a very good leaving group, such as a triflate, is present in the anomeric position, it is possible to form a β-C-glycoside without any Lewis acid activation in an intramolecular Friedel-Crafts ring closure (Scheme 18) [125].
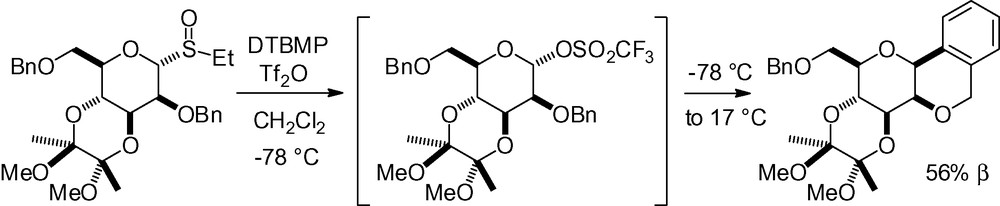
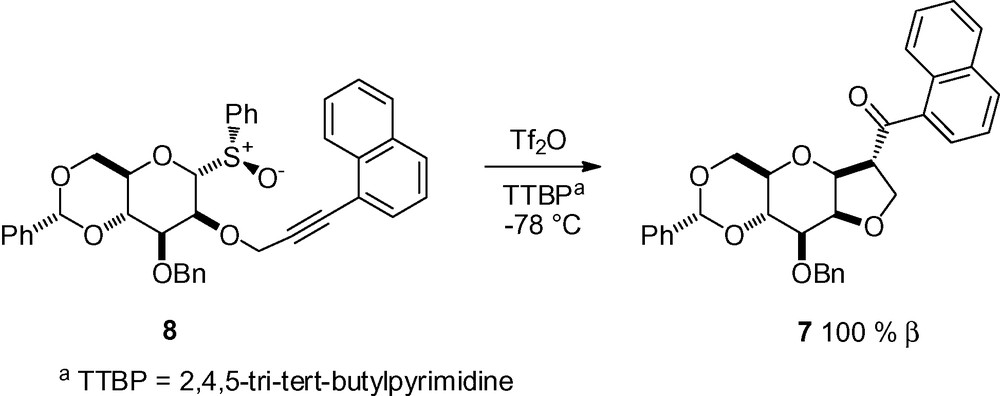
This same group observed the formation of ketone 7 in undetermined yield, as a major by-product of glycosylation reaction performed with sulfoxide 8 as the glycosyl donor. This product resulted from cyclization of the protecting group in position 2 onto the activated glycosyl donor with concomitant oxidation of the benzylic position (Scheme 19) [126]. No further explanation was given for this reaction.
3.1.1.5 Substitution with an allyl silane
Since the pioneering work of Lewis et al. [127], the Lewis acid catalyzed addition of allysilanes to diversely substituted glycopyranoside derivatives is one of the most popular ways to form C-glycosides and usually gives the corresponding α-C-glycoside with good selectivity. This reaction has been applied to numerous mannoside derivatives such as the peracetylated mannose 9 (Table 11) or activated 2,3,4,6-O-benzylmannosides (Table 12).
Allyl trimethylsilane addition to peracetylated mannosides. .
| Reaction conditions | Yield (%) | Ref. |
| BF3·OEt2 TMSOTf, CH3CN | > 76 (α/β = 8/1) | [57] |
| BF3·OEt2 TMSOTf, CH3CN | 68 (α/β = 19/1) | [128] |
| BF3·OEt2 TMSOTf, CH3CN | 55 (α/β = 6/1) | [60] |
| BF3·OEt2, CH3CN | 68 (α/β = 4/1) | [129] |
| TMSOTf, CH3CN | 39 (α/β = 4/1) | [130] |
| BF3·OEt2 TMSOTf, CH3CN | 75–77 (α/β = 6.8/1) | [40] |
Allyl trimethylsilane addition to 2,3,4,6-O-benzylmannosides. .
| Z | Reaction conditions | Yield (%) | Ref. |
| OMe | TMSOTf CH3CN | 87 (α/β > 15/1) | [67,131,132] |
| OMe | TMSI CH3CN | 68 α | [131,132] |
| Cl | TMSOTf | 76 α | [132] |
| TMSOTf, CH2Cl2 | 93 α | [121,123] | |
| TMSOTf/−78 °C CH2Cl2 | 78 (α/β = 1/1) | [103] | |
| BF3·OEt2 | 85 α | [133] | |
| BF3·OEt2 CH3CN | 79 (α/β > 10/1) | [127] | |
| Tf2O TBDMP | 55 (α/β = 3/1) | [122] | |
| SPh | BSP TTBP/−60 °C CH2Cl2 | 70 (α/β = 2/1) | [134] |
Protecting group variations have been described on the starting O-mannoside with an acetate or methoxy ether in the anomeric position. In each case, the corresponding α-allyl derivative is exclusively obtained [36,73,135]. Allylation has even been reported with a substrate possessing a bulky TBDMS group in position 2, and shows the compatibility of the reaction conditions with these functional groups [136]. The use of transient trimethylsilyl protection in a one-pot allylation reaction has also been described in an approach to glycosyltransferase inhibitors [137].
In the case of the constrained mannose derivative 10, β-selectivity was observed. The authors noted that this selectivity was highly dependent on the presence of the benzylidene acetal and was also the result of tight ion pairs in the transition state, as in the case of the corresponding O-β-mannosides (Scheme 20) [134].
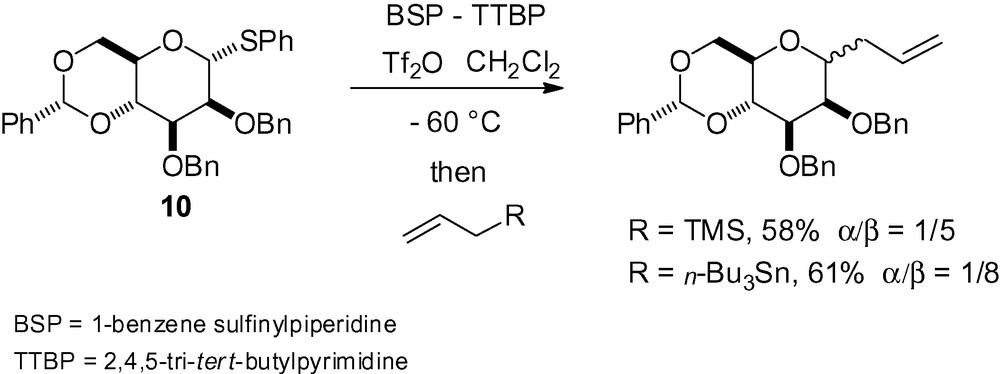
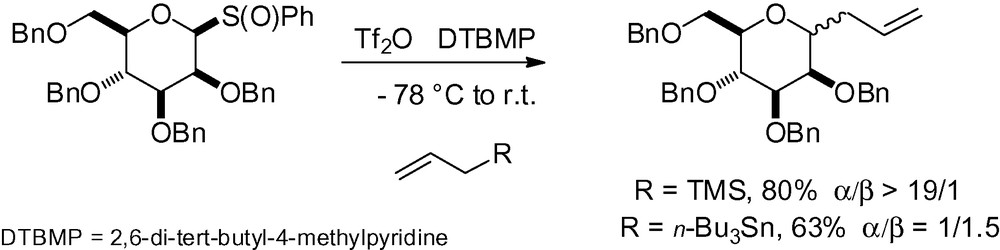
McGarvey et al. also published an extensive study dealing with the stereoselective synthesis of C-allyl glycosides from sulfoxide donors [138]. Interestingly, when the conformation of the mannose derivative is not restricted by a benzylidene acetal, selectivity is dependent on the reaction conditions and the source of the allyl donor (Scheme 21).
The use of substituted allylsilanes is also possible in order to introduce side chain diversity (Table 13).
Reaction of substituted allyltrimethylsilanes with mannose derivatives. .
| Z | R1 | R2 | Reaction Conditions | Yield (%) | Ref. |
| Cl | H | Br | TMSI, CH3CN | 65 α | [131,132] |
| OMe | H | Br | TMSI, CH3CN | 45 α | [131,132] |
| OMe | H | Me | TMSOTf, CH3CN | 81 (α/β = 8/1) | [131,132] |
| OAc | OAc | H | BF3·OEt2, ClCH2CH2Cl | 81 (α/β = 10/1) | [139] |
| OAc | OAc | Me | BF3·OEt2, ClCH2CH2Cl | 65 (α/β = 5/1) | [139] |
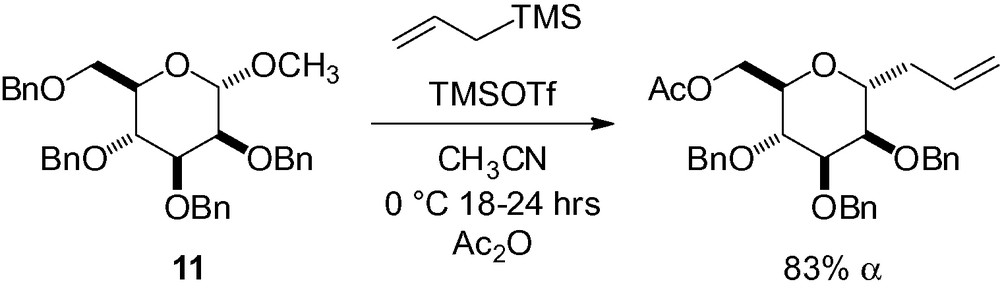
With the methyl 2,3,4,6-tetra-O-benzylmannopyranoside derivative 11, allylation in the anomeric position, selective protecting group removal in position 6, and subsequent acetylation is possible in good yield (Scheme 22) [50,53,140,141].
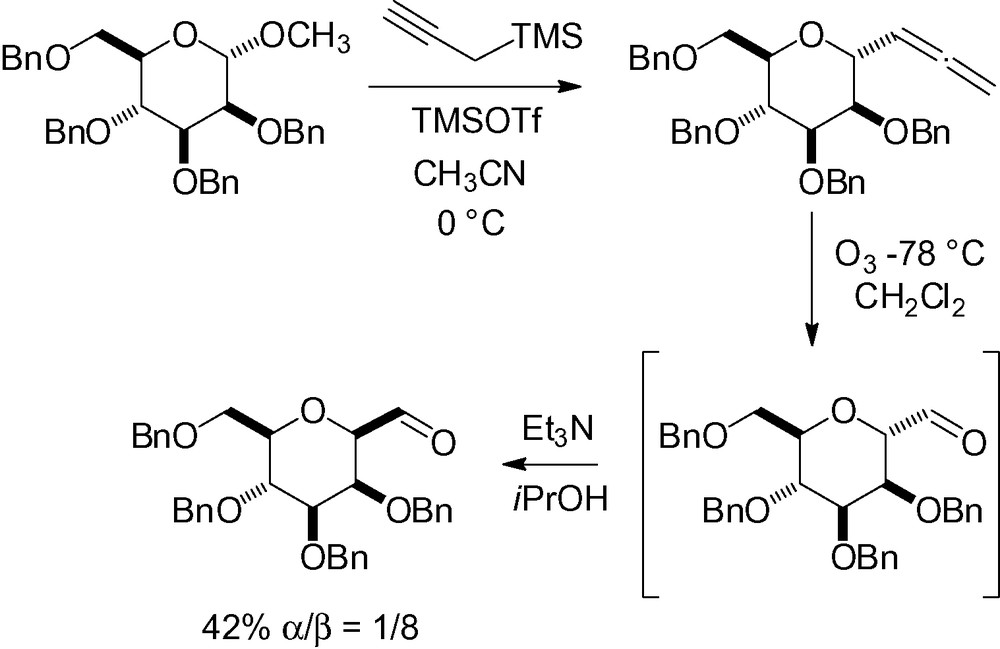
The extension of this methodology to propargyltrimethylsilane has proven to be very useful because the intermediate allene is stable and can be isolated [140]. Further transformation gives the corresponding anomeric aldehyde by simple ozonolysis at low temperature (Scheme 23) [142]. Epimerization of the α-linked C-mannoside aldehyde to the β-isomer is possible under mild basic reaction conditions.
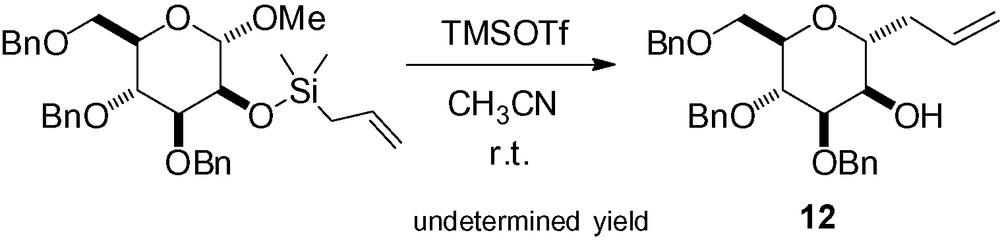
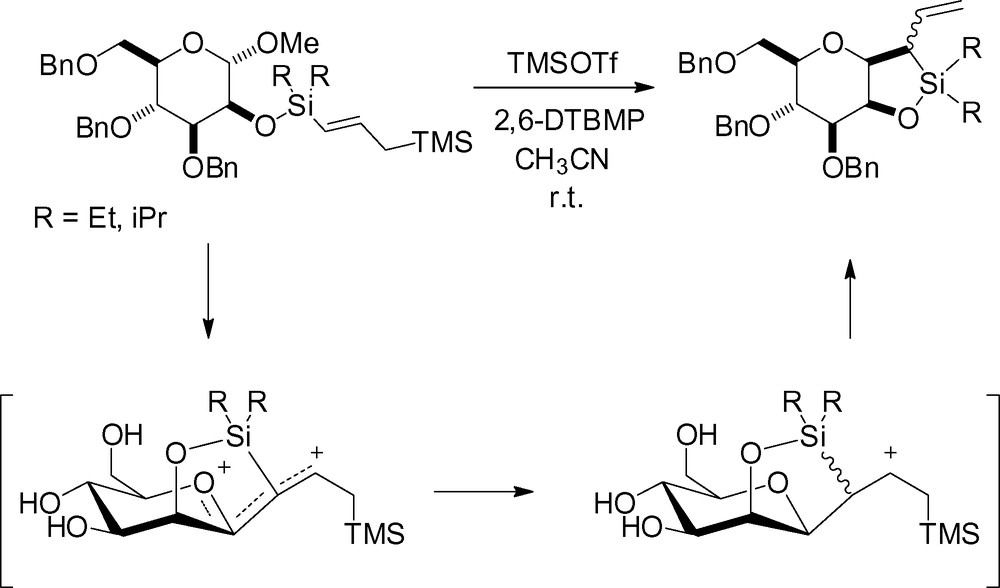
Beignet et al. have recently described an interesting strategy for the synthesis of β-C-allylmannosides. Their idea, based on the intramolecular aglycon delivery developed for the synthesis of β-O-mannosides, was to use a temporary silicon tether in position 2 to deliver the allyl group in a stereoselective manner. Interestingly, activation with BF3·OEt2, tin (IV) chloride or TMSOTf at –78 °C in CH2Cl2 resulted in cleavage of the silyl ether in the starting material. In acetonitrile, treatment with TMSOTf at r.t. gave the α-C-glycoside 12, resulting from cleavage of the tether followed by an intermolecular reaction (Scheme 24) [136].
The authors postulated that the necessary transition state for the desired transformation was unfavorable. Relocating the tether to the γ position of the allylsilane and subsequent reaction gave the desired cyclic β-C-mannoside derivatives albeit in undetermined yield (Scheme 25).
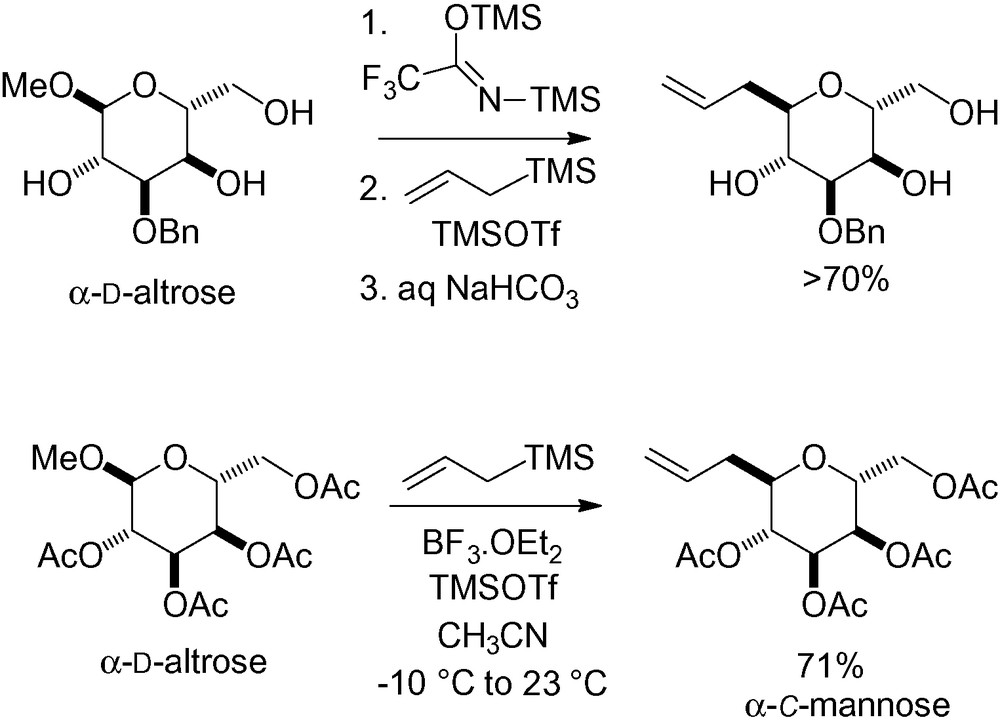
Another original way to obtain the d-C-mannose skeleton relies on the use of the analogous allylation reaction with d-altrose derivatives. Work on the anomeric carbon in this series is equivalent to a disguised study on the “C-5” of d-mannose (Scheme 26). This work was described in the synthesis of okadaic acid [143,144] and zooxanthellatoxin [39].
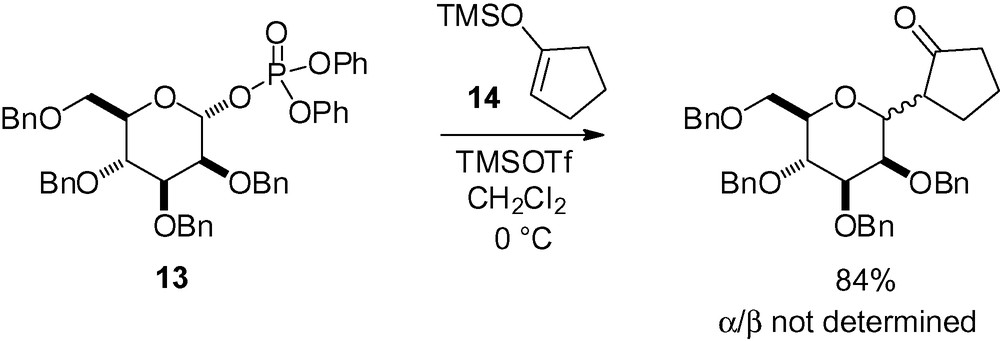
Enol silanes can also add to the anomeric position of protected d-mannosides to give keto derivatives after hydrolysis. For example, coupling of the phosphate derivative 13 to the cyclopentanone-derived trimethylsilylenol ether 14 gave an interesting C-mannosyl derivative as a mixture of diastereomers in 84% yield (Scheme 27) [121].
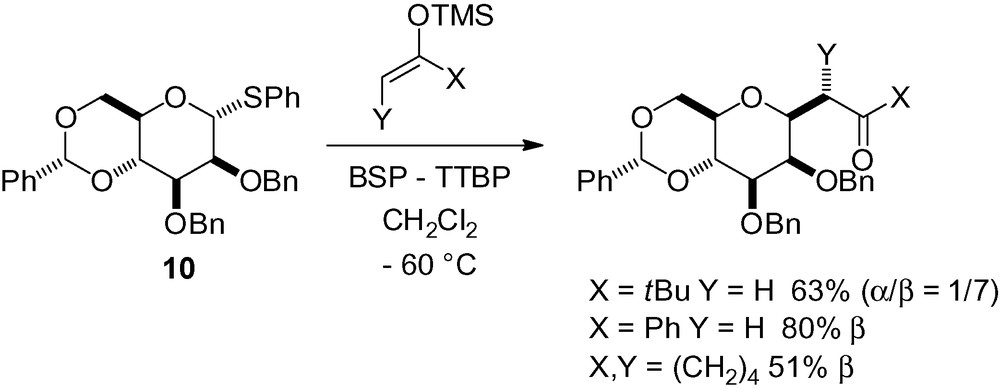
Along with the previously described allylsilane additions, Crich and Sharma also reported the reaction of several different silyl enol ethers with thiomannoside 10 (Scheme 28) [134]. The β-C-mannosides were obtained with good overall selectivity and yield.
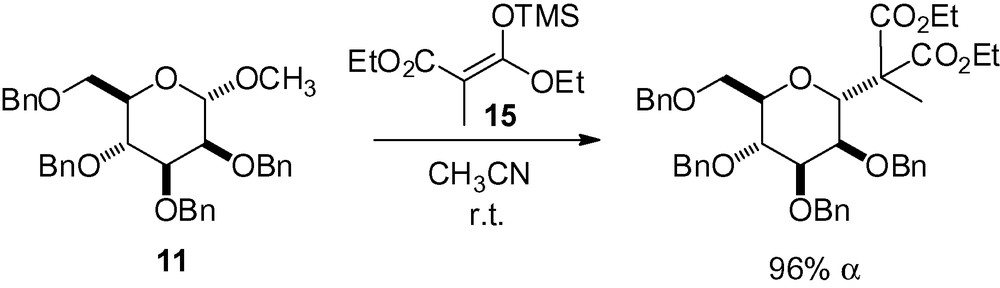
Addition of the stable silylketene acetal 15 to the simple mannoside derivative 11 was performed as part of the synthesis of E and P selectin antagonists (Scheme 29) [145]. Surprisingly, the same reaction with a less complex enol ether failed.
It is clear that the preparation of C-mannosides by coupling of an electrophilic anomeric carbon and a nucleophilic carbon is possible in a multitude of different ways. Both α- and β-selectivity is possible and depend on the substrate and the reaction conditions. It is, however, easier to synthesize α-d-C-mannosides in general.
3.1.2 Coupling between a radical anomeric carbon and an unsaturated moiety
In this category, two general types of reaction exist, one in which a radical is added to a CC unsaturated bond, and the other where addition occurs with a C-heteroatom unsaturated bond. It is important to note that the majority of the reactions developed from radical anomeric carbohydrates occur with α-selectivity (Fig. 9).
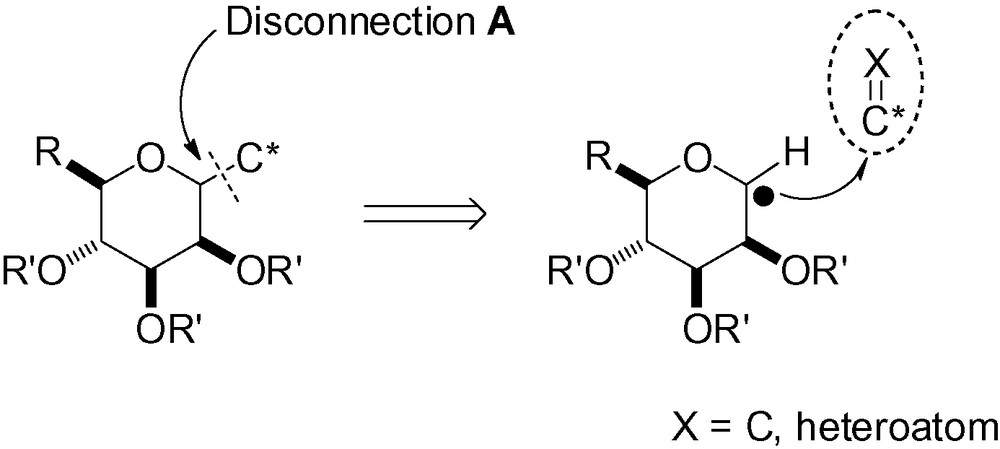
Disconnection A.
3.1.2.1 Reaction with alkenes and alkynes
A radical species in the anomeric position can be generated in different ways. This type of radical is electron-rich, and consequently, the CC unsaturated bond must be activated by an electron-withdrawing group in order to respect the nucleophilic/electrophilic radical effect thus allowing a good SOMO/LUMO overlap [77,146–148]. Glycosyl bromides are natural candidates for this type of reaction as the corresponding glycosyl radicals can be generated easily. Addition to methyl acrylate has been reported in varying yields, and depends on the activator and the solvent system used (Table 14).
Mannosyl radical addition to methyl acrylate. .
| Reaction Conditions | Yield (%) | Ref. |
| n-Bu3SnH, hν, Et2O | 45 α | [62] |
| n-Bu3SnH, AIBN, toluene | > 24 α | [149] |
| Ni(COD)2, Zn, DMA | 76 α | [150] |
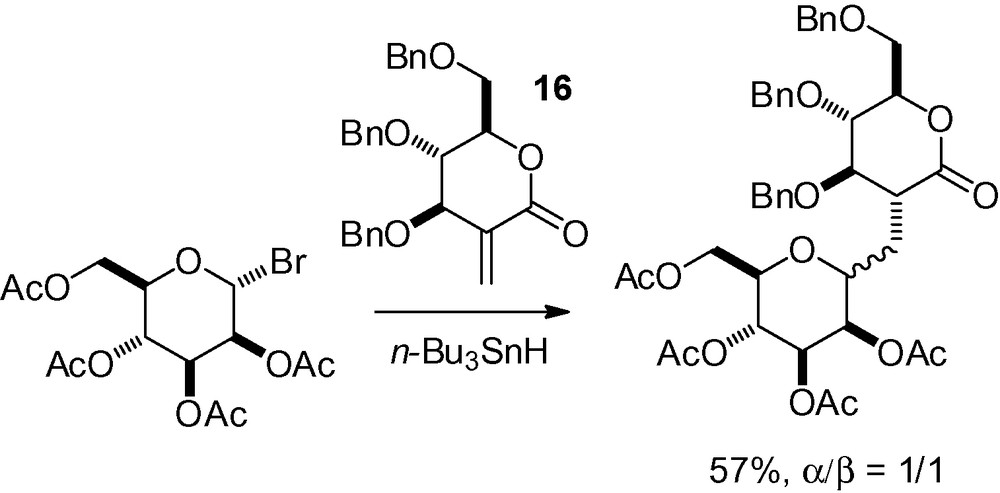
Other Michael additions are possible with these mannosyl radicals. Early on, Giese et al. applied this strategy to the synthesis of methylene bridged C-disaccharides [151]. In their work, reaction of the lactone 16 with 2,3,4,6-tetra-O-acetyl-d-mannopyranosyl bromide gave a 1/1 mixture of the desired compound (Scheme 30).
Radical addition to α,β-unsaturated ketones has proven to be effective, despite the possible minor side-reactions due to direct attack on the carbonyl function (Scheme 31) [51,58,59].
α,β-unsaturated phosphonates are good candidates for this radical coupling, giving an original access to Horner-Emmons reagents (Table 15).
Radical addition to α,β-unsaturated phosphonates. .
| Z | Reaction Conditions | Yield (%) | Ref. |
| H | n-Bu3SnCl, NaBH3CN AIBN, t-BuOH | 68 α | [152] |
| H | n-Bu3SnH, Et2O | 38 (α/β > 49/1) | [153] |
| COOMe | TTMSSa | 47 (α/β > 49/1) | [154] |
a TTMSS = tris(trimethylsilyl)silane.
Radical addition to acrylonitriles is also possible in the presence of different activators (Table 16).
Radical addition to acrylonitriles. .
| Reaction Conditions | alkene | Yield (%) | Ref. |
| n-Bu3SnH, hν, Et2O | 53–68 α | [62,155,156] | |
| n-Bu3SnH, AIBN, hν, Et2O | 34 α | [156] | |
| hν, THF | 53 α | [157] |
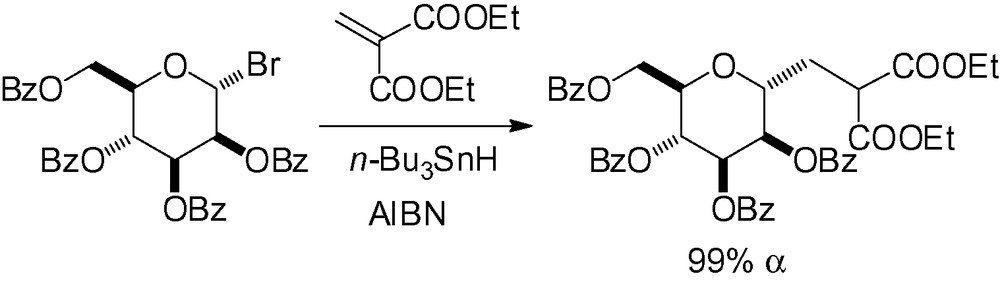
When the alkene is further activated with two electron-withdrawing groups, excellent yields can be obtained with a nucleophilic mannosyl radical (Scheme 32).
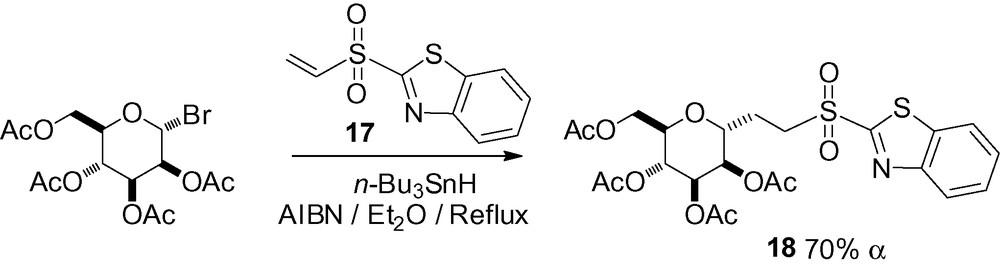
A last example of a Michael addition to the benzothiazolyl vinyl sulfone 17 gave the complex sulfone 18, a potential Julia-Kociensky reagent (Scheme 33) [158]
C-allylation of protected mannosyl bromides with allylic sulfides and sulfones has been reported using a procedure published in the galactose series [159], but no yields were given [34,160]
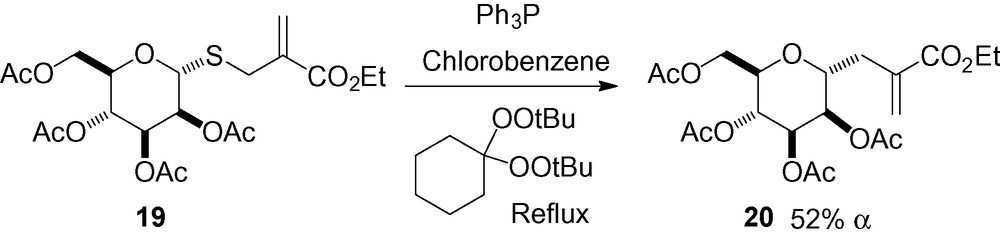
As part of a study of radical-chain desulfurisation of various α-(alkylthiomethyl) acrylates, the allylic sulfide derivative 19 was reacted with triphenylphosphine in the presence of a peroxide initiator (Scheme 34). Radical desulfurisation followed by allylic rearrangement of the reactive intermediate gave the C-mannoside 20 in moderate yield [161].
Glycosylcobaloximes, which are synthesized from the corresponding glycosyl bromides, undergo radical addition to various alkenes (Table 17). The ratio of addition (alkane) vs substitution (alkene) products was governed by the electron-withdrawing ability of the starting alkene [162].
Reaction of mannosylcobaloximes with alkenes. .
| Y | Z | Product | Yield (%) | |
| (alkane/alkene) | 15 °C (hν) | 80 °C (Δ) | ||
| H | CN | (> 98: 2) α | 51 | 75 |
| H | CO2CH3 | (15: 85) α | 27 | 40 |
| H | C6H5 | (< 2: 98) α | 65 | 47 |
| OEt | CN | (< 2: 98) α | 60 | – |
Readman et al. reported a radical based synthesis of C-glycosides from the corresponding bromides in the presence of nickel and manganese catalysts [163]. The deoxy sugar 21, which could be isolated, reacted with several different electron deficient radical acceptors to give the corresponding alkanes (Table 18).
Nickel catalyzed synthesis of C-mannosides. .
| X | Y | Yield (%) |
| CO2Me | H | 62 α |
| CO2Me | CH3 | 48 α |
| CO2n-Bu | H | 53 α |
| CN | H | 49 α |
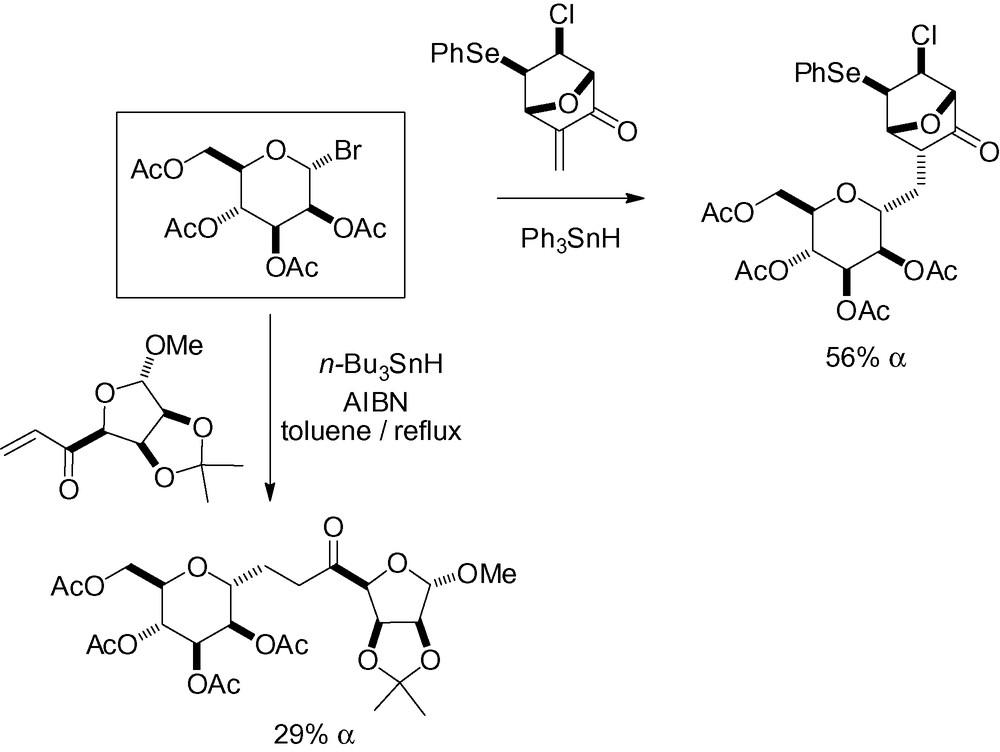
Anomeric radicals can also be generated from selenium derivatives [164,165]. When the olefin is attached in position 2, intramolecular radical cyclization occurs, trapping the less stable β-radical species, and giving a stereocontrolled bicyclic framework with excellent selectivity in certain cases (Table 19).
Intramolecular radical cyclization reactions with selenium derivatives. .
| R | Reaction Conditions | Yield (%) | Exo/endo |
| H | Benzene (0.1 M), Δ | 92 | 9/91 |
| H | Toluene, −30 °C, hν | 92 | 3/97 |
| CH3 | Benzene (0.1 M), Δ | 90 | 38/62 |
If an alkene is linked to the carbohydrate in position 2 via a temporary tether, α-glucose or β-d-mannose derivatives can be synthesized depending on the tether length [166]. In this example, a silica tether was used and the alcohols were obtained after cylization and Tamao oxidation. The ratio of products was influenced by the reaction solvent, temperature, and reagent addition (Table 20).
Synthesis of C-mannosides with vinylsilyl tethers. .
| R | Substrate concentration (M) | Temperature (°C) | Yield (%) | Product ratio (1° OH:2° OH:reduced SM) |
| CH3 | 0.01 (benzene) | 80 | 90 | 6:74:20 |
| CH3 | 0.002 (chlorobenzene) | 80 | 75 | 36:62:2 |
| CH3 | 0.002 (chlorobenzene) | 130 | 53 | 62:38:0 |
| Ph | 0.01 (benzene) | 80 | 67 | 22:64:14 |
| Ph | 0.002 (chlorobenzene) | 80 | 63 | 57:43:0 |
| Ph | 0.002 (chlorobenzene) | 130 | 74 | 86:14:0 |
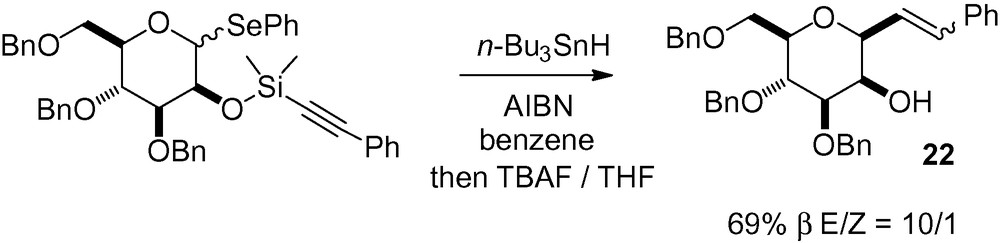
Extending this methodology to an alkynylsilyl derivative gave the β-C-mannoside 22 in good yield (Scheme 35) [167,168].
Vauzeilles et al. described an elegant synthesis of a C-disaccharide using the mannose derivative 23 (Scheme 36) [169]. In this case, the major reaction product was the α-C-mannoside, in contrast to the examples cited previously.
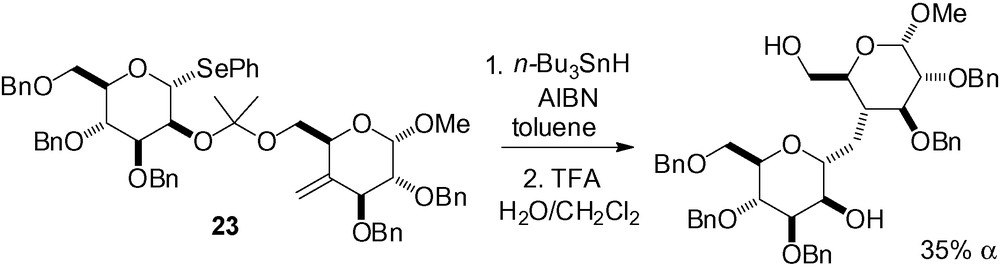
The generation of an anomeric radical from a glycosyl aryl sulfone is possible by treatment with SmI2 in the presence of a polar aprotic solvent. This methodology, applied with a silicon tether approach in position 2, gave access to 1,2-cis glycosides without the use of tributyltin hydride [170,171]. Analogous results were achieved as compared to the selenium series. A pyridine on the anomeric sulfone was essential for good yields as this increased the lifetime of the radical, and thus improved cyclization efficiency (Table 21).
Samarium iodide induced C-mannoside formation. .
| Ar | R | R′ | Yield (%) | E/Z |
| Ph | CHCH2 | Et | 37 β | – |
| Ph | CCPh | CHCHPh | 27 β | E |
| 2-Pyr | CHCH2 | Et | 80 β | – |
| 2-Pyr | CCPh | CHCHPh | 64 β | 10/1 |
| 2-Pyr | CCTMS | CHCHTMS | 61 β | 10/1 |
| 2-Pyr | CCnC6H13 | CHCHn-C6H13 | 25 β | E |
3.1.2.2 Reaction with carbonyls and imines
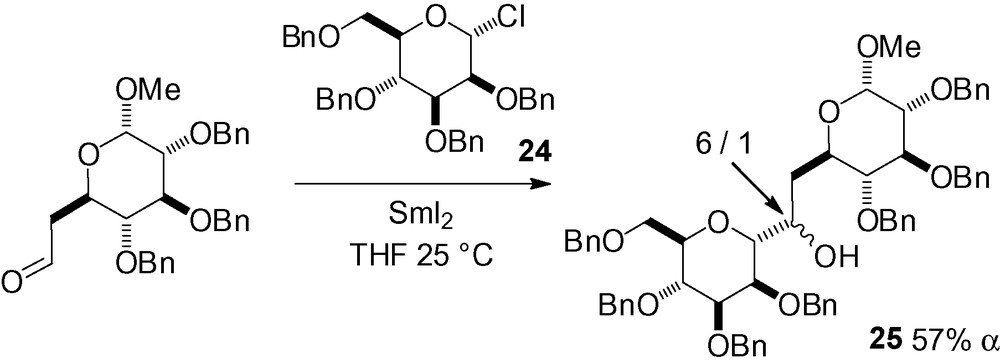
Carbohydrate-derived radicals are also able to react with unsaturated C-heteroatom multiple bonds. Samarium iodide has proven to be the reagent of choice in mannosyl addition to aldehydes and ketones as demonstrated by the large number of examples in the literature. In a first example, the glycosyl chloride 24 reacted with SmI2 and a carbohydrate-derived aldehyde to cleanly give the α-C-mannopyranoside 25 as a mixture of diastereoisomers at the newly created stereocenter (Scheme 37) [172].
Glycosyl iodides can also be used in a radical process with ketones or aldehydes. The unstable iodide is generated in situ with trimethylsilyl protecting groups or with more conventional benzyl groups (Table 22). Reaction then takes place in the presence of samarium iodide and a carbonyl compound to give the α-C-glycoside exclusively in good yield [173].
SmI2-induced coupling of mannopyranosyl iodides with carbonyl compounds. .
| Protecting groups | Carbonyl compound | Yield (%) |
| TMS | Cyclohexanone | 85 α |
| TMS | Pentan-3-one | 58 α |
| TMS | Isobutyraldehyde | 71 α |
| TMS | n-octanal | 72 α |
| Bn, Ac | Cyclohexanone | 68 α |
| Bn, TMS | Cyclohexanone | 85 α |
Mannosyl phosphates also generate radical species in the presence of SmI2 [5,174]. These radicals smoothly add to ketones and aldehydes (Table 23). Addition to carbon dioxide gave the corresponding carboxylic acid in the anomeric position in 56% yield.
SmI2-induced coupling of mannopyranosyl phosphates with carbonyl compounds. .
| Z | Carbonyl compound | Yield (%) |
| Bn | Cyclopentanone | 81 α |
| Ph | Cyclopentanone | 85 α |
| Bn | Acetone | 72 α |
| Ph | Acetone | 74 α |
| Bn | Isobutyraldehyde | 68 α (11.7/1 ratio of epimers) |
| Ph | Isobutyraldehyde | 68 α (10.2/1 ratio of epimers) |
| Ph | CO2 | 56 α |
The use of samarium iodide and glycosyl sulfones for the stereoselective synthesis of 1,2-trans C-glycosides has been extensively studied by Skrydstrup et al., Mazéas et al. and Jarreton et al. [75,175,176]. The reactive radical intermediates, which add efficiently to alkenes and alkynes, can also add to aldehydes and ketones, even complex ones, because of the mild reaction conditions. A series of mannosyl sulfones were synthesized and the hydroxyl protecting group in position 2 was varied in order to determine which one gave the best results (Table 24) [175,176].
Protecting group variation in the samarium iodide promoted addition to cyclohexanone. .
| R | Yield (%) |
| Bn | 82 α |
| H | 13 (β) |
| Me | 78 α |
| Ac | 0 |
| COOBn | 0 |
| CONHn-Pr | 0 |
| MEM | 45 α |
| THP | 56 α |
| TMS | 86 α |
| TBDMS | 80 α |
It is clear that protection of the hydroxyl group is necessary, and that carbonyl functions are not tolerated. The best results were obtained in the case of a benzyl group or a silyl derivative (TMS, TBDMS). To further extend the scope of the reaction, a second study was then performed with the TMS and TBDMS derivatives and their addition to a variety of different carbonyl derivatives (Table 25) [75,175,176].
Synthesis of α-C-mannosides from carbonyl compounds. .
| R | Carbonyl compound | Yield (%) |
| TMSa | Cyclohexanone | 86 α |
| TMSa | 3-pentanone | 80 α |
| TMSa | Isobutyraldehyde | 77 α (13:2)b |
| TMSa | Octanal | 82 α (9:2)b |
| TMSa | Benzaldehyde | 10 α (2:1)b |
| TBDMS | Cyclohexanone | 80 α |
| TBDMS | Cyclohexanecarboxaldehyde | 82 α (4:1)b |
| TMDMS | Nonanal | 71 α (5:1)b |
a RH in the final product.
b Ratio of diastereomers
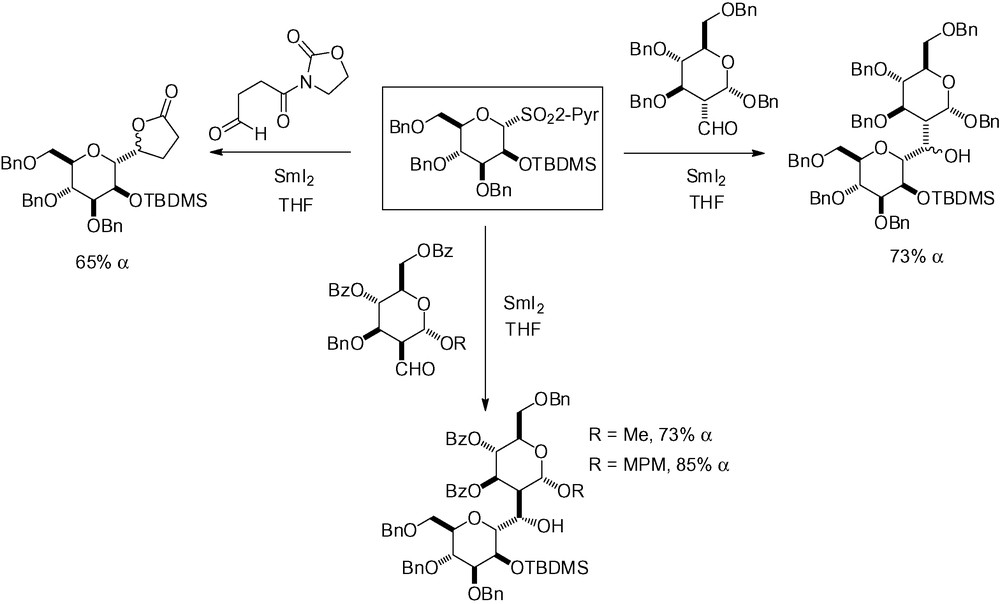
The choice of a TMS or a TBDMS protecting group in position 2 depends on whether or not the hydroxyl group needs to remain protected after the addition step as the TMS group is eliminated during the aqueous work-up. The greater stability of the TBDMS group has made it popular in reactions with more complex aldehydes, in particular for the synthesis of C-disaccharides (Scheme 38) [175,177–179].
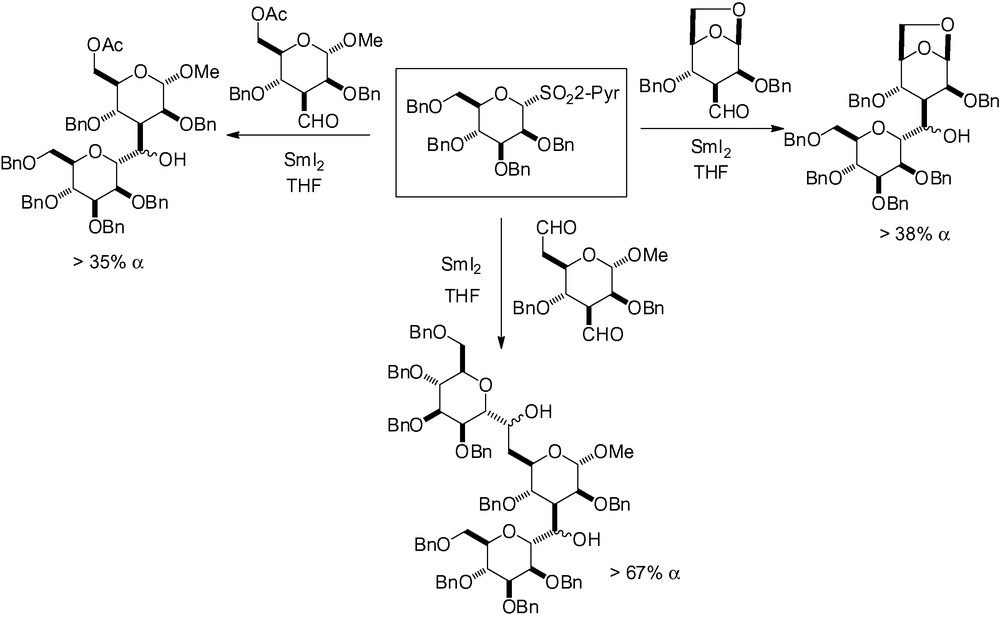
Benzyl protection in position 2 has also been shown to be efficient when used with complex aldehydes. Mikkelsen et al. and Krintel et al. have successfully used this methodology for the synthesis of the C-trisaccharide analogue of the core region α-d-man-(1→3)-[α-d-man-(1→6)]-d-man of the asparagine-linked oligosaccharides for the assembly of the three monosaccharide units in a single step (Scheme 39) [45,180].
As seen in Scheme 40, the use of catalytic nickel(II) iodide in the reductive samariation reactions can substantially increase reaction yields especially in the gluco- and galactopyranosyl series [181,182].
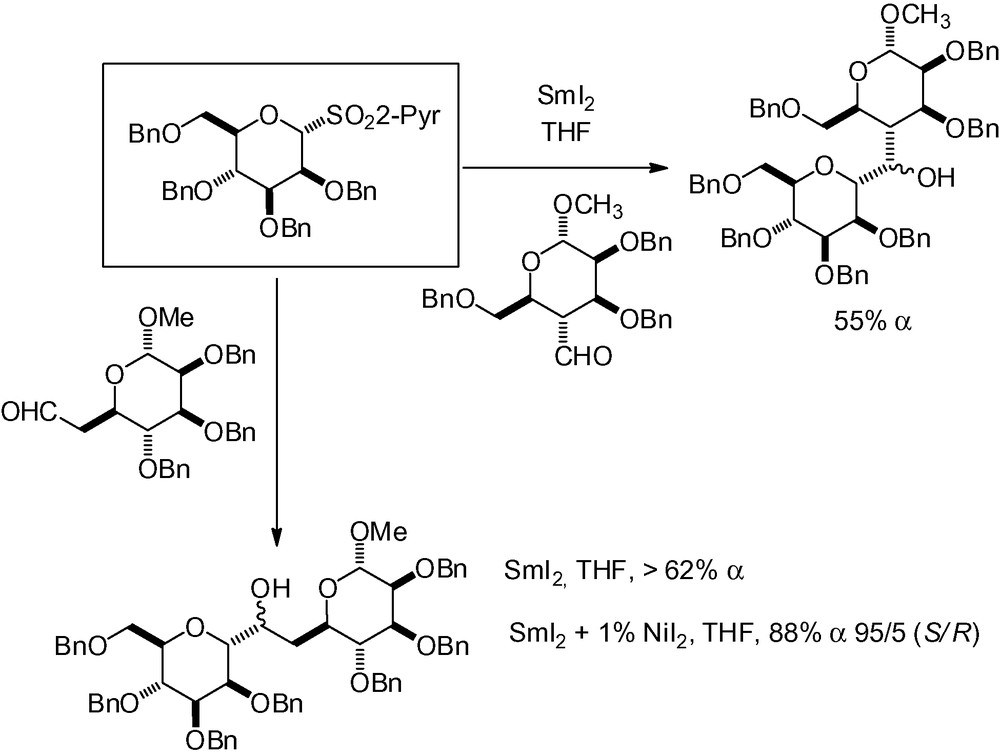
A two-step/one-pot dimethyl dioxirane oxidation-SmI2 reduction sequence has been reported with different glycals [183]. Use of this sequence with the “mannosyl” derivative 26 and addition to acetone and isobutyraldehyde gave the corresponding α-C-mannopyranosides as the major reaction products with a free OH in position 2 (Scheme 41).
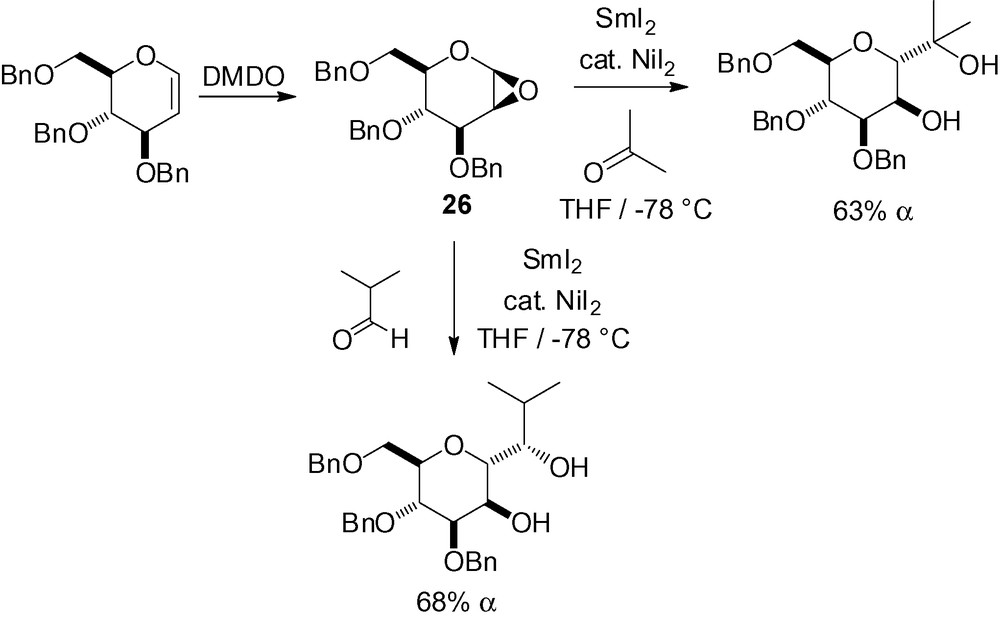
Radical addition to CN double bonds are not as common, and only two examples have been described in the literature. In the first one, intramolecular addition to an imine gave compound 27 in ≈ 45% yield (Scheme 42) [184].
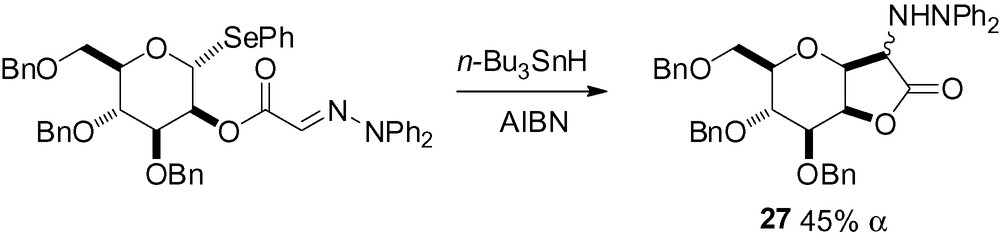
In the second, addition of an anomeric mannosyl radical to t-butylisocyanide in the presence of TTMSS gave the corresponding α-nitrile in 40% yield [185].
3.1.3 Coupling between a nucleophilic anomeric carbon and an electrophilic carbon
To the best of our knowledge, only one example of this kind has been reported in the literature. This is not surprising because the generation of a carbanion in the anomeric position logically results in an elimination reaction which forms a glycal (Scheme 43). This side reaction was sometimes observed in radical addition when samarium species were used.
If the hydroxyl group in position 2 remains unprotected, glycosyl phenyl sulfoxides can react, through a phenylsulfinyl–lithium exchange, to generate an anomeric carbanion [186]. Subsequent reaction with a carbon electrophile is then possible (Scheme 44). The reaction was stereoselective and proceeded with retention of the configuration at the anomeric center.
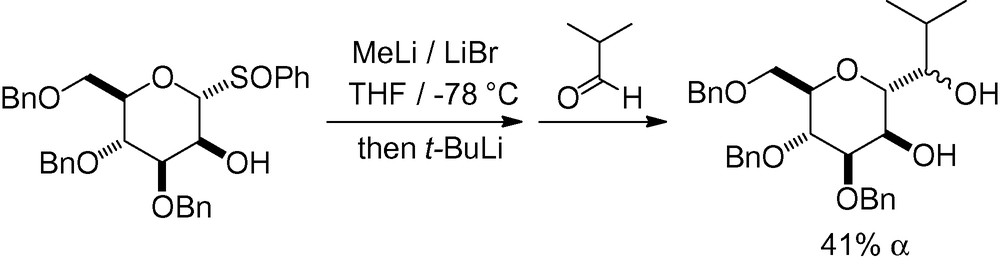
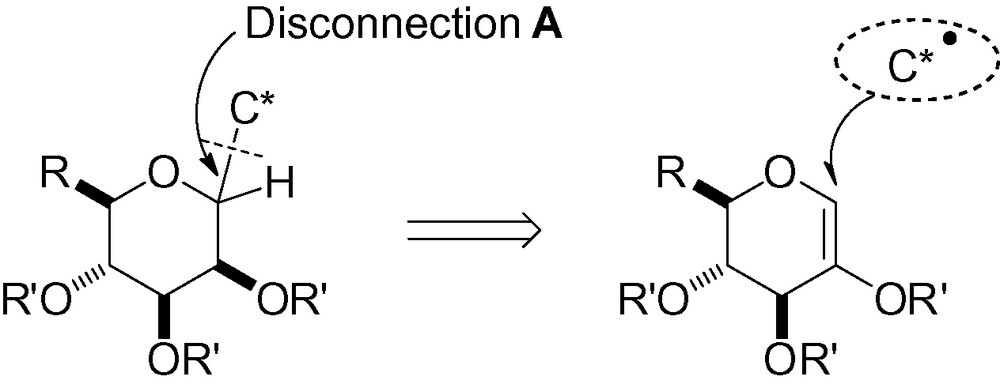
3.1.4 Coupling between an unsaturated carbohydrate and a radical carbon
Another little used approach is the photochemical generation of a carbon radical, followed by addition to a 1,2-unsaturated carbohydrate, namely a glycal (Scheme 45).
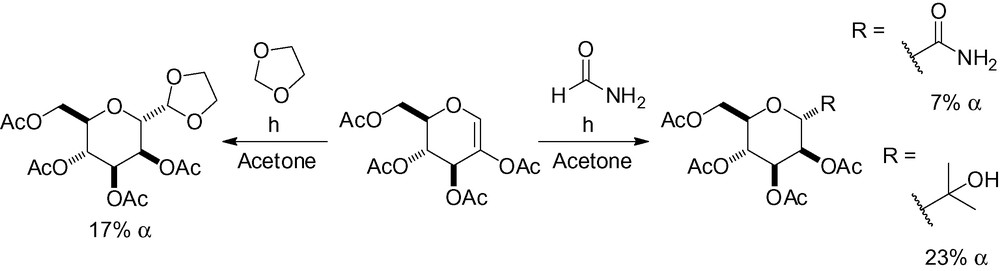
This methodology is not very selective and suffers from low yields (Scheme 46) [187–189]. d-C-glucopyranosides are often the major reaction products because of the reduced steric hindrance in the transition state.
3.1.5 Miscellaneous
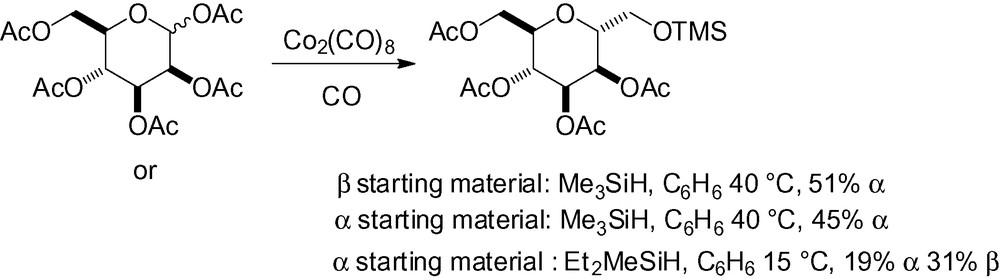
This last example is clearly not related to any of the general chapters, and C-glycoside formation results from a CO2(CO)8-catalyzed reaction with hydrosilanes and carbon monoxide (Scheme 47). The reaction mechanism probably involves a cobalt complex activation with carbon monoxide insertion promoted in the outer sphere of the cobalt. The choice of the reducing agent proved to be fundamental in order to control α/β stereoselectivity.
3.2 Disconnection B
3.2.1 Nucleophilic reduction of an anomeric ketal with a hydride donor
As largely illustrated in the text, nucleophilic attack in the d-mannose series occurs preferentially from the α-face, and α-C-mannopyranosides are almost exclusively the major diastereomers obtained. Consequently, if the desired carbon side chain is already in the anomeric position along with the anomeric oxygen group, hydride attack should prefer the α-face, following the same principle. Reduction of an anomeric ketal would thus lead to a β-C-mannopyranoside (Scheme 48).
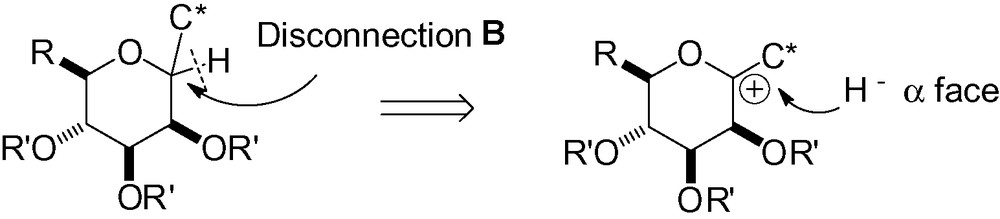
Inspired by the seminal work of Lewis et al. [127], Terauchi et al. effectively applied this strategy to the synthesis of β-C-glycosides based on conformational restriction [190]. In the case of unrestricted mannosides, low selectivity was observed except when RPh (Scheme 49).
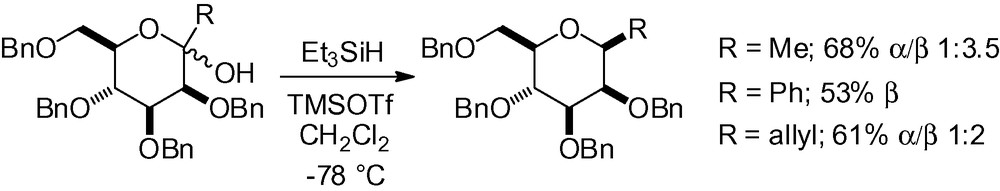
It was hypothesized that this came from the equilibrium between different conformations. When the substrates were conformationally restricted in the 4C1-chair form by a 3,4-O-cyclic diketal or a 4,6-O-benzylidene protecting group, excellent β-selectivity was observed (Scheme 50).
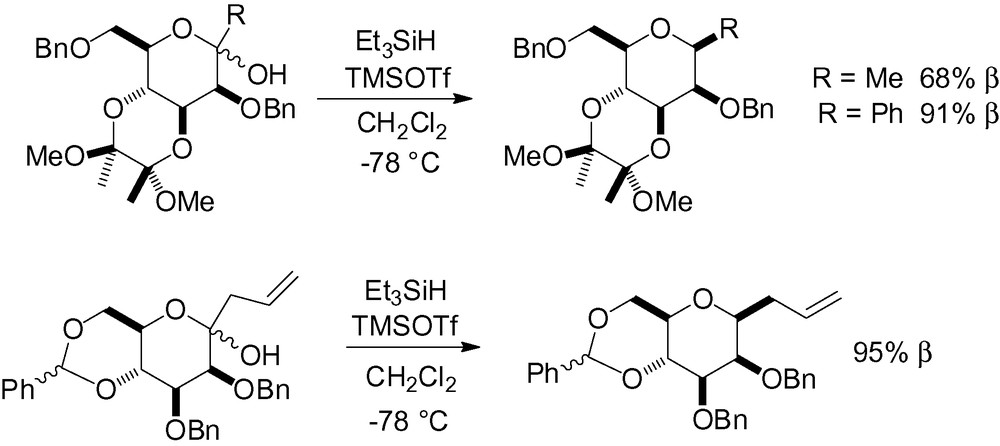
Incorporation of an allyl side chain is very attractive, because it can be subsequently cleaved to generate a functionalized aldehyde. This reaction has been reported in the literature with different Lewis acids and gives an allyl mannoside in 61–95% yield with selectivities ranging from 1:1 (α/β) to 100% β [67,127,190].
An acetate protecting group in position 2 is possible, but a low yield and poor selectivity was observed [136] (Scheme 51).
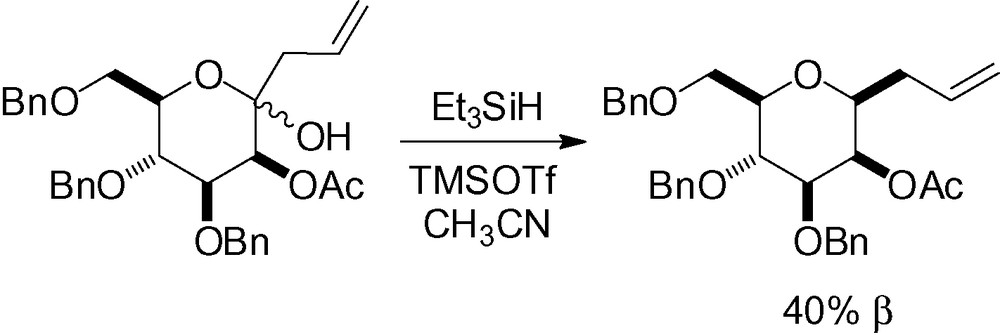
In the presence of an alkyne side chain, stereoselectivity and efficiency have proven to be difficult to reproduce [74,191]. The use of (TMS)3SiH as a reducing agent solved this problem giving good yields and excellent stereoselectivity. This was possible because of a less reactive reagent, and a later transition state [74] (Table 26).
Reduction of ketals with silanes. .
| R | T (°C) | Yield (%) | α/β |
| Et | −40 | 56–77 | 1/2 to 2/5 |
| iPr | 0 | 67 | 1/99 |
| TMS | 0 | 76 | β |
Reduction is also possible when the starting substrate contains an ester function (Scheme 52) [127].
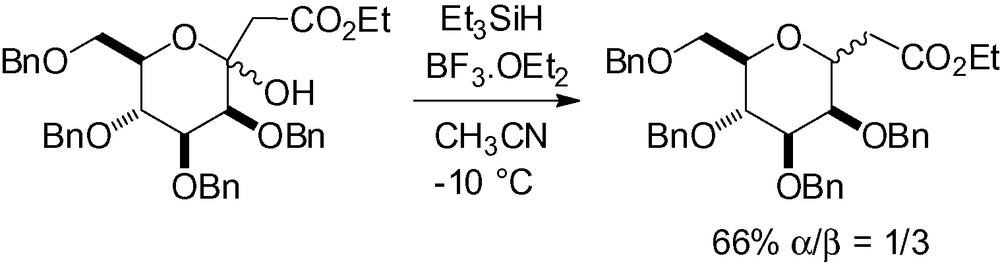
The introduction of a thiazole in the anomeric position of a mannosyl lactone, followed by deoxygenation of the resulting ketal to give the corresponding β-C-mannopyranoside was first reported by Dondoni and Scherrmann in 1994 (Scheme 53) [192].
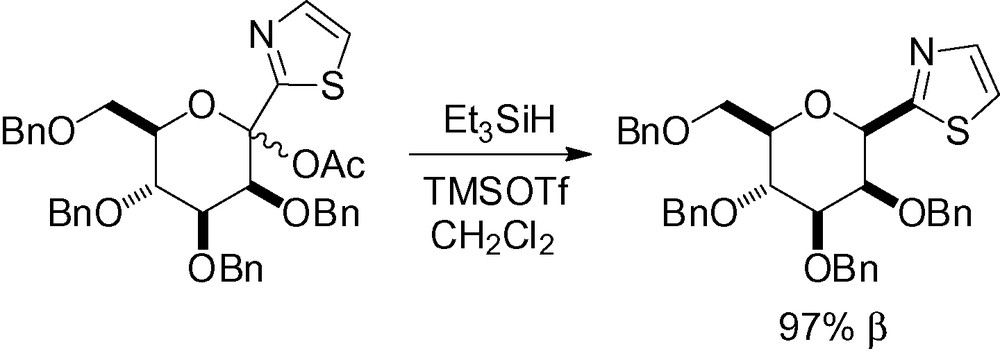
Subsequent unmasking of the formyl group then gives a valuable synthon for numerous chemical transformations [193–197]. When deoxygenation was performed with SmI2 in the presence of ethylene glycol, 75% of the α product was obtained after chromatographic purification [198]. The same methodology was used with 2-lithiobenzothiazole followed by deoxygenation to give the β-C-mannopyranoside in good yield [199].
In a study related to the synthesis of C-mannosyltryptophan, this transformation gave a surprisingly low yield, presumably due to the equilibrium with the 1C4 conformation [69] (Scheme 54).
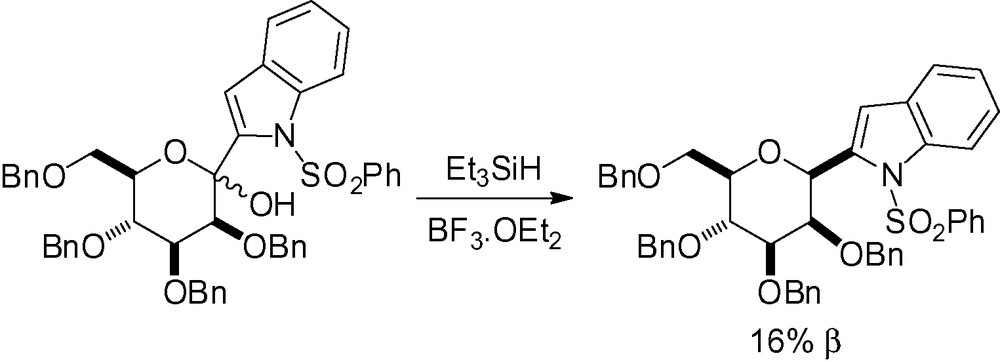
This strategy is so powerful that even highly functionalized compounds can be reduced in good yields [200,201] (Scheme 55). Surprisingly, in the second case, only the α-C-mannopyranoside was isolated [201].
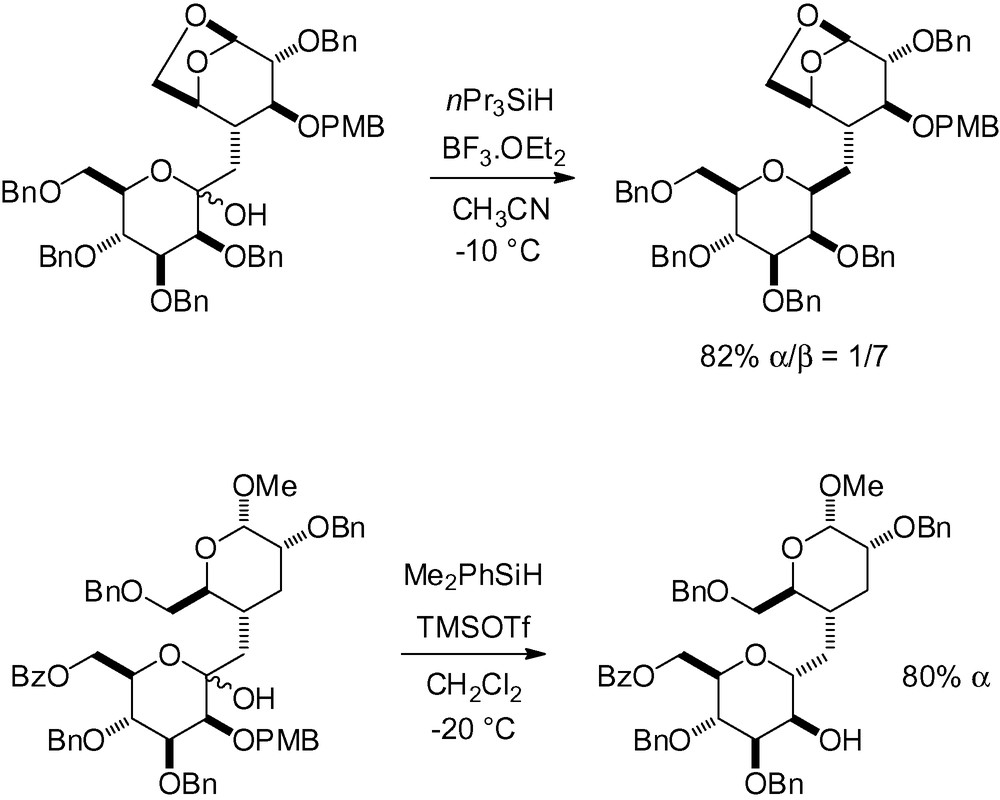
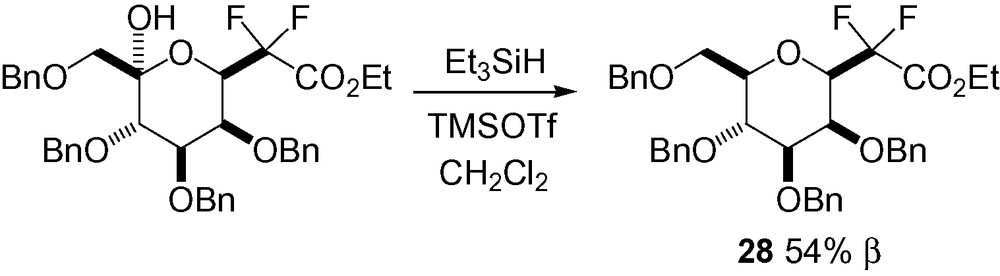
Karche et al. recently reported the synthesis of β-CF2-d-mannopyranoside 28 where reduction of the ketol in position 5 of the mannoside starting material gave access to the desired skeleton in moderate yield [42] (Scheme 56).
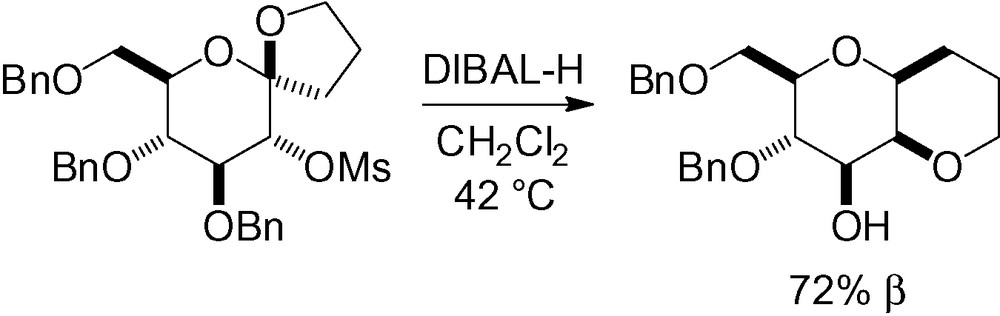
The last example of this paragraph is very unusual in that the β-C-mannose product is obtained from a protected glucose with an anomeric spiroketal [202] (Scheme 57). DIBAL-H plays a double role in the reaction, first, the reductive opening of the spiroketal followed by recyclization through an SN2-type displacement of the mesylate. Secondly, a de-O-alkylation reaction in position 3 was promoted by the excess of DIBAL-H [203].
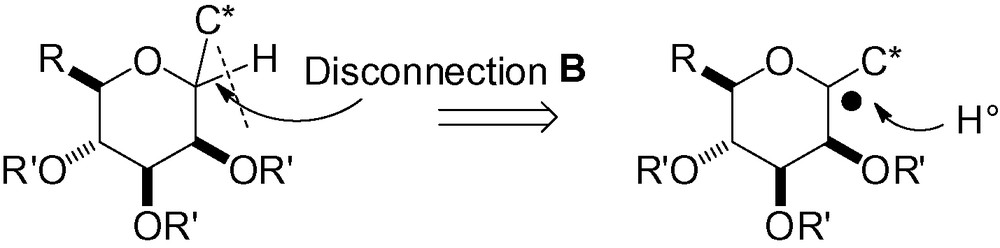
3.2.2 Radical reduction of an anomeric carbon-heteroatom with a hydrogen donor
The same principle of the preceding section can be used if the desired bond, normally a carbon-heteroatom one, can be cleaved and reduced, but this time with a radical mechanism (Scheme 58).
Radical reduction of anomeric carbonhalogen bonds has been reported, but for the most part yields are low and mixtures are obtained (Scheme 59) [204–206]
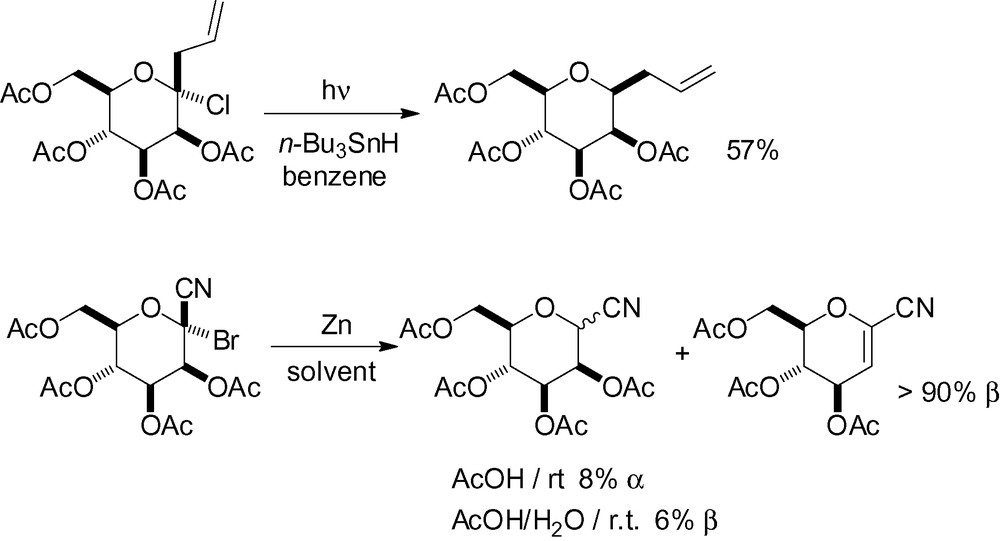
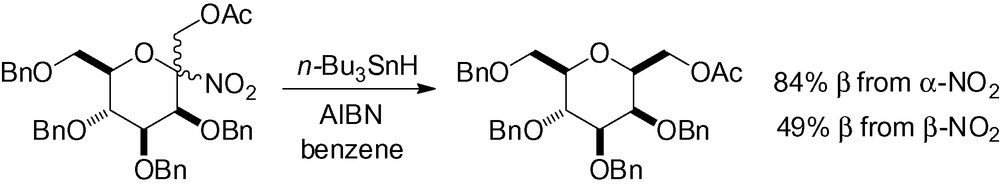
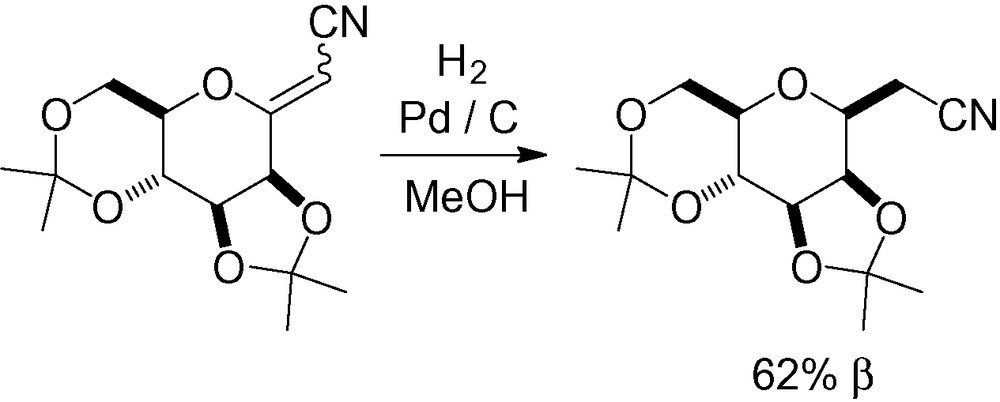
An anomeric carbonnitrogen bond can also be reduced radically. This procedure was shown to be very efficient, giving the β-C-mannopyranoside in both cases (Scheme 60) [207].
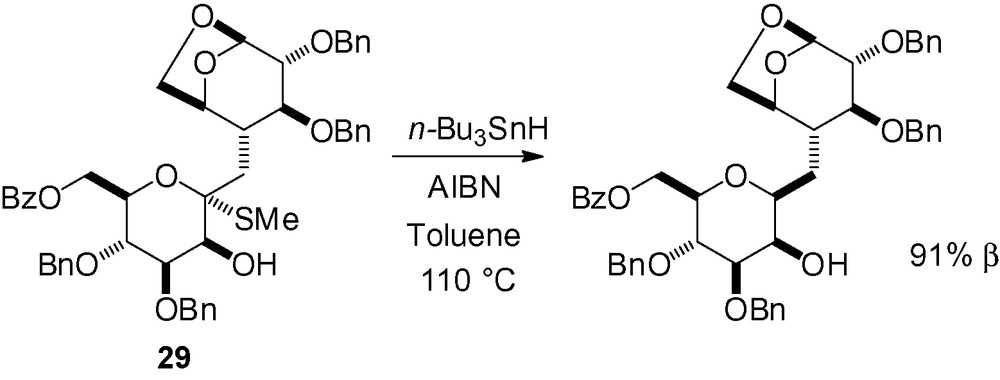
Thioketal reduction is also possible. Radical reduction of the thio derivative 29 gave the corresponding β-C-mannopyranoside in excellent yield (Scheme 61) [201].
3.2.3 Hydrogenation of an exocyclic unsaturated carbon in the anomeric position
Taking advantage of the significant steric hindrance of the β-face in the manno series, an idea emerged where an exocyclic glycal in the anomeric position could simply be reduced to give the corresponding β-C-mannopyranoside (Scheme 62). This attractive approach suffers from the fact that it remains difficult to easily prepare this type of compound, and examples in the literature are limited.
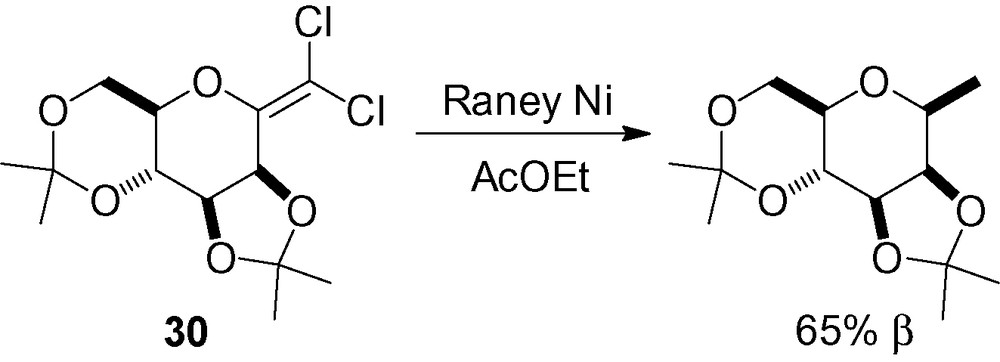
As part of their synthesis of an analogue of guanosyldiphosphofucose, Carchon et al. described an interesting reduction of the dichloromannoside 30 (Scheme 63) [208]. The dichloro derivative was prepared from the corresponding lactone [209,210].
A simple carboncarbon double bond can also be reduced under a hydrogen atmosphere in the presence of catalytic palladium (Scheme 64). This strategy was used on compounds resulting from the iridium catalyzed isomerization of C-allyl glycosides [211], and with derivatives resulting from a Ramberg-Bäcklund reaction [212].
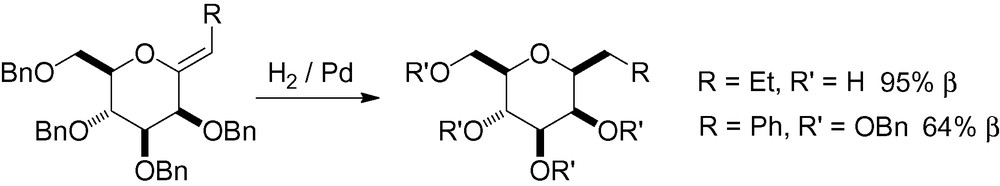
Synthesis of conjugated double bonds in the anomeric position with electron-withdrawing groups is possible using Wittig-Horner olefinations. These compounds can then be catalytically hydrogenated to give the desired β-C-mannosides (Scheme 65) [213].
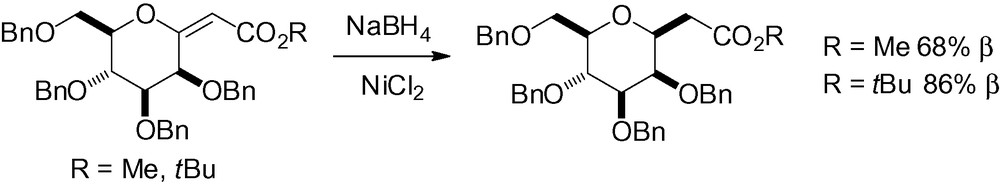
In most cases, however, catalytic hydrogenation is not compatible with benzyl type protecting groups, as they are concomitantly cleaved. In their study on the synthesis of (E)-mannosylidene derivatives using the Wittig reaction, Coumbarides et al. tested the use of sodium borohydride in the presence of nickel (II) chloride [214]. The desired β-C-mannosides were obtained in good yield (Scheme 66).
3.2.4 Hydroboration of an exocyclic unsaturated carbon in the anomeric position
Hydroboration of an exocyclic glycal gives access to β-C-mannopyranosides with further functionalization on the carbon group in the anomeric position (Table 27). Few examples, however, have been described in the literature for this transformation. In the last entry, the reactive borane derivative is directly coupled in a Suzuki cross-coupling reaction to give new C-disaccharides linked with a 3-carbon tether.
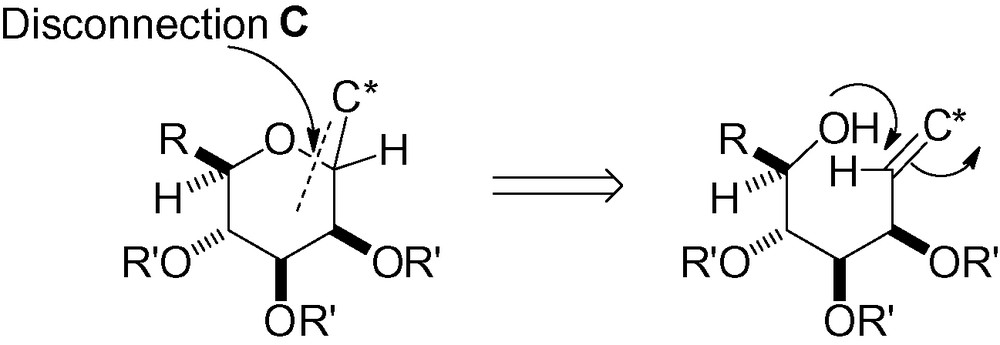
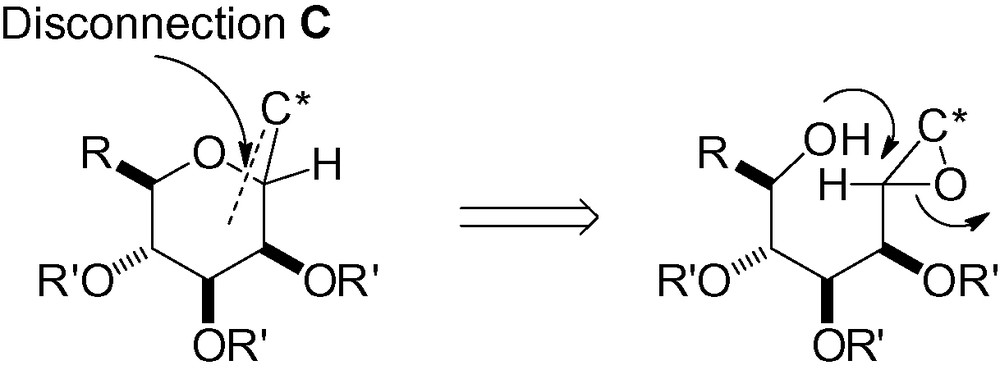
3.3 Disconnection C
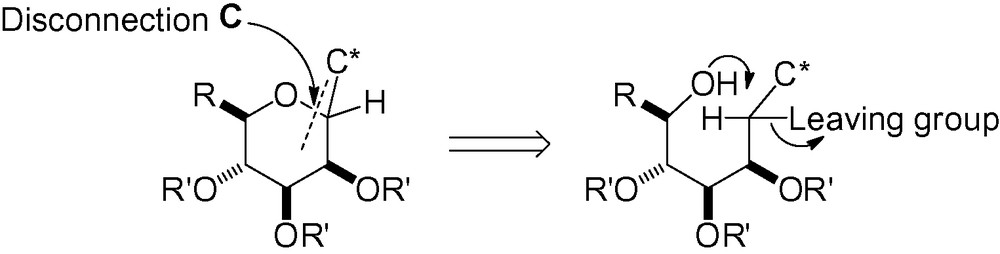
3.3.1 Intramolecular reaction between an alcohol and a leaving group
Attack of the hydroxyl group in “position 5” with a leaving group in the “anomeric” position can lead to the desired C-mannoside skeleton (Scheme 67).
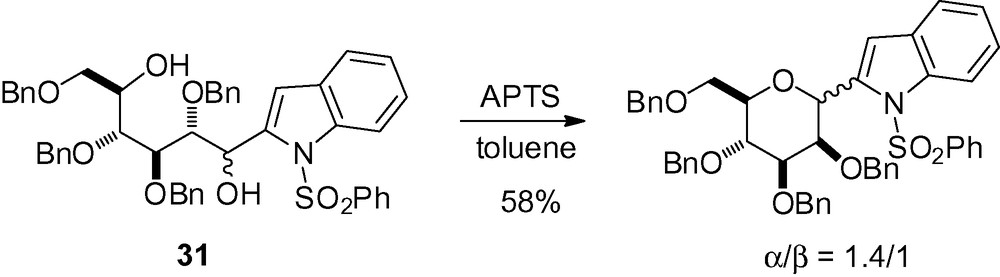
In the course of studies toward the synthesis of α-C-mannosyltryptophan, cyclization of the indole derivative 31 in the presence of acid gave a mixture of cyclized product (Scheme 68) [69].
3.3.2 Intramolecular reaction between an alcohol and an unsaturated moiety
Ring closure, generating the target C-mannopyranoside, can be achieved by addition of an hydroxyl group to a CC double bond, both unactivated or conjugated to an electron-withdrawing group (Scheme 69).
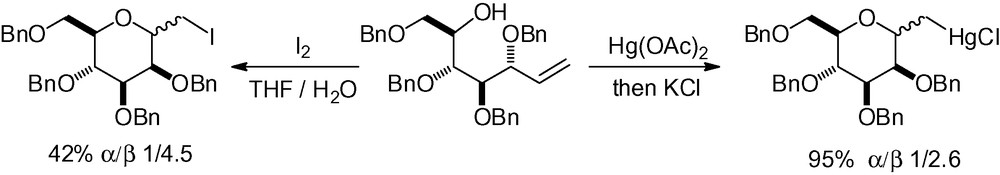
The simplest double CC bond is a methylene. This type of alkene is not activated and needs an electrophile in order to react with the free hydroxyl group. The use of iodine or mercury (II) diacetate, inspired by the work of Pougny et al. [217] has been described in the literature, although with limited stereoselectivity (Scheme 70) [218,219].
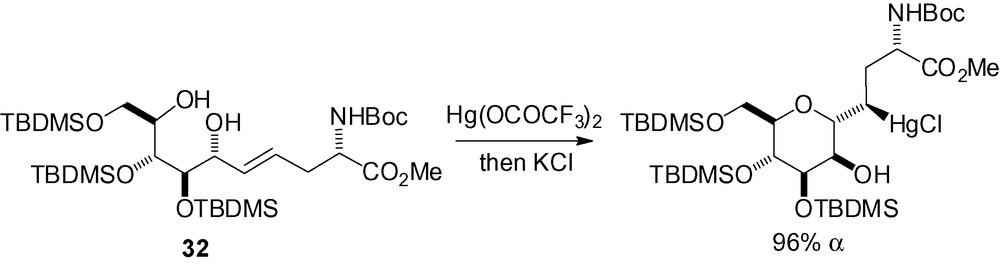
Compound 32 readily reacted with mercury (II) trifluoroacetate to give the α-C-mannoside in excellent yield (Scheme 71) [220].
If the alkene is conjugated with an electron-withdrawing group, cyclization is easier because the double bond is activated. If α-C-mannopyranosides are equilibrated under basic conditions, the corresponding β-C-mannopyranosides are isolated, thus showing they are clearly the thermodynamically favored stereoisomers (Scheme 72) [73].
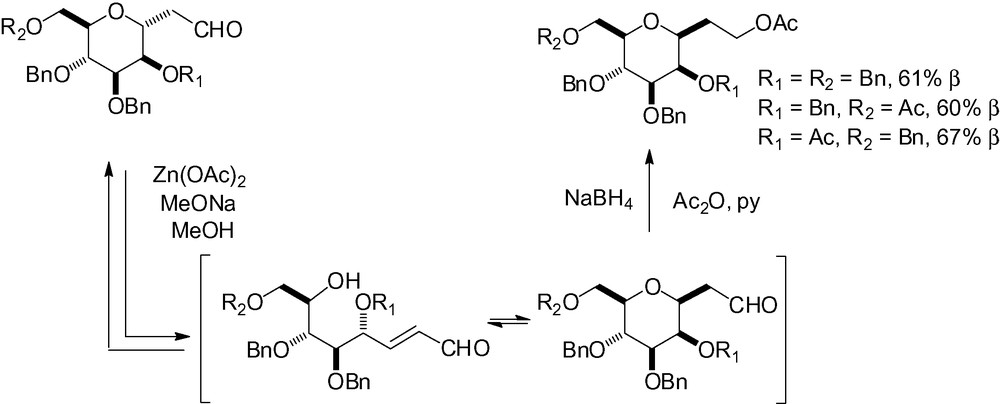
Condensation of 2,4-pentanedione with unprotected d-mannose in an aqueous alkaline solution gives good access to β-C-mannoside ketones via a Knoevenagel condensation (Scheme 73) [221,222].
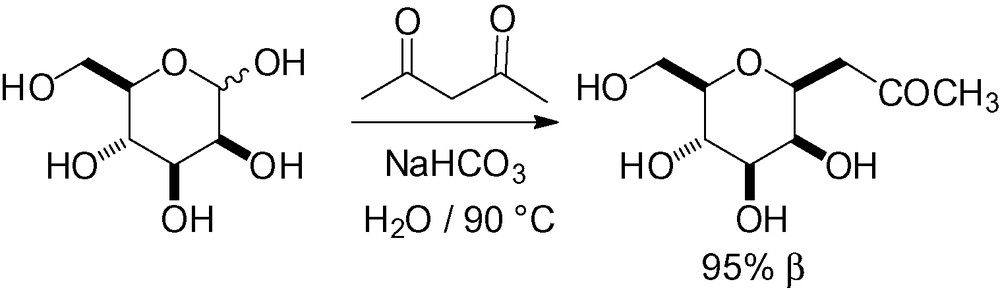
In the same manner, Meldrum's acid has been reacted with various unprotected carbohydrates under thermodynamic conditions [223,224]. When applied to d-mannose, the major reaction product was the 3,6-anhydro-2-deoxy-aldono-1,4-lactone 33, but a small amount of lactone 34 was also isolated (Scheme 74).
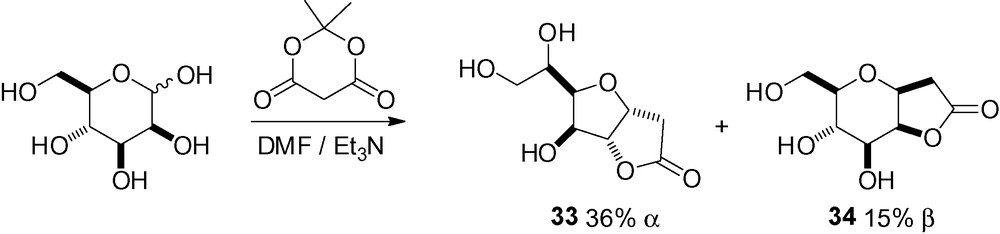
Starting from free d-mannose, reaction of the masked aldehyde function with pyrimidine derivatives [225] or 1,3-dimethylbarbituric acid [226–228] followed by heating, directly gave the desired β-C-mannopyranosides (Scheme 75).
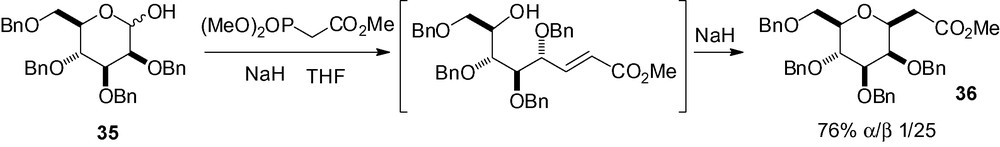
A Wittig-Horner reaction was successfully applied to 2,3,4,6-tetra-O-benzyl-d-mannopyranose 35 giving the β-C-mannoside 36 in good yield and selectivity (Scheme 76) [229]. A mixture of α- and β-anomers was initially obtained, but basic equilibration gave the thermodynamically more favored β-derivative. Kaila et al. have used this intermediate for the synthesis of sialyl LewisX mimics as E-selectin inhibitors [48,49].
Two other examples of Wittig reactions have been reported in the literature using compound 35. The use of BrCH2CO2Me/n-Bu3P in the presence of zinc gave the desired product in low yield with no selectivity (49%, α/β = 1/1) [230]. With an arsenic derived reagent (Ph3AsCHCO2Me), low yield and selectivity were also observed in favor of the α-stereoisomer (22%, α/β = 3.4/1) [231].
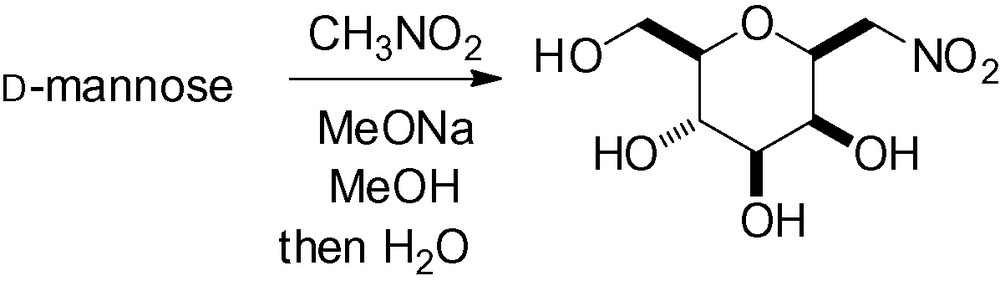
The introduction of nitromethane in the anomeric position gives a useful chemical synthon which can be further transformed into different functional groups [232–234] such as aldehydes [235,236], aldehyde oximes [237–240], and carboxylic acids [241]. d-mannose, for example, reacts with nitromethane under basic conditions and, because of the thermodynamic reaction conditions, the β-nitromethyl stereoisomer is the major reaction product (Scheme 77) [242–245].
This reaction has also been applied to a partially protected mannose derivative (4,6-isopropylidene) to give the desired β-C-mannoside in 50% yield [246].
A one-step Horner–Emmons/Michael reaction has been reported for the preparation of d-mannosyl methanephosphonate derivatives. This gives access to interesting structures ready for further Horner-Emmons couplings, or which can be directly used as mimics of phosphated carbohydrates (Table 28).
Synthesis of d-mannosyl methanephosphonates. .
| R | R1 | R2 | R3 | R4 | Base | Yield (%) | α/β | Ref |
| Me | Bn | Bn | Bn | Bn | LiHMDS, 55 °C | 33 | – | [247–249] |
| Me | Me2C | Me2C | tBuOK/THF | 80 | 2.8/1 | [247–249] | ||
| Me | Me2C | Bn | Bn | tBuOK/THF | 85 | 10/1 | [247–249] | |
| Et | Me2C | Me2C | NaOH 50% CH2Cl2 | 44 | 1/2.7 | [63,250] |
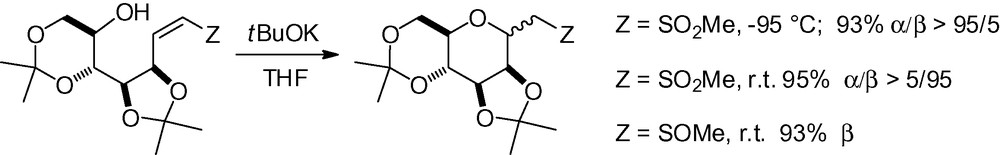
In a recent study, an efficient method was described for the stereocontrolled addition of an alcohol to a vinyl sulfoxide or sulfone resulting in a cyclized product [251]. The α-C-mannopyrannoside was almost exclusively obtained under kinetic control at –95 °C and the β-C-mannopyrannoside under thermodynamic control at r.t. (Scheme 78).
3.3.3 Intramolecular reaction between an alcohol and an epoxide
The last possible C disconnection is ring closure through epoxide opening (Scheme 79). The stereochemistry of the epoxide and the “attacking” hydroxyl group dictates the stereochemical course of the cyclization.
As a result, if the epoxide is not formed in a stereocontrolled manner, intramolecular cyclization then gives rise to a mixture of pyranosides (Scheme 80) [252].
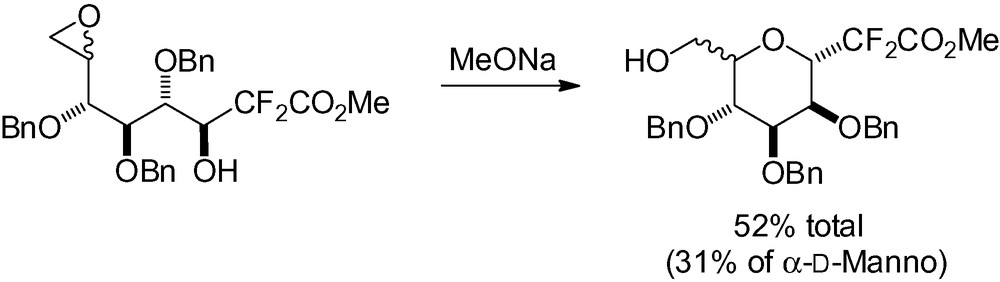
During their synthesis of the B-C ring of ciguatoxin, Oka et al. isolated an interesting reaction by-product [253]. Sharpless epoxidation of compound 37 occurred to give the β-C-mannopyranoside as the major reaction product after concomitant deprotection of the benzyl group (Scheme 81). The expected epoxide was a minor product isolated in only 17% yield.
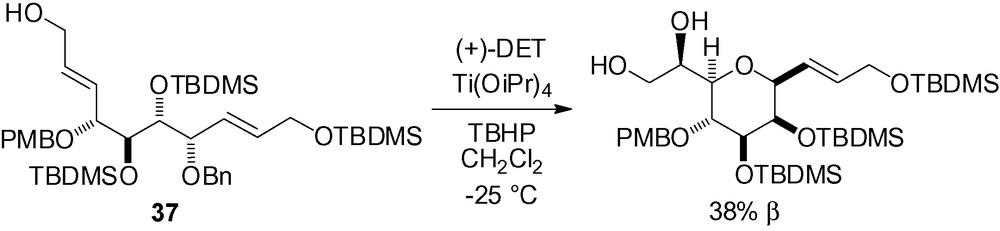
Reaction of a stabilized sulfur ylide with 2,3,4,6-di-O-isopropylidene-d-manno-pyranose 38 gave an intermediate epoxide which could be isolated in excellent yield [61]. Further treatment with NaH gave the cyclized product quantitatively (Scheme 82).
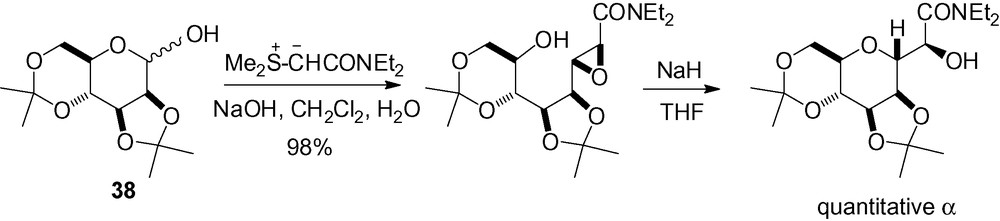
In the course of their work on the synthesis of 1,4-linked C-disaccharides, Kishi et al. explored the acid catalyzed intramolecular epoxide opening. Access to either the α- or the β-C-mannopyranoside was possible by modifying the configuration of the stereogenic center as seen in Scheme 83. In both cases, protection of the hydroxyl group α to this center with an acyl group increased the reaction yield. In the second example, an electron-rich acyl group was necessary to avoid furanoside formation.
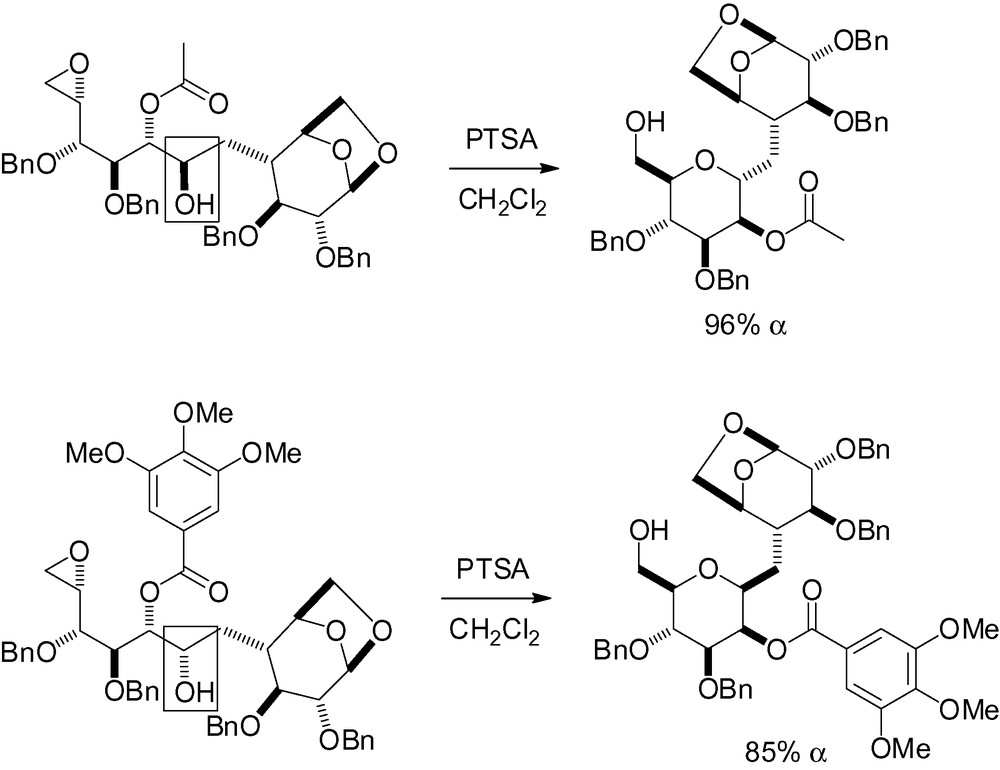
This same methodology was used for the stereocontrolled synthesis of an α,α-C-disaccharide as part of a study of the synthesis and conformational analysis of α,α-, α,β- and β,β-C-trehaloses (Scheme 84) [254].
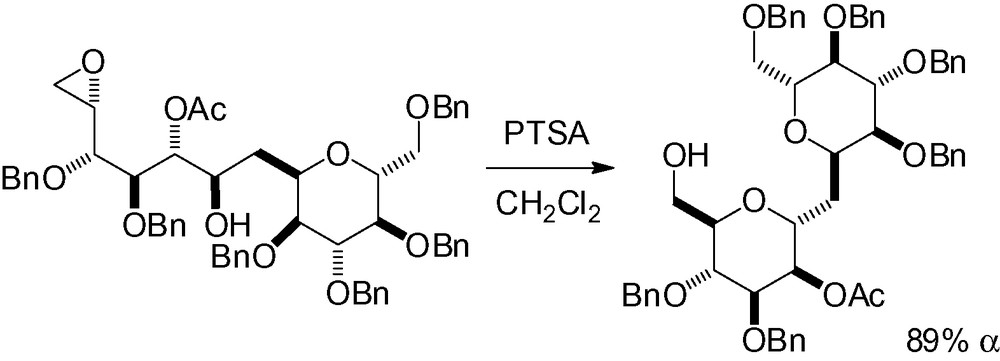
The different strategies that have been presented thus far stem directly from a mannose derivative. One of the riches of carbohydrate chemistry is that it is also possible to start from a different carbohydrate, and envisage further modifications to produce the desired d-mannose framework. This is the subject of the last chapters of this monograph.
3.4 Disconnection D
3.4.1 cis-dihydroxylation of a sugar glycal
In order to access the desired d-manno skeleton, osmylation of an unsaturated carbohydrate with a CC double bond in the 2,3 position is possible (Scheme 85).
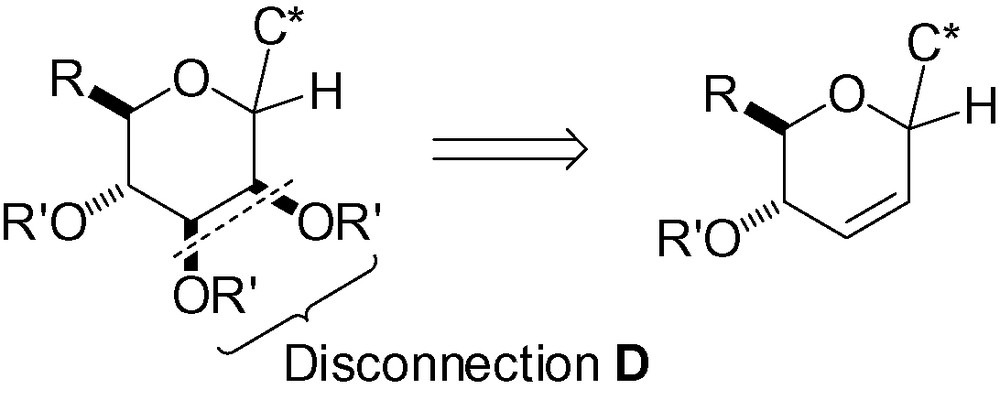
In order to obtain the desired “manno” stereochemistry in the cis-dihydroxylation reaction, C-glycal substrates must have an α configuration in the anomeric position. Osmylation then occurs on the upper β-face, and reaction efficiency depends on the steric hindrance of the surrounding groups. This was demonstrated in the study carried out by Fahka and Sinou in the epoxidation and dihydroxylation of C-Phenyl-Δ2,3-glycopyranosides [255]. When the starting substrate has a β configuration, the β-C-mannopyranoside can still be obtained as the major reaction product if a bulky protecting group is present on the α-face (Scheme 86). As part of their synthesis of 2,3-unsaturated-C-aryl glycopyranosides, Price et al. also reported that dihydroxylation is efficient even with simple acetate protecting groups [256].
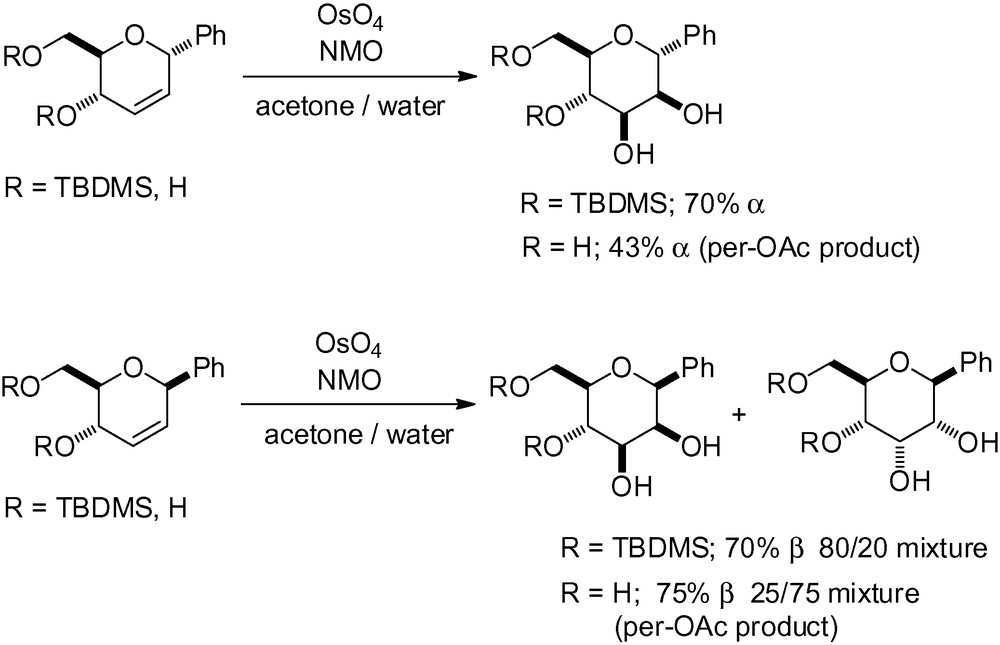
The osmylation reaction has proven to be quite general with diverse aromatic or aliphatic carbon side chains and a wide variety of protecting groups as can be seen in Table 29. Dihydroxylation has even been carried out in the presence of potassium permanganate.
Dihydroxylation reactions of various 2,3-unsaturated-C-glycopyranosides. .
| R1 | R2 | C* | Reaction conditions | Yield (%) | Ref. |
| TBDMS | Ac | OsO4, DABCO acetone/H2O | 82 α (de > 20/1) | [257] | |
| TBDMS | TBDMS | OsO4, NMO acetone/H2O | 30 α | [258] | |
| CHPh | CN | OsO4, NMO dioxane/H2O | α | [259–261] | |
| C(CH3)2 | OsO4, NMO THF/H2O | > 55 α | [262] | ||
| Ac | Ac | OsO4, NMO t-BuOH/Acetone/H2O | 48 α | [33] | |
| Ac | OsO4, NMO t-BuOH/Acetone/H2O | 55 α | [33] | ||
| Ac | Ac | OsO4 | 68 α | [35] | |
| TBDPS | H | CF2CO2Et | OsO4, NMO CH2Cl2 | 62 α | [263] |
| CHPh | K2OsO4, K3Fe(CN)6, MeSO2NH2, K2CO3/H2O, t-BuOH/THF | 50 α | [264] | ||
| Ac | Ac | KMnO4, EtOH/H2O | > 55 α | [265] |
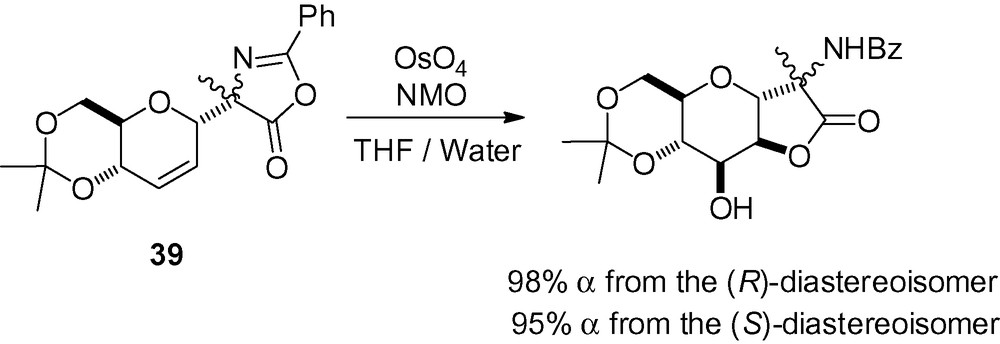
A last example involves the dihydroxylation of compound 39 in a practical stereoselective synthesis of α-d-C-mannosyl-(R)-alanine [76]. The α-C-anomeric bond was formed using a Claisen rearrangement, and the mannosyl lactone was obtained in excellent yield by treatment with osmium tetroxide (Scheme 87).
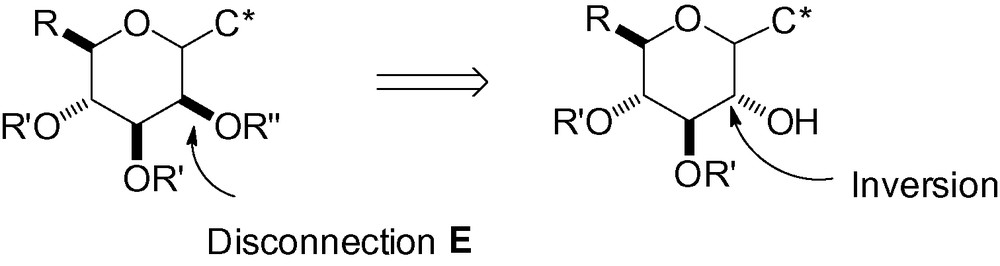
3.5 Disconnection E
Although d-mannose is not considered to be an expensive sugar starting material, d-glucose still remains cheaper. As a result, chemistry involving the inversion of the stereogenic center in position 2 has been developed and many examples exist in the literature. This attractive approach takes advantage of the abundant chemistry developed in the d-glucose series with inversion to the d-mannose family in the final steps of the synthetic sequence (Scheme 88).
3.5.1 Reduction of a ketone in position 2
An oxidation/reduction sequence is one of the first ways to perform the desired inversion in position 2. This method relies on the ability of the reducing agent to give the inverted alcohol in a stereoselective manner. Hydride delivery occurs on the lower α-face giving the β-C-mannoside preferentially (Table 30).
C-mannosides through an oxidation-reduction sequence using d-glucose. .
| R1 | R2 | R3 | C* | Oxidation | Reduction | Yield (%) | Ref. |
| Bn | Bn | Bn | DMSO/Ac2O | LiAlH4/−40 °C | 65 β | [266] | |
| Bn | Bn | Bn | DMSO/Ac2O | NaBH4/CH2Cl2/MeOH | – | [267,268] | |
| Bn | Bn | TIPS | Dess-Martin | L-Selectride/THF/0 °C | 85 β | [269] | |
| Bn | Bn | Bn | DMSO/Ac2O | NaBH4/CH2Cl2/MeOH | 54 β | [66,268,270] | |
| Bn | Bn | Bn | DMSO/Ac2O | NaBH4/CH2Cl2/MeOH | 55 β | [66,268,270] |
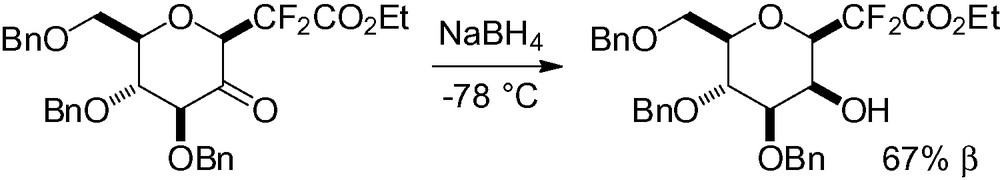
Similar conditions were also applied to a CF2 glycoside, reduction at low temperature gave the desired d-manno sugar (Scheme 89) [271].
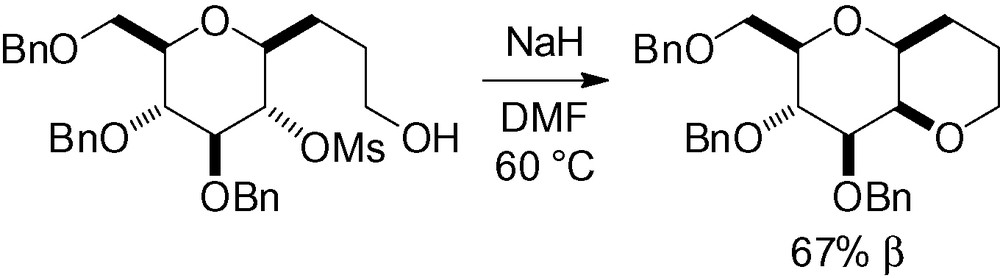
3.5.2 SN2-type inversion in position 2
In spite of the relative simplicity of this approach, few examples exist in the literature because this reaction is, in reality, very inefficient. Activation of the hydroxyl group is required and, to the best of our knowledge, only one example of this type has been described in the literature (Scheme 90) [203].
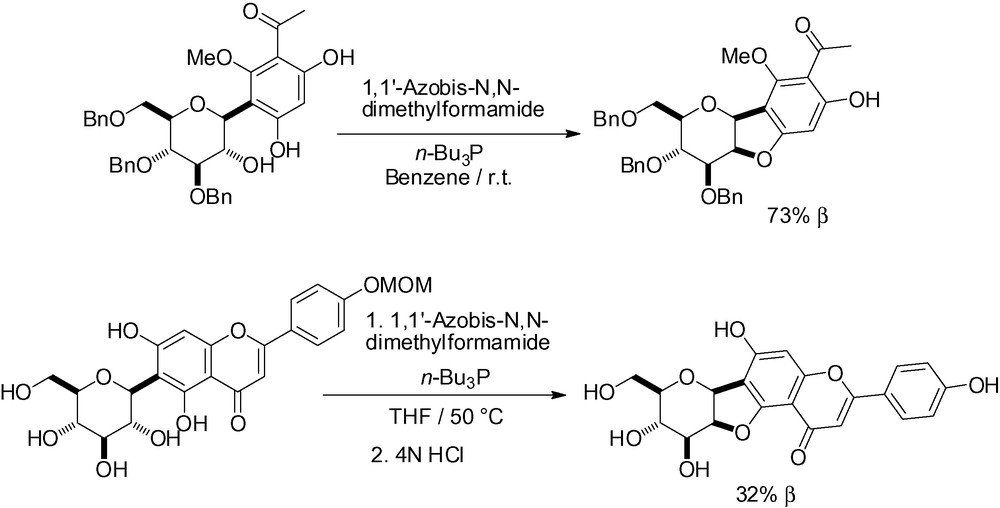
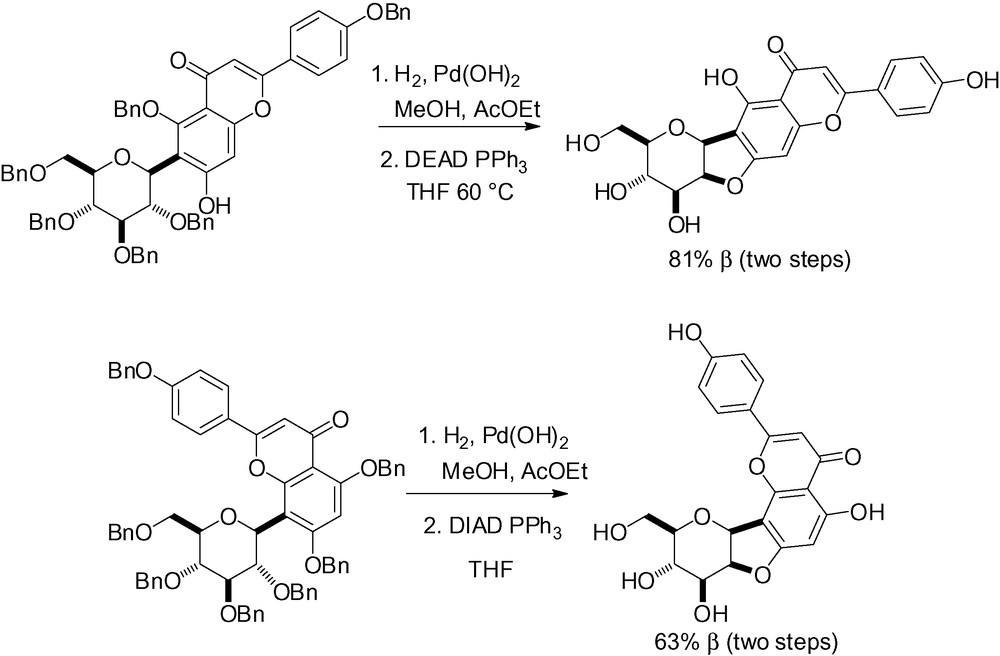
More elegant and efficient in terms of yield and selectivity, the intramolecular Mitsunobu reaction has been successfully applied to structurally unique flavonoid type substrates (Scheme 91) [65,272] as well as for the synthesis of chafurosides A and B (Scheme 92) [273].
3.6 Miscellaneous
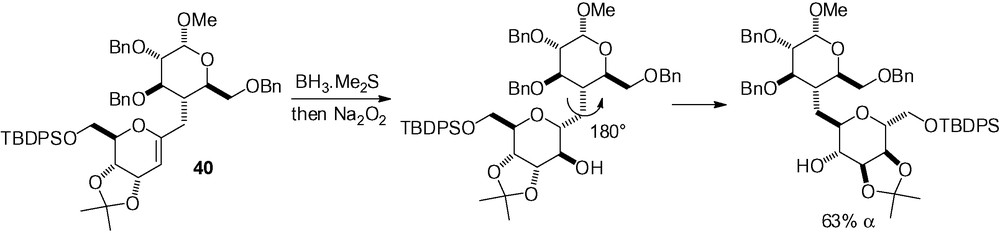
This last section unites several disparate examples which all lead to a d-mannose skeleton. In 2003, Denton and Mootoo reported the synthesis of the C-glycoside of methyl α-d-altropyranosyl-(1→4)-α-d-glucopyranoside by the stereoselective hydroboration of the disaccharide 40 (Scheme 93) [274]. A more unconventional view is possible by simple rotation of the d-altrose moiety around the disaccharide bond to give the d-mannose framework because of the absence of a true “anomeric” carbon.
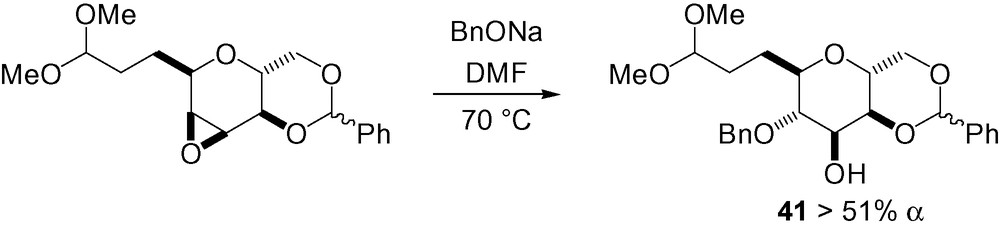
This same reasoning can be applied to compound 41 as part of the “B-segment” of Okadaic acid. Regioselective diaxial opening of the starting epoxide gave the α-C-mannose derivative (Scheme 94) [275].
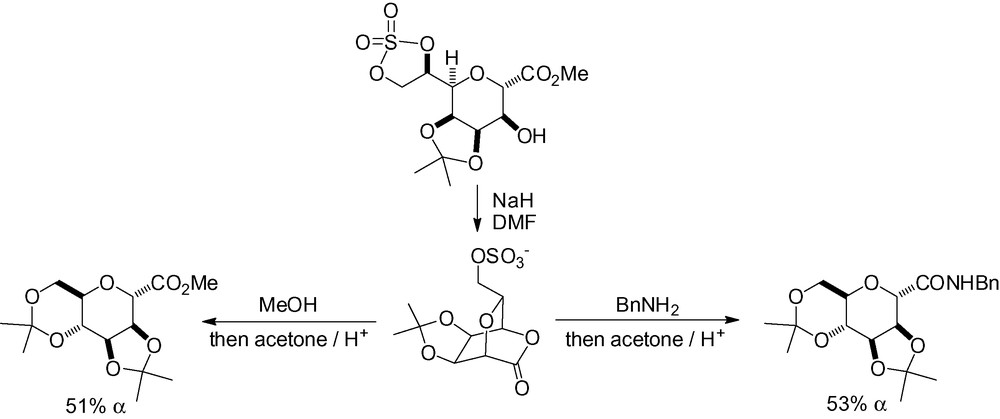
Skead et al. have reported the synthesis of various tetrahydrofurans, tetrahydropyrans and cyclohexanes from cyclic δ-lactone sulphates (Scheme 95) [276].
In a last series of examples, Gerber and Vogel described the synthesis of various β-C-mannosides from the cyclic lactone 42 in good yields (Table 31) [277,278]
Synthesis of β-C-mannosides from lactone 42. .
| R | Yield (%) |
| CH2OC-Ph | 63 β |
| CH2CHCH2 | 76 β |
| CN | 60 β |
| SPh | 66 β |
| OBn | 82 β |
4 Conclusion
In spite of the numerous methods presented in this article, the stereoselective synthesis of d-C-mannopyranosides remains a challenge in organic chemistry. There is no unique recipe for the synthesis of α versus β-C-mannosides, one of the problems encountered being the presence of the axial hydroxyl group in position 2. Selectivity results from a combination of substrate control, reaction conditions and steric environnement, among others. Fine tuning these factors has lead to general trends and good to excellent selectivity in many cases, but not all.
The use of an electrophilic or a radical anomeric carbon usually follows anomeric control, giving an α-C-mannoside. When a β-C-mannoside is desired, the well documented reduction of an anomeric ketal with a hydride donor is probably the best route to follow, even if reduction of an exocyclic unsaturated carbon can give good results. Subsequent modification of suitably designed sugar derivatives are also interesting, for example the cis-dihydroxylation of a sugar glycal or Mitsunobu inversion in position 2 of a d-glucose derivative.
The purpose of this review is to help the reader navigate among the multitude of methods available, and to choose a convenient strategy when C-mannopyranosides are needed.