

1 Introduction
Tetrathiafulvalenes (TTF) and functional derivatives represent a well-known wide family of electroactive sulfur rich compounds [1], extensively used for various applications in molecular electronics and optoelectronics [2]. For many of these applications, the occurrence of multiple redox states is an important prerequisite. One of the straightforward strategies to address this issue is the use of dimeric TTF, in which, depending on the degree of the through-bond or through-space interactions between the redox active units, intramolecular mixed valence species can be evidenced in solution and/or in the solid state [3]. A particularly interesting family of bis(TTF) is that containing either one heteroatomic linker connecting the TTF moieties in close vicinity, such as the series X(TTF)2, with X = S or Se [4], Te [5], PPh, SiMe2 or Hg [6], Me2Si–SiMe2 [7], and SbMe [8], or two heteroatomic bridges as in the rigid derivatives X2(TTF)2, with X = S [9], Te [10], PPh [11], SiMe2 and GeMe2 [12] (Scheme 1).

The fused derivatives (o-DMTTF)2(SiMe2)2, (o-DMTTF)2(GeMe2)2 [12], and (o-DMTTF)2(PPh)2 [11] have been described by us, together with the (o-DMTTF)2(S = PPh)2 compound and coordination bimetallic complexes based on the last 1,4-dihydro-1,4-diphosphinine system [13]. In these compounds we have isolated, in solution or solid state, mixed valence species and demonstrated by spectroscopic and theoretical investigations extensive electron delocalization over both TTF units. One of the possible modifications of this type of rigid dimeric system consists of the variation of the lateral substituents on both TTFs, while keeping the same heteroatomic bridges, in order to extend the scope of these valuable precursors. We have thus decided to replace the lateral methyl substituents by thiomethyl ones in the bis(TTF)-diphosphinine fused system, since it is known that additional sulfur atoms attached to the TTF skeleton can engage in intermolecular contacts in the solid state. Moreover, the thiomethyl substituents are expected to insure a better solubility of the rigid system. We describe herein the synthesis and single crystal X-ray structure of the bis(TTF) [(MeS)2TTF]2(PPh)2, preceded by the description of the solid state structure and a theoretical study of the precursor (MeS)2TTF.
2 Results and discussion
2.1 Structural and theoretical investigations on the dithiomethyl-tetrathiafulvalene precursor
The precursor we have used for the preparation of the bis(TTF) system was the dithiomethyl-tetrathiafulvalene (MeS)2TTF (Scheme 2), prepared using the published procedure involving the phosphite mediated cross coupling of the bis(thiomethyl)-dithiolethione half with the dithiolone-dicarboxylate half, followed by the double decarboxylation of the TTF-diester [14]. The spectroscopic data of the compound were in agreement with those described in the literature [15]. In the course of our investigations we have succeeded to crystallize this precursor, for which the solid-state structure was unknown. Large size orange single crystals of (MeS)2TTF were grown after several hours from a solution of (MeS)2TTF, in a small volume of diethyl ether, poured in a large excess of hexane. The compound crystallizes in the orthorhombic system, non-centrosymmetric space group P212121, with one independent molecule in the unit cell (Fig. 1).

Synthesis of 1.
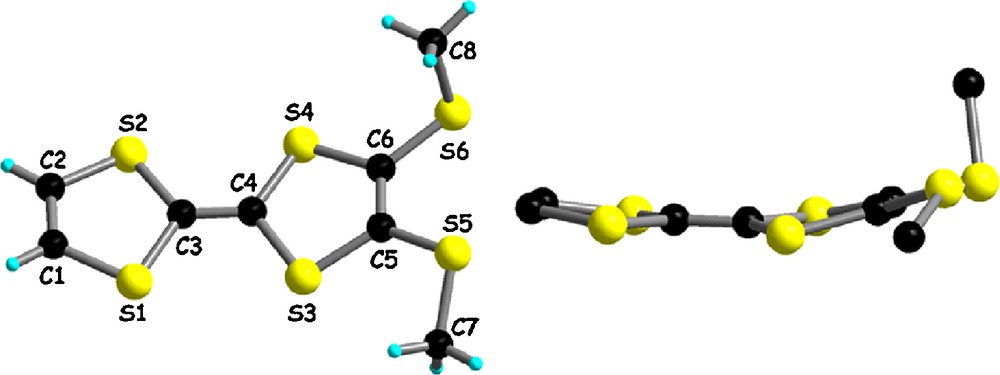
Molecular structure of (MeS)2TTF (a) and side view of the molecule (b).
All the bond lengths and angles are in the usual range of neutral TTFs, together with the moderate folding of the dithiole ring adjacent to the thiomethyl substituents, amounting to 15.8° around the hinge. Interestingly, the thiomethyl substituents arrange in a nonsymmetric manner, with the methyl group defined by C8 located above the TTF mean plane (dihedral angle C8S6C6C5 of 103°), while the other methyl group defined by C7 lies practically in the TTF plane (dihedral angle C7S5C5C6 of 161°). In order to evaluate whether this conformation observed in the solid state corresponds to an equilibrium geometry in the gas phase, we have undertaken theoretical calculations at the DFT level, with the B3LYP hybrid functional [16] and the 6-31(+)G(d) basis set. The geometry optimizations provided two energy minima (checked by frequency calculations which were all positive) corresponding to relative trans and cis orientations of the thiomethyl substituents with respect to the TTF plane (Fig. 2).

Optimized geometries for (MeS)2TTF: trans (a) and cis (b); DFT/B3LYP/6-31(+)G(d).
The two minima are isoenergetic (ΔEtrans–cis = 0.39 kcal mole−1) and are characterized by the folding of the dithiole ring adjacent to the substituents, as observed in the experimental solid-state structure. Nevertheless, the orientation of the thiomethyl substituents in both equilibrium conformations is different with respect to the experimental one, since in the trans geometry the methyl groups are located above and below the TTF plane (the dihedral angles C15S13C3C2 and C16S14C2C3 are 70.7° and 111.3°, respectively), while in the cis geometry, with the methyl groups on the same side of the TTF plane, the angles are wider (125.1 and 123.2°), probably because of the larger steric hindrance. Therefore, it is very likely that the rotation barrier of the two substituents is very small and the packing effects are responsible for the observed experimental conformation. Note that in the monosubstituted (MeS)TTF the methyl group is also arranged above the TTF plane in the optimized geometry [17].
2.2 Synthesis, structural analysis and electrochemistry of [(MeS)2TTF]2(PPh)2 1
The preparation of 1 has been achieved in a one-pot protocol, previously applied in the preparation of (o-DMTTF)2(PPh)2 [11], upon bis(lithiation) of the precursor (MeS)2TTF with LDA, followed by trapping the bis(lithiated) TTF with PhPCl2 (Scheme 2).
The cis/trans ratio in the compound purified by column chromatography, as determined by 31P NMR, is 4:1, with the chemical shift for the cis isomer appearing at −21.1 ppm, and the one for the trans isomer at −25.4 ppm. The cis and trans isomers of 1 arise from the relative orientation of the phenyl rings on the phosphorous atoms with respect to the central six-member ring. The chemical shifts for the two isomers have been assigned by comparison with the ones for the analogue (o-DMTTF)2(PPh)2, for which the preferential crystallization of the cis isomer from a toluene solution of the isomeric mixture could be achieved [11]. By applying the same crystallization conditions in the case of 1, we could not grow any crystal, probably because of its better solubility. However, when diethyl ether was layered onto the top of a solution of 1 in a small volume of THF, very small and thin plate-like orange crystals have been obtained within several hours. Variation of the crystallization conditions did not afford any improvement of the crystal quality. Single crystal X-ray analysis on a small crystalline plate allowed the collection of a rather fair data set when taking into account the size of the measured crystal. This explains the mediocre quality of the crystal structure, with a large value for the reliability factor R. However, the gross connectivity between atoms could be undoubtedly established; however, because of the relatively large esd's values, no fine analysis or comparison of the bond lengths and angles will be further performed. The compound crystallizes in the triclinic system, centrosymmetric space group P–1, with one cis isomer and one half trans isomer, located on an inversion center, in the asymmetric unit (Fig. 3 for the cis and Fig. 4 for the trans isomers).

Molecular structure of the cis isomer in the crystalline structure of 1.
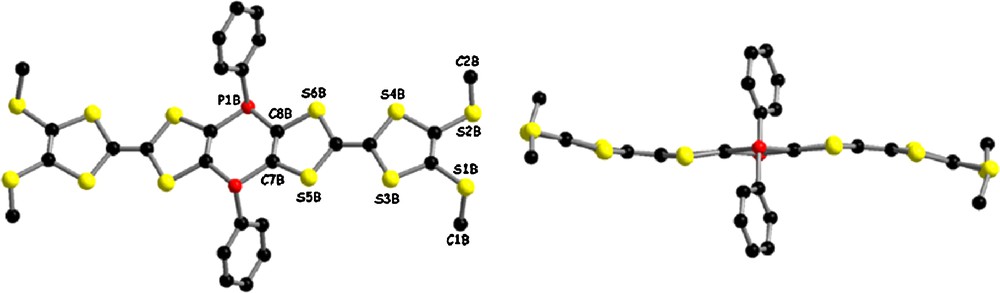
Molecular structure of the trans isomer in the crystalline structure of 1.
As can be observed in Fig. 3, the solid-state conformation of the cis isomer does not present any symmetry element. The most striking difference between the two TTF units consists in the different arrangement of the SMe substituents, since C1A and C2A methyl groups are in relative cis position, while C15A and C16A are in trans, although the dihedral angle for the latter is rather wide, providing a conformation close to that observed in the precursor (MeS)2TTF (vide supra). The central diphosphinine ring is folded around the hinge by about 38.6°, to be compared to 30.4°, the folding angle measured in the case of the (o-DMTTF)2(PPh)2 analogue [11]. The other component found in the crystal structure of 1 is the trans isomer, which conformation is symmetric with respect to the inversion center (Fig. 4).
The methyl substituents are trans each other on both sides of the molecule, thus corresponding to the optimized trans conformation found in (MeS)2TTF. The central six-member ring is planar. Interestingly, this represents the first crystal structure of a trans isomer of the free ligand in this series of compounds, since for (o-DMTTF)2(PPh)2 only the cis isomer could be crystallized so far. In the crystalline packing the two isomers are not isolated, but form mixed stacks along c, with an alternation of a dyad of cis isomers, corresponding each other through the inversion center, and a trans isomer twisted by about 35° with respect to the neighboring cis isomers, a likely consequence of the steric hindrance due to the phenyl rings (Fig. 5).

Alternate stacking of trans/cis/cis isomers of 1 along c.
Cyclic voltammetry measurements on the cis/trans mixture of 1 reveal the splitting of both oxidation waves, resulting into a pair of two closely spaced one-electron processes, corresponding to the successive formation of cation–dication species (ΔE = 60 mV), and then, at higher potential, formation of trication–tetracation species (ΔE = 80 mV) (Fig. 6). These wave separations are somewhat smaller than those measured for (o-DMTTF)2(PPh)2, which were both of 120 mV [11], but in that case only the cis isomer was measured and not the mixture. However, these results suggest through-bond communication between the two-redox active units, as previously emphasized in other heteroatom containing rigid dimers [11,12].

Cyclic voltammetry of 1 (ref. Ag/AgCl, 0.1 M TBAPF6 in CH2Cl2/MeCN, 0.1 V s−1). Insert: The deconvoluted curves, better illustrating the peak-to-peak separation of the one-electron processes. .
The oxidation potential values are similar to those measured for (o-DMTTF)2(PPh)2 and show that the new rigid bis(TTF) system can serve as precursor for radical cation salts, keeping in mind that intramolecular mixed valence species could be in principle obtained.
3 Conclusion
A new rigid bis(TTF) system consisting in fused (MeS)2TTF donors to a central 1,4-dihydro-1,4-diphosphinine ring has been successfully prepared and structurally characterized as a mixture of cis and trans isomers. This represents the second example of this type of rigid bis(TTF) with phosphino linkers, and the first one where the trans isomer could be analyzed by single-crystal X-ray diffraction. The (MeS)2TTF precursor has been also structurally characterized. Theoretical calculations at DFT level afforded two energy minima for this donor, with cis or trans mutual orientations of the SMe substituents. Both orientations have been found in the solid state structure of the bis(TTF) donor. The electrochemical behaviour of [(MeS)2TTF]2(PPh)2, showing four-one electron processes in cyclic voltammetry, demonstrates electronic communication between the TTF units, and, therefore, the isolation of mixed valence species can be envisaged.
4 Experimental
4.1 General
Reactions were carried out under argon. THF was distilled from Na/benzophenone. NMR spectra were recorded on a Bruker Avance DRX 500 spectrometer operating at 202.39 MHz for 31P. Chemical shifts are expressed in parts per million (ppm). The following abbreviations are used: s, singlet. The (MeS)2TTF was synthesized according to the published procedure [14].
4.2 Synthesis of [(MeS)2TTF]2(PPh)2 1
LDA (lithium diisopropylamide) (11 ml of a solution 0,79 M in hexane; 8,7 mmol) was added drop wise to a solution of ortho-dithiomethyl-TTF (MeS)2TTF (1,184 g; 4 mmol) in 60 ml dry THF at −78 °C. The mixture was stirred for 4 hours, following which PPhCl2 (0.57 ml; 4. 2 mmol) was added drop wise. After returning to room temperature during the night, the solution was partially evaporated and the compound was precipitated by adding hexane. The precipitate was washed with methanol, dissolved in toluene and purified by chromatography on a silica column, eluant toluene/hexane 1:1. Yield 222 mg (0.3 mmol, 15%) of brown solid. 31P NMR (THF): δ = −21.1 (s, cis), −25,4 (s, trans).
4.3 X-Ray structure determinations
Details about data collection and solution refinement are given in Table 1. X-ray diffraction measurements were performed on a Bruker Kappa CCD diffractometer for 1, and on a Stoe Imaging Plate System for (MeS)2TTF, both operating with a Mo-Kα (λ = 0.71073 Å) X-ray tube with a graphite monochromator. The structures were solved (SHELXS-97) by direct methods and refined (SHELXL-97) by full-matrix least-square procedures on F2 [18]. All non-hydrogen atoms of the donor molecules were refined anisotropically, and hydrogen atoms were introduced at calculated positions (riding model), included in structure factor calculations but not refined. Crystallographic data for the two structures of (MeS)2TTF and 1 have been deposited with the Cambridge Crystallographic Data Centre, deposition number CCDC 761647 (DTM-TTF) and CCDC 761648 (1). These data can be obtained free of charge from CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (e-mail: deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk).
Crystallographic data, details of data collection and structure refinement parameters.
| (MeS)2TTF | 1 | |
| Formula | C8H8S6 | C28H22P2S12 (1 cis +0.5 trans) |
| Mw | 296.50 | 1207.67 |
| Cryst syst | orthorhombic | triclinic |
| Space group | P212121 | P-1 |
| a (Å) | 8.8699(11) | 14.641(8) |
| b (Å) | 10.3346(11) | 14.681(5) |
| c (Å) | 13.6162(14) | 15.218(8) |
| α (deg) | 90 | 89.39(3) |
| β (deg) | 90 | 72.90(4) |
| γ (deg) | 90 | 60.19(4) |
| V (Å3) | 1248.15 | 2676.2 |
| Z | 4 | 2 |
| T (K) | 293(2) | 293(2) |
| dcalcd, g cm−3 | 1.578 | 1.499 |
| μ (mm−1) | 1.054 | 0.845 |
| Total number of measured intensities | 7722 | 8128 |
| Abs correction | multi-scan | multi-scan |
| Number of unique data | 2419 | 4608 |
| Observed reflections | ||
| (I > 2σ(I)) | 1988 | 2361 |
| Number of refined variables | 127 | 565 |
| λ (Å) | 0.71073 | 0.71073 |
| R(Fo)a | 0.0356 | 0.1227 |
| 0.0472 | 0.2174 | |
| GoF | 0.988 | 1.095 |
4.4 Theoretical calculations
The optimized geometries have been obtained with the Gaussian03 [19] package at the DFT level of theory. The B3LYP functional [16] with the 6-31 + G* basis set has been used. Vibrations frequency calculations performed on the optimized structures at the same level of theory yielded only positive values.
4.5 Cyclic voltammetry measurements
Cyclic voltammetry measurements were performed using a three-electrode cell equipped with a platinum millielectrode of 0.126 cm2 area, a silver wire pseudoreference electrode, and a platinum wire counter electrode. The potential values were then readjusted with respect to the Ag/AgCl electrode, using ferrocene as internal reference. The electrolytic media involved a 0.1 mol L−1 solution of (n-Bu4N)PF6 in a CH2Cl2/MeCN mixture. All experiments have been performed at room temperature at 0.1 V s−1. Experiments have been carried out with an EGG PAR 273A potentiostat with positive feedback compensation.
Acknowledgements
Financial support from the Ministry of Education and Research (grant to I.D.) is gratefully acknowledged. This work was also supported by the CNRS.