1 Introduction
In recent years, the methods of the so-called “green chemistry” have considerably modified research in organic synthesis [1]. Indeed, the use of new catalysts, novative physical methods of activation, new reagents, strategies, solvents and even starting materials is currently described and presented as representative of sustainable approaches. In Europe, the setting up of the Registration, Evaluation, Authorization and Restriction of Chemicals (REACH) regulation aims to safeguard the appropriate handling of already in use and forthcoming marketed substances, to decrease the risk associated to the industrial production and to limit significantly the negative impact on environment.
Only two decades after the publication of the atom economy principle and E factor (Environmental Impact Factor) by Trost [2] and Sheldon [3] and only few years after the publications of the 12 principles of green chemistry and of green chemical engineering by Anastas and Warner [4], these new concepts are taught in every chemical department of universities and chemical engineering schools. This success could be explained by several parameters in brief: new legal restriction of the use of chemicals (REACH), increase of the raw material price, increase of the control on effluents and wastes and maybe more importantly, the demand of consumers. Results of research in these fields have already had a positive effect on both ecological and economical points of view (decrease of the waste production and of the use of permanent organic pollutants, for example). Conversely, the new legislation appeared often to be a constraint and a serious limitation on the discovery process. Many of the solutions proposed for increasing the productivity of discovery process such as combinatorial chemistry, solid phase synthesis, and development of novative and selective (and generally more sophisticated) reagents, have negative effect on the waste production and ecological impact in respect with the chemical transformations. Practically, the technology used for the two phases of innovation (i.e., the discovery and the production processes) seems to differ more and more sharply, making transposition from laboratory to pilot plant and factory more difficult.
In this article, we report one example developed in our laboratory of a new cleaner (greener) chemical transformation which was originally studied in order to decrease the ecological impact of a particular chemical reaction (aryl-aryl C–C bond formation) and that would provide a new insight for the access of higher molecular diversities depicted in biologically active molecules.
Aryl-aryl bond formation is one of the most important tools of modern organic synthesis, due to its large representation in natural products and pharmaceutical or agrochemical specialities [5]. In recent years, direct arylation reactions to substitute C(sp2)–H bond has raised considerable attention, providing an efficient alternative to classical nickel and pallado-catalyzed cross-coupling reactions [6–10]. All these important reactions required the preparation of organometallic reagents such as boronic acids, organo-magnesium, -tin or -zinc derivatives. Efforts have been made recently to limit the use of toxic reagents by the use of grafted reagents (on polymers or ionic liquids). In the direct activation of the C–H bond, the traditional activated metal species is substituted by a non-substituted arene, avoiding preliminary functionalization always associated with additional step reaction, wastes, solvents, purifications and time. This reaction is therefore similar in result, but not in a mechanistic outlook, to the Heck reaction which allows also the formation of C(sp2)–C(sp2) bond between aryl halide and activated alkene.
Direct arylation, sometimes called “C–H activation”, is illustrated in the recent reviews on the topic ([11] and references cited therein) and can be regarded as a new synthetic tool, in respect of the principles of green chemistry (Principle 2: Atom economy; Principle 8: Reduce Derivatives; Principle 9: Catalysis).
Over the past decades, significant contributions have been brought to develop efficient and atom-economical methods for direct arylation, with use of palladium and ruthenium catalysts. Less expensive and less investigated copper, iron and nickel metals have been already promoted. Coupling aryl halide partners have been enlarged to sulfonates, phosphonates, mesylates, tosylates, anhydrides and carboxylic acids, revealing a fruitful potential [12]. Functionalization through direct arylation is for the moment mainly reserved to small molecules, and the future studies should open the way to a better mechanistic understanding. Palladium-catalyzed direct arylation is proposed to undergo through four different mechanisms, strongly dependent with the substrates, catalytic systems and bases (Scheme 1): (i) C–H activation with oxidative addition of the metal (Ru, Ir, Rh or Pd) directly in the strong C–H bond (90–105 kcal/mol) (pathway 1); (ii) electrophilic aromatic substitution (pathway 2); (iii) concerted metalation deprotonation (pathway 3); (iv) Heck-type cross coupling with anti/syn-β-hydride elimination (pathway 4).
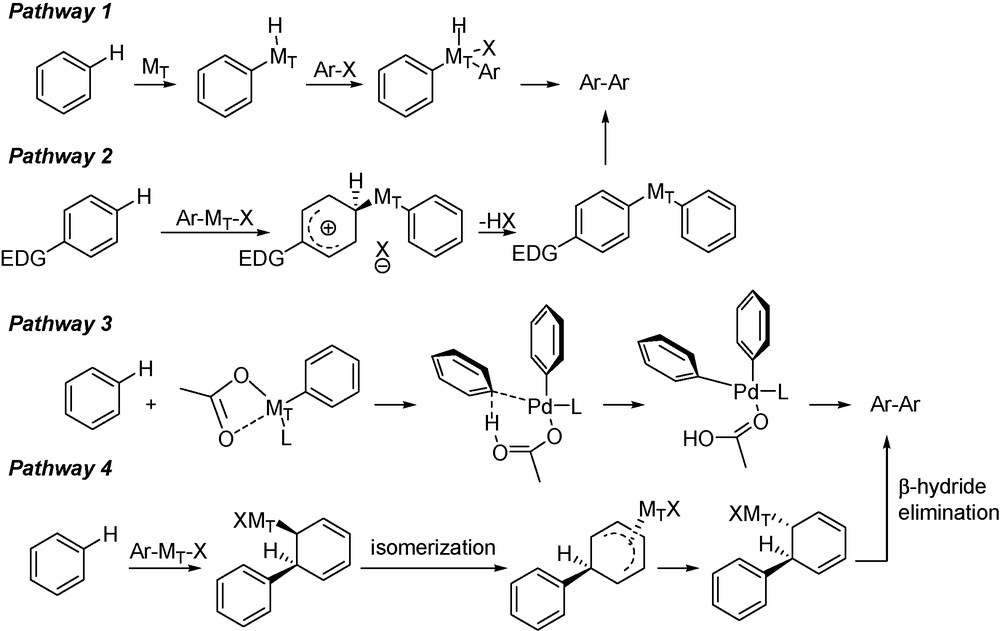
Supposed mechanisms of direct arylation.
2 Development of the direct arylation on thiophene and benzo[b]thiophene derivatives
Since 13 years ago, the laboratory has studied the direct arylation of thiophene heterocycle in order to find a new polymerization process of thiophene. At that time, the processes of production of poly(alkyl-thiophene) required either a very large quantity of iron [13] or the use of Grignard, zinc, tin or boron reagents which are scarcely compatible with the material production. Therefore, the industrial development of such fascinating material was strongly limited.
2.1 Direct arylation on thiophene
In an ongoing project to prepare functionalized thiophenes as suitable monomers for the synthesis of regioregular polymers with high electrical conductivity, the direct arylation of activated thiophenes was performed using a direct arylation reaction. In a more general pattern, the substitution reaction of activated thiophenes in C(2), C(3) or C(5) positions by aryl halides was obtained in moderate to good yield. Reactions were performed in a mixture of acetonitrile and water (9/1) in the presence of tetra-n-butylammonium bromide, potassium carbonate and palladium acetate as a catalyst. This ligandless procedure was globally more efficient on 2-substituted thiophenes (yields ranging: 30–81%) than on 3-substituted thiophenes (30–35%) (Scheme 2). The nature and the position of the substituent, as well as the stability of the substituted thiophenes, influence dramatically the scope of the reaction [14].
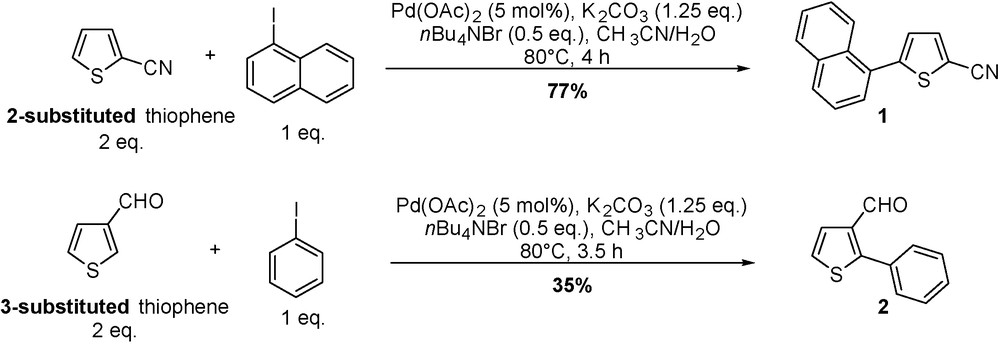
Arylation of activated thiophenes catalyzed by palladium.
These preliminary results prompted us to evaluate the catalytic system in order to identify the best reaction conditions, minimizing side reaction product rising from Ullmann cross-coupling of aryl halide [15]. The polymerization of 2-halogeno-3-alkylthiophenes was thus developed by a slight change of the previously reported arylation conditions: palladium acetate (5 mol%), K2CO3 (2.5 eq.), n-Bu4NBr (1 eq.) in DMF at 80 °C for 48 h [16]. As an example of step-by-step synthesis, the synthesis of tetrathienyl oligomer 6 was performed in four steps and 46% overall yield with two steps of direct arylation (Scheme 3) [17].
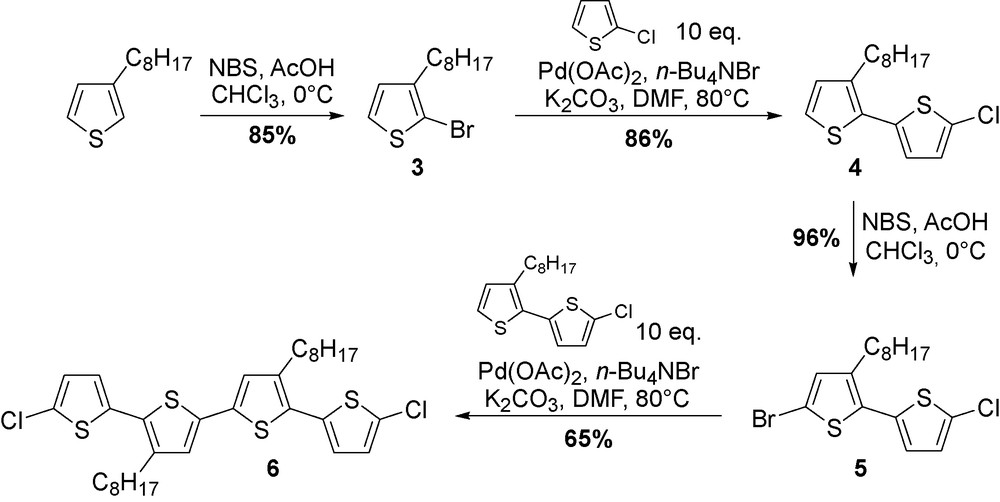
Synthesis of thiophene oligomers.
Although the direct polymerization of 2-bromo-3-alkyl thiophene was efficiently performed using our new method of C–H activation, the methodology did not reach the economic requirement for further industrial development. We decided to use this new reaction to synthesize a large diversity of new molecules and evaluate some of their biological properties thanks to the chimiotheque of our institute. This strategy relies on the relative novelty of this reaction that normally should expedite the access to original structures with the hope of meeting more success in new “hit” research.
2.2 Direct arylation on benzo[b]thiophene
Extension of the direct arylation procedure to 3-substituted benzo[b]thiophenes provided a new route to 2-arylbenzo[b]thiophenes that have been reported to display potential biological activities for the treatment of hypolipemia and estrogen-dependent diseases. The potentiality of these substrates has been already pointed out by Miura [18] and Ohta [19] in the presence of palladium salts, triphenylphosphine, and in the case of Miura, an over stoichiometric amount of copper salt. The reaction was found to perform relatively fast whatever the electrodonating or the electrowithdrawing group at C(3) position, providing the 2-arylsubstituted derivatives 7 and 8. The scope was enlarged to a wide number of aryl halides affording the corresponding derivatives in moderate to good yield (40–77%) (Scheme 4) [20]. The selectivity of the reaction was also moderate due to the major side reaction leading to symmetrical Ullmann-type coupling adducts up to 30%.
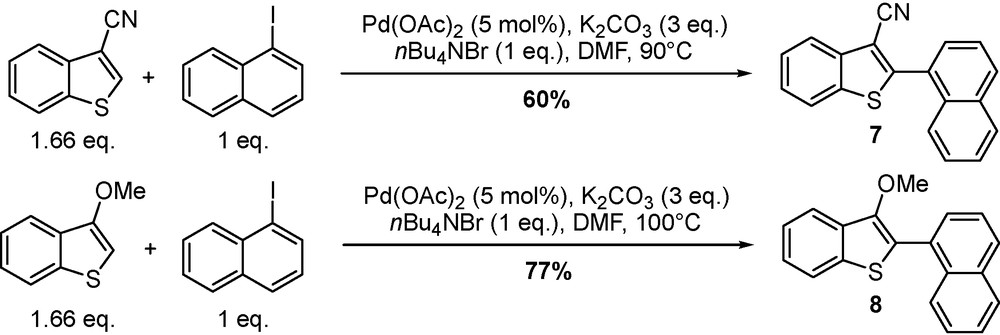
Synthesis of 2-arylbenzo[b]thiophenes.
Highly functionalized 2-arylbenzo[b]thiophenes 11 (analogues of Arzoxifene 12) were prepared by a sequence of nucleophilic aromatic substitution followed by a one-step Heck-type coupling (Scheme 5) [21]. The sulfoxide adduct of 3-bromobenzo[b]thiophene was subjected to nucleophilic substitution with miscellaneous phenolates in good yield. We assumed that the oxidation into the corresponding sulfoxide would favor the nucleophilic displacement of bromide by decreasing the charge density. Subsequent reduction of compounds 9 regenerated the benzo[b]thiophene scaffold 10 that was coupled with various halides in palladium-catalyzed direct arylation.
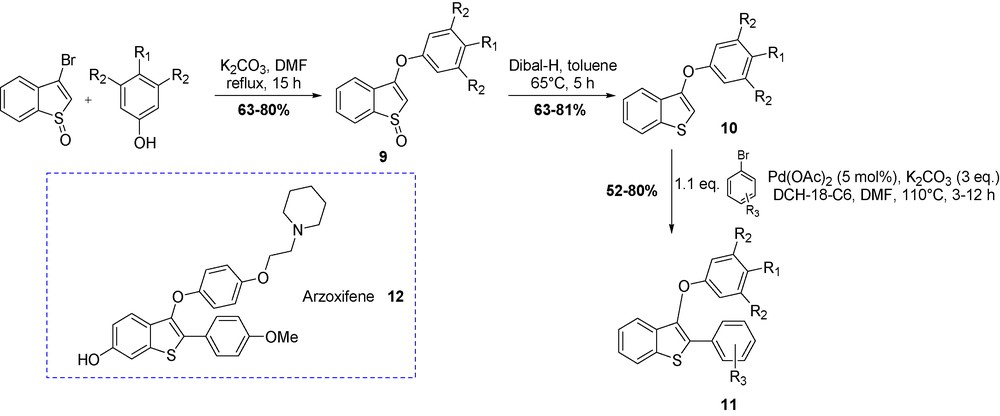
Synthesis of Arzoxifene analogues 11.
The functionalized benzo[b]thiophene scaffold was used as a template for more complex heterocyclic systems such as quinolines and tetrahydroquinolines (Scheme 6). In the presence of Pd(OAc)2/PPh3 as a catalytic system and potassium carbonate as a base, 2-(2′-nitro-4′-methoxyphenyl)-benzo[b]thiophene 13 was obtained in 62% yield from benzo[b]thiophene. Acylation at C(3) position under Friedel and Craft conditions afforded compound 14 in excellent yield. Subsequent hydrogenation afforded aniline intermediate which immediately cyclized into the 2,3-diarylated tetrahydroquinoline 15 [22]. The desulfurization procedure has been already performed efficiently with Raney nickel. In our case, we turned our attention to other nickel systems which are known for their high efficiency and chemoselectivity in the desulfurization of polyaromatic sulfur-containing compounds.
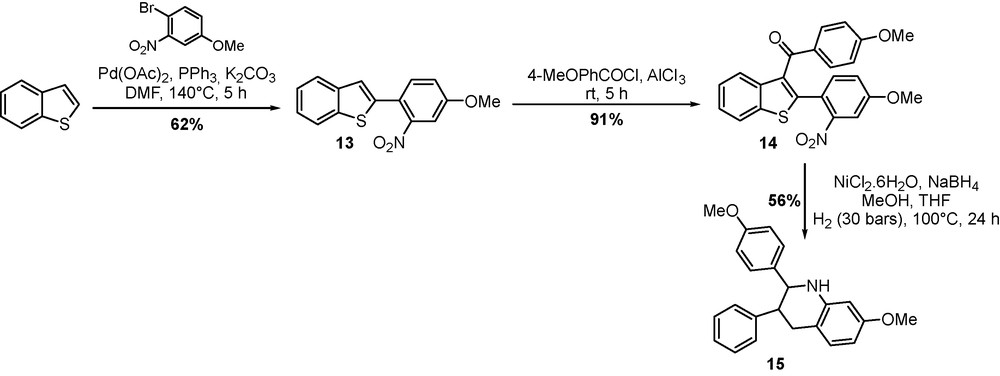
New synthetic route to highly substituted tetrahydroquinoline 15.
The formation of the benz[c]azepine ring is based on a direct arylation of 3-cyanobenzo[b]thiophene with a survey of commercially functionalized aryl halides, followed by a Pictet-Spengler cyclization. Previously obtained by aromatic nucleophilic substitution of the commercially available 3-bromobenzo[b]thiophene, 3-cyanobenzo[b]thiophene was subjected to the previously reported Heck-type reaction conditions with 3-bromo-N,N-dimethylaniline (Scheme 7). Subsequent reduction of compound 16 with BH3-THF complex afforded primary amine 17 in good yield which upon treatment with benzaldehyde provided intermediate imine. The Pictet-Spengler reaction of the intermediate imine, mediated by trifluoroacetic acid furnished cyclized derivative 18 in 70% yield [23].
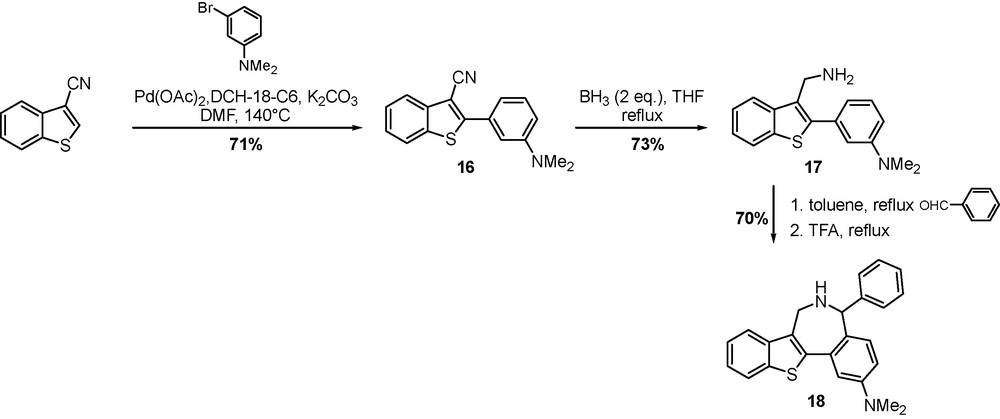
Synthesis of benz[c]benzothiophene[2,3-e]azepine 18.
2.3 Fluorescent rhodamine dyes
Rhodamine dyes have been used in various biological applications from biology to material science. Their excellent photophysical properties (long absorption and emission wavelength, high fluorescence quantum yield, large extinction coefficient, high light stability) render them extremely attractive as fluorophores and chromophores. Benzo[b]thiophene and indole containing rhodamine dyes were synthesized by a sequence involving a direct arylation key step. Direct arylation between benzo[b]thiophene and p-methoxy-o-nitrophenyl bromide proceeded in moderate yield in the presence of the palladium catalyst affording compound 19 (Scheme 8). Reduction of the nitro-group was followed by a Pictet-Spengler cyclization with benzaldehyde [24]. Intermediate benzothieno[3,2-c]quinoline 21 was converted into rhodamine 22 derivative following the procedure described by Liu et al. [25].
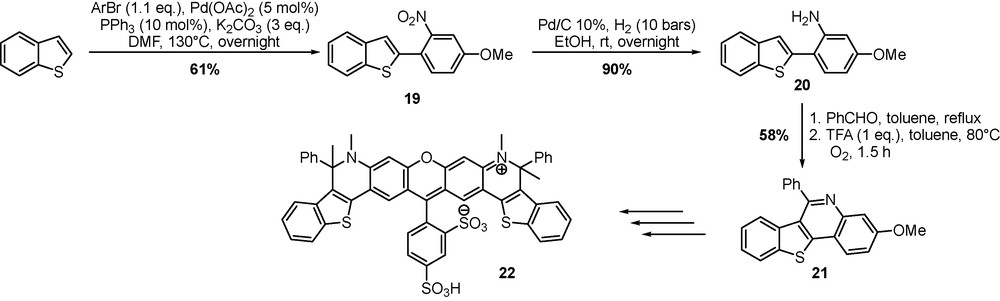
Synthesis of fluorescent rhodamine dyes.
Several photophysical characteristics including absorption, emission maxima, fluorescence quantum yields, and lifetimes were measured in methanol and DMSO and compared to standard Rhodamine 101, raising a novative methodology for the access to a new family of rhodamines.
A benzothieno[3,2-c]quinoline library of compounds type 21, was also prepared with modulation of the substituents on phenyl rings [26].
3 Application to molecules of therapeutic interest
Direct arylation methodology was used as a synthetic tool for the construction of more elaborated heterocyclic systems, keeping in mind the potentiality of biological applications. Recently, the group has focused on different biological mechanisms and taken advantage of the successful expertise acquired on heterocyclic chemistry. Some biological targets have been identified amongst them: alkaline phosphatase, pump NorA, heat shock protein 90.
3.1 Alkaline phosphatases inhibitors
Osteoarthritis and associated diseases are characterized by the presence of basic calcium phosphate in knee joints. The development of inhibitors of alkaline phosphatases should open the way for the treatment of such pathologies. Levamisole or l-tetramisole is a known tissue non-specific alkaline phosphatase (TNAP) inhibitor, used for rheumatoid arthritis but displaying major side effects, limiting thus its therapeutic use. The direct arylation reaction was applied to several strategies to synthesize a library of benzothiophene derivatives as depicted in Scheme 9.
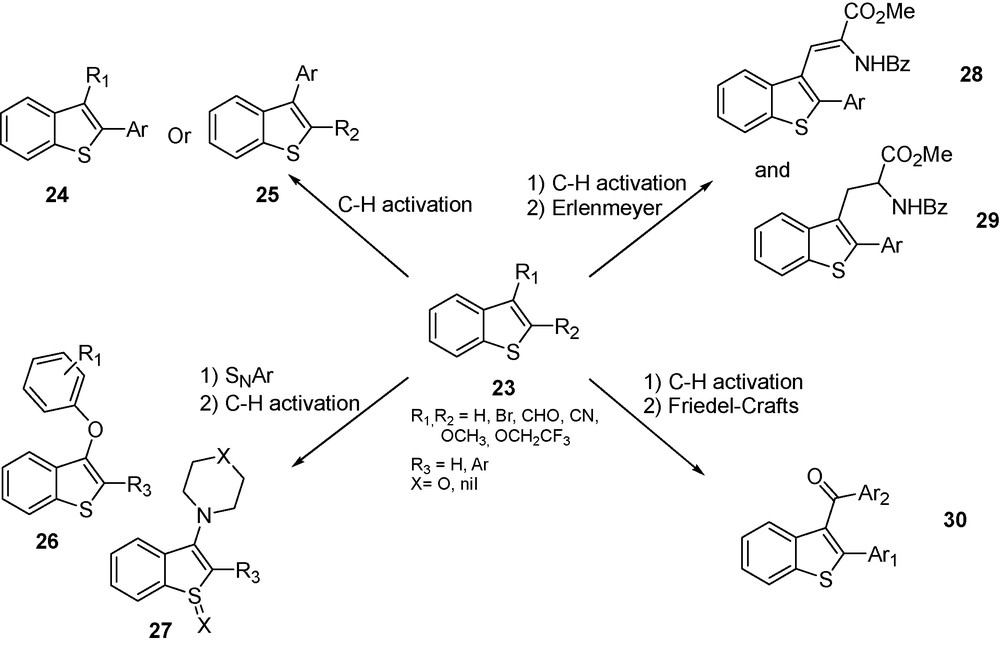
General synthesis of benzo[b]thiophene derivatives.
The related inhibitory properties with inhibition constants Ki or percentage of inhibition of promising compounds have been measured and compared to levamisole 31, giving weight to this drug therapy strategy for osteoarthritis [27]. Compounds 32, 33 displayed a similar range of activity in comparison with levamisole 31. Compound 30a and derivatives rising from this series exhibited lower activity both on bovine intestinal alkaline phosphatase (BIAP) and TNAP activities (Scheme 10).
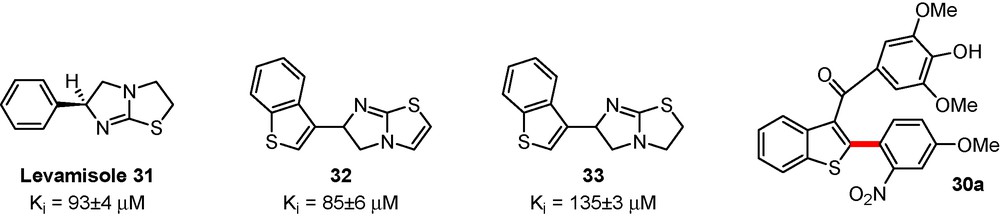
Novel inhibitors of alkaline phosphatases produced in the group.
3.2 NorA pump inhibitors
From a general point of view, bacteria employ a number of resistance mechanisms to counter antibacterial challenges. The overexpression of transmembranar protein-based efflux pumps can pump out a wide range of antibacterial agents from within the cells, thus corrupting their therapeutic efficiency (associated to a low antibacterial concentration inside the cell). In Staphylococcus aureus stream, NorA pump can efflux structurally heterogeneous antibacterials such as Berberin and ciprofloxacin. One strategy to struggle against resistant bacteria is to combine the administration of a known antibacterial agent with a NorA pump inhibitor that could bypass the problem of resistance. Reserpin is the first inhibitor of NorA pump, inducing a restoration of antibiotic activity of norfloxaxin (Scheme 11). The restriction associated to the use of this molecule arises from the neurotoxic properties induced at effective concentration and was dropped in early clinical phases.
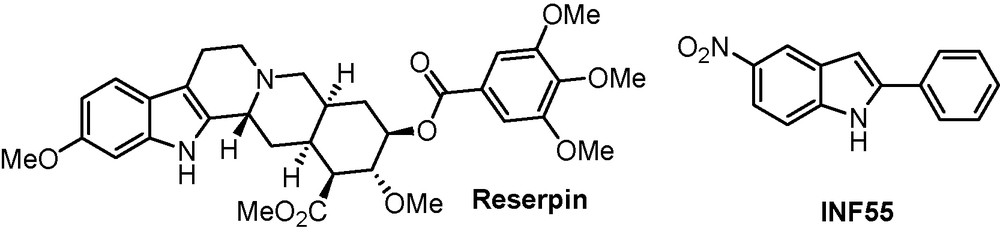
Known inhibitors of NorA pump.
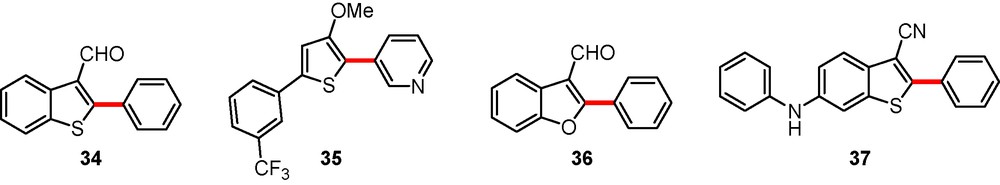
The laboratory developed a novative and straightforward synthesis of a new generation of 2,5-diarylthiophenes and 2-aryl-benzo[b]thiophenes as structural analogues to INF55 leading to the design of new NorA pump inhibitors. The exact mechanism of inhibition of NorA pump still remains unknown, which is a major drawback in the rational design of new inhibitors bearing the 2-arylbenzo[b]thiophene skeleton. Molecules 34, 35, 36 and 37 which were assayed in combination with ciprofloxacin against resistant bacteria harbouring an efflux pump may be considered as potential bacterial efflux pump inhibitors (Scheme 12) [28]. Amongst them, compound 37 displayed a subminimal inhibitory concentration (MIC) lower than reserpin (MIC = 20 mg/L) which is a promising result.1
3.3 Hsp90 inhibitors
Heat shock protein 90 (Hsp90) is one of the most abundant cellular proteins. Although its functions are still to be characterized, it appears to serve as a chaperone for a growing list of cell signaling proteins, including many tyrosine and serine/threonine kinases, involved in cell proliferation and/or survival. The recent discovery of natural products which are able to inhibit Hsp90 function has allowed for both identification of its client proteins and for a better understanding of its role in their activity. Accumulating data have suggested that targeting Hsp90 in cancer cells may be of clinical benefit.
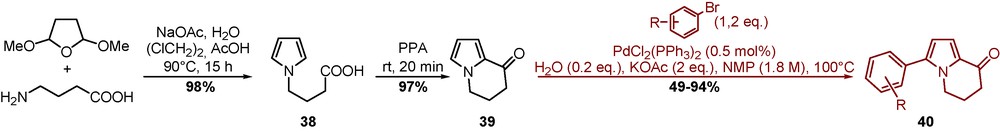
The pyrrole ring is one of the most common heterocycle in organic chemistry present in a wide range of molecules of therapeutic interest and is considered as an important building block in the synthesis of natural products. In particular, the 8-oxo-5,6,7,8-tetrahydroindolizine skeleton was reported as a key intermediate in the synthesis of indolizidine building blocks and natural indolizidine alkaloids such as the (+)-monomorine, indolizidine 209D, or polygonatines A, B and kinganone. Recently, 3-aryl-8-oxo-5,6,7,8-tetrahydroindolizines 40 were described as inhibitors of Hsp90 (Scheme 13) [29].
Synthetic strategy to tetrahydroindolizidinone.
The tetrahydroindolizinone scaffold 39 was obtained by a sequence of the Clauson Kaas reaction followed by PPA-mediated intramolecular cyclization of carboxylic acid 38. Site-selective sp2 direct arylation of compound 39 allowed the introduction in C-3 position of aryl and heteroaryl substituents (Scheme 13). The reaction performed with electrowithdrawing aryl bromides provided substituted derivatives 40 with satisfactory yields, whereas electrodonating groups penalized the coupling in terms of reaction times and yields [30].
In our ongoing project of finding new Hsp90 inhibitors, the access to 8-oxo-3-acetylenic-5,6,7,8-tetrahydroindolizines via Sonogashira coupling was also investigated [31].
4 Conclusion
The results obtained by using a relatively new technology (thiophene C–H activation) during these last 10 years, provided promising opportunity in different fields of researches of biologically active molecules. This reaction allowed us the preparation of somewhat more than 400 different molecules, which can appear to be a very small quantity compared to what is generally claimed with the use of parallel, combinatorial and other high-throughput synthesis. Despite this limited number of unseen molecules, new hits have been identified in the fields of NorA pump and heat shock protein 90 inhibitors, tissue non-specific alkaline phosphatases and some promising antiproliferative activities. Moreover, we were also able to synthesize new rhodamines for protein labeling. We developed in our laboratory the direct arylation of three types of heterocycles targeting biological applications. The development of the direct arylation answers more or less the principles of green chemistry as this methodology can avoid the preparation of organometallic species involving especially the greater use of solvents and the formation of more wastes.
This fortunate approach, to some extent, exposed that maybe one solution facing the difficulties in finding new and more efficient drug candidates would be the emergence of new chemical technology and reactions. Indeed, on the contrary to the high-throughput approach which requires the use of well-known reactions, this new methodology should give access to more original structures. The study initiated by the sustainable development requirement should be envisioned not only as a constraint but rather as an opportunity for new methodology potentially useful not only for the pilot development and production but also for the discovery process.
1 Unpublished results.