

1 Introduction
The strong π-accepting capacity of phosphaalkene ligands has made them an innovative tool in coordination chemistry and catalysis [1,2]. Most relevant applications in catalysis have involved the use of bidentate phosphaalkene ligands such as Yoshifuji's 1,2,-diaryl-3,4-diphosphinidene cyclobutene ligands DPCB-Y (A, Scheme 1) [3] and Brookhart's phosphaalkene-sulfide and phosphaalkene-imine ligands (B) [4,5]. The exceptional efficiency of the DPCB-Y system is explained by inherent enhanced π-acceptor properties of the PC bond, [6] which is reinforced through π-conjugation with remote aromatic substituents [1–3].
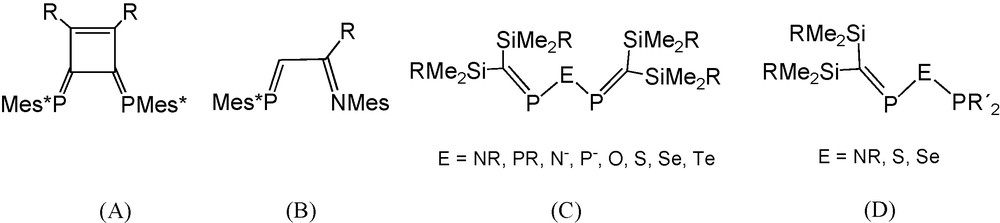
Types of bidentate phosphaalkene ligands allowing π-conjugation.
In type A and B ligands, π-conjugation is made possible by sp2 hybridised organic scaffolds. Alternatively, single heteroatoms with p-type lone pairs could play the role of a connector between the two PC systems, creating heteropentadienide-like 5c-6π electron systems in a new class of small bite angle ligands, which are the “PC-unsaturated” relatives (C) of classic imino- and imidobisphosphanes, related phosphinous anhydrides, and their heavier congeners with bridging elements from the third and fourth period (R2P)2E (E = NR, N−, PR, O, S, Se, etc.).
An alternative case would be the insertion of the potentially π-interacting heteroatom between a phosphaalkene moiety and another coordinating phosphorus atom leading to “hybrid” ligands (D).
Examples of type C and D ligands (R2CP)2E and R2CPEPR2′ are extremely rare in the literature. In the group of R. Appel the triphosphane-related compound [(Me3Si)2CP]2PtBu [7] was characterised and used as a chelate ligand toward M(CO)4 acceptors (M = Cr, Mo, W), and E. Niecke et al. reported the lithium salt Li(THF)x{[(Me3Si)2CP]2P} [8] containing a 2,3,4-triphosphapentadienide anion, which represents the first case of a π-donor heteroatom-bridged bis-phosphaalkene anion allowing electronic communication between the π-systems (PC bonds) of two neighbouring phosphaalkene moieties. Thermal and protolytic decomposition reactions of the anion were studied and anionic complexes with M(CO)4 acceptors were compared with R. Appel's uncharged compounds [7–9]. In a study on the reactivity of P-phosphanylphosphaalkenes (Me3Si)2CPPR2 towards chalcogens, type D molecules (Me3Si)2CPEPR2 (E = S, Se; R = iPr, tBu) were recognized to be products of the monoinsertion of sulfur and selenium into the PP bonds [10]. With help of a tiny 31P-NMR singlet signal exhibiting 77Se satellites in solutions containing the crude selenium compound (Me3Si)2CPSePiPr2, the selenium-bridged bisphosphaalkene [(Me3Si)2CP]2Se was fortuitously recognized [10,11]. Subsequently this compound and the related sulfide [(Me3Si)2CP]2S were synthesised and cycloaddition, chalcogen addition, and coordination reactions were studied [11]. In the context of possible application in catalysis, however, the O- and N-bridged “PC-unsaturated” compounds (type C) related to classic “saturated PNP” ligands (Ph2P)2NR appear to be particularly attractive goals. This expectation gave the incentive to focus on novel type C N- and O-bridged bisphosphaalkenes [12,13] and on type D phosphanylaminophosphaalkenes (Me3Si)2CPN(R)PR′2 [14].
In this overview, recent results on type C structures [(RMe2Si)2CP]2E (E = O, NR, N−) will be presented in Section 2 and properties of type D ligands (RMe2Si)2CPN(R)PR2 and related compounds will be discussed subseqently (Section 3). A discussion of the heavier P-, As-, S-, Se-, and Te-bridged analogs is added to each section.
2 Heteroatom-bridged bisphosphaalkenes [(RMe2Si)2CP]2E (type C structures)
2.1 Imino- and imido-bridged bisphosphaalkenes, E = NR
2.1.1 N-alkylimino-brigded bisphosphaalkenes [Ph(Me3Si)CP]2N(nC3H7)
(1), the first N-alkyl-2,4-diphospha-3-azapentadiene reported in the literature, is thermally unstable [15]. For related compounds [(Me3Si)2CP]2NR it was necessary to turn to alkali metal chloride elimination procedures based on the deprotonation of aminophosphaalkenes 2 such as (Me3Si)2CPN(H)tBu, [6] followed by coupling of the in situ-prepared anion [(Me3Si)2CPNtBu]− 3a [16,17] with P-chlorophosphaalkene (Me3Si)2CPCl (4a; Scheme 2). Since it turned out that the outcome of these reactions, i.e. the insufficient stability of the products, precluded the isolation of single-crystalline solids, precursors with bulkier silyl groups at carbon were developed. These reactions are based on the new starting material (iPrMe2Si)2CPCl 4b [18,19]. To improve crystallisation properties of thermolabile N-alkyl products, N-1-adamantyl derivatives were also used [13].
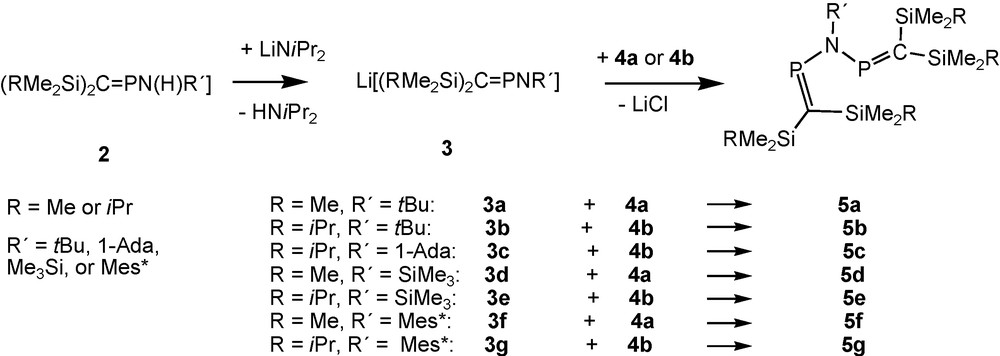
Formation of alkyl-, silyl- and arylimino-bridged bisphosphaalkenes.
Metalated aminophosphaalkenes Li[(RMe2Si)2CPNtBu] (3a: R = Me; 3b: R = iPr) react with the corresponding P-chlorophosphaalkenes at low temperatures, furnishing solutions of the desired tbutylimino-bridged bis-phosphaalkenes 5a, 5b (Scheme 2), which exhibit singlet signals in 31P-NMR [13]. Raising the temperature to 0 °C and above leads to formation of products from rearrangement reactions indicated by the occurence of one or more “growing” 31P-NMR signals that appear as AX-type patterns.
In the case of [(Me3Si)2CP]NtBu 5a, one of the new AX patterns (δ 31P = 358.4 and 55.1, J = 9 Hz; species I in Scheme 3) indicates the presence of a phosphaalkene-type 31P nucleus coupling with a tri- or tetra-coordinated phosphorus atom. The second AX pattern (δ 31P = 135 and 7.7, J = 76 Hz; species II) involves two nuclei that are both not parts of phosphaalkene functions, and a third AX pattern (δ 31P = 351.9 and 38.5, J = 107 Hz; species III) has to be assigned to another 31P(C) nucleus coupling with another tri- or tetra-coordinated phosphorus atom that is part of a 31P-C-H function (2JPH = 13 Hz) [13].
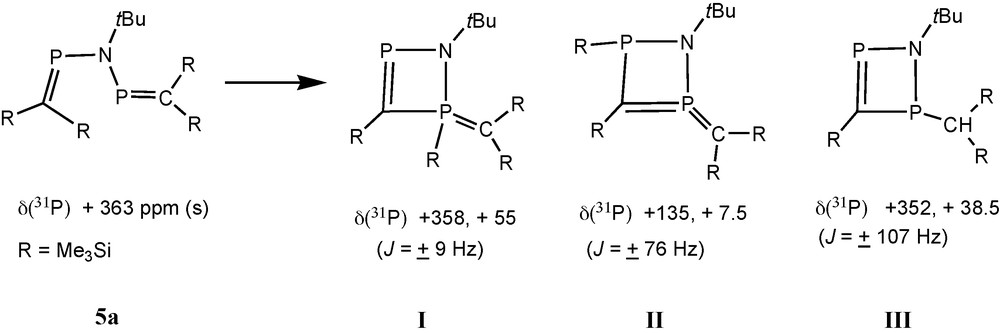
Proposed structures of products from decomposition of 5a.
Addition of 4a to a reaction mixture containing decomposing 5a leads to consumption of 5a and of rearranged species I with formation of a new compound 6 exhibiting an AMX pattern in 31P-NMR (δ 31P = 330.8, -17.1 and -24.3), involving one 31P(C) phosphorus atom coupling with two inequivalent “non-PC” 31P nuclei in a diphosphirane unit. Compound 6 is also formed from the straightforward 1:2 reaction of 3a with 4a. Single crystals of 6 were isolated from the latter reaction Eq. (1).
An X-ray crystal structure determination revealed that 6 is a bicyclic C2N2P3 species containing an endocyclic PC bond [1.679(3) Å] “in conjugation” with an exocyclic ylidic P(+)C(−)(SiMe3)2 function [1.688(3) Å] involving one of the two bridgehead phosphorus atoms that form a diphosphirane unit [13]. We assume that the motif of a (pp)π PC double bond “in conjugation” with an exocyclic ylidic P(+)C(−) bond from compound 6 is also present in species I (Scheme 3), which is a precursor to 6.
6 is derived from 4a by the reaction with 5a, or by the reaction with species I (Scheme 3) by elimination of one equivalent of Me3SiCl. Formation of rearranged products I–II from 5a involves trimethylsilyl group migration, and III is apparently a product from protolytic PSi bond cleavage, followed by proton migration to the basic ylid carbon atom.
When in a “cross” experiment anion 3a reacted in an NMR tube with the bulkier chlorophosphane 4b, or anion 3b reacted with 4a, the mixed-substituted bisphosphaalkene (Me3Si)2CP(NtBu)PC(SiMe2iPr)2 7 (AB pattern, δ 31P = 365.3 and 361.8 ppm, 2JPP = ± 18 Hz) was accompanied by only one rearranged species (AX pattern, δ 31P = 136.7 and 7.3 ppm, J = 78 Hz) that relates to the proposed species II from the “symmetric” experiment. This result indicates that the silyl group migration pathway leading to the exocyclic ylid function is closed when two bulkier iPrMe2Si groups are introduced at the carbon atom of one of the participating PC bonds (Schemes 3 and 4) [13].
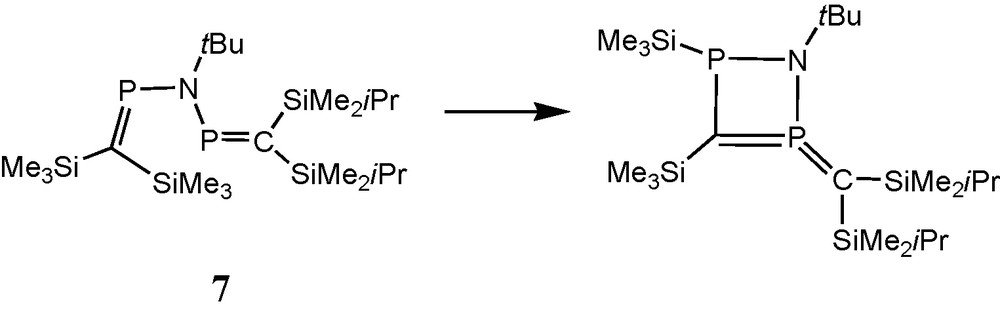
Proposed structure of a major product from the cyclisation of the unsymmetric bisphosphaalkene (Me3Si)2CP(NtBu)-PC(SiMe2iPr)2 (7).
In the case of tbutylimino-bisphosphaalkene 5b (with only iPrMe2Si groups at carbon) decomposition is significantly slower that in the case of 5a, but isolation of pure 5b was still precluded by decomposition. Low-temperature single crystal growing was enabled by switching to the corresponding N-1-adamantyliminophosphaalkene anion 3c, thus furnishing N-1-adamantyliminobis-phosphaalkene 5c, which was X-rayed as solid tetrahydrofuran solvate at low temperature. In solution, 5c exhibits a 31P-NMR singlet resonance signal, but in the solid, the two PC moieties of each independent molecule are inequivalent. Different adamantyl group orientations relative to the common “helically distorted” CPNPC skeleton in solid 5c of the three independent molecules coincide with different contacts to the solvating THF molecules (Fig. 1). The conformations of the non-planar C-P-N(R)-P′C′ backbones (in molecule #1: torsion angles C11-P1-N-P2 and C22-P2-N-P1) of the three independent molecules are similar [molecule #1: −40.9(2) and −55.6(2)°, molecule #2: −41.1(2) and −54.2(2)°, molecule #3: −48.4(2) and −48.5(2)°], the PC moieties being directed out of the NP2 planes about half-way between orthogonal [20] and planar. Within the non-planar CPNPC moieties, PC (1.664–1.670 Å) and PN bonds (1.712–1.737 Å) appear essentially undisturbed by conjugative effects. In this respect, 5c is comparable to the 2-[6-bis(trimethylsilyl)amino]pyridylimino-bridged bisphosphaalkene (Scheme 5, see also Section 2.1.3) [20].

Arrangement of the three independent molecules of THF-solvated 5c. Selected bond lengths [Å] and angles [°] of molecule #1: P1-C11 1.6670(3), P2-C22 1.6606(3), P1-N 1.712(2), P2-N 1.737(2), C23-N 1.524(13), P1-N-P2 126.77(13). P1-N-C23 119.36(18), P2-N-C23 113.72(17), C11-P1-N-P2 -40.9(2), C22-P2-N-P1 55.6(2). Hydrogen atoms are omitted for clarity. Atoms are drawn as 50% thermal ellipsoids.
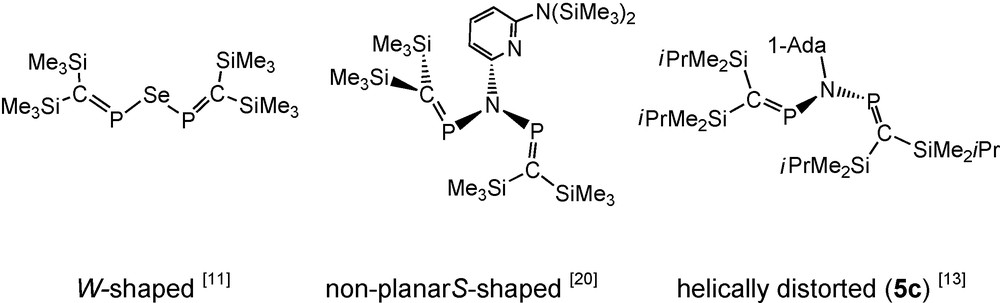
Conformations of uncharged heteroatom-bridged bisphosphaalkenes.
2.1.2 N-silyl-bridged bisphosphaalkenes
[(Me3Si)2CP]2NSiMe3 (5d), made from Li[(Me3Si)2CPNSiMe3] (3d) with 4a, is a thermally unstable oil, [9b,17,21] but bulkier (iPrMe2Si)2CP groups [19] stabilise type C compounds. The reactions of the bulkier aminophosphaalkene 2e with lithiumdiisopropylamide (LDA) in THF solution (Scheme 2), followed by reactions of the lithium salt 3e with P-chlorophosphaalkenes (RMe2Si)2CPCl (R = iPr: 4a; Me: 4b; Ph: 4c) at temperatures below −40 °C furnish solutions of N-trimethylsilylimino-bridged bisphosphaalkenes 5e and the unsymmetric compounds 8a, 8b (Scheme 6).
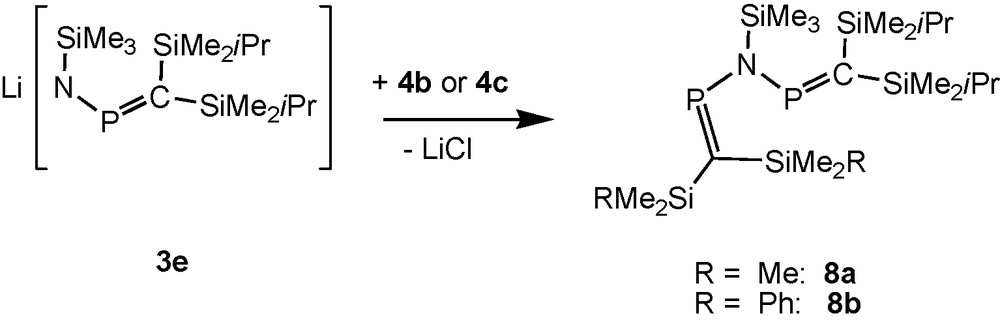
Formation of mixed substituted silyliminobis(phosphaalkenes).
The 31P NMR spectrum of 5e exhibits a single resonance whereas the mixed substituted compounds 8a, 8b show AM patterns [8a: δ 31P = 365.4 and 361.7 ppm, 2JPP = 19.6 Hz; 8b: δ 31P = 370.6 and 365.6 ppm, 2JPP = 19.7 Hz]. The NMR equivalence of the two 31P nuclei of 5e in solution would be consistent with either a symmetric structure or with a less symmetric ground state conformation (Scheme 5), which still allows free rotation of the PN bonds on the NMR time scale at room temperature in solution [19]. All the B3LYP/6-31 + G* optimized structures of the [(R3Si)2C = P]2N(SiR3) molecules (R: H, Me) were non-planar. For R = H the minima were located within an 0.9 kcal mol−1 energy range, including the S- and W-shaped forms (Scheme 5). The fact that several conformers exist with similar energies on the rotational potential energy surface is an indicative of their low energy interconversion. For R = Me only the non-symmetrical S-shaped structure could be optimized, in agreement with the X-ray structure (see below). Similar behaviour was observed in case of the di(phosphavinyl)ether analogues, [12] indicating the effect of steric encumbrance in determining the final structure. The X-ray crystal structure determinations of solid 5e and 8a at low temperature reveal (as for 5c, see above) the presence of helically distorted structures with two inequivalent PC groups (5e: torsion angles C11-P1-N-P2 and C22-P2-N-P1 -52° and -45°), but within the solid unsymmetric compound 8a all (iPrMe2Si)2CP groups and all (Me3Si)2CP groups are equivalent. The PC and PN bonds distances in 5e and 8a do not indicate significant conjugation within the CPNPC moieties [13,19,20].
2.1.3 N-aryl-bridged bisphosphaalkenes
A 31P-NMR-study on N-Mes*-bridged bisphosphaalkenes reveals that the reaction mixtures from metalation of (RMe2Si)2CPN(H)Mes* (R = Me or R = iPr) with LDA, followed by reactions of the lithium salts with chlorophosphaalkenes (3f with 4a and 3 g with 4b), each exhibit in 31P-NMR a singlet signal accompanied by an AM-pattern of [(Me3Si)2CP]2NMes*, 5f: δ = 352 (s), 329 (d) and 323 (d) ppm, 2JPP = 7.4 Hz and of [(iPrMe2Si)2CP]2NMes*, 5 g: δ = 352 (s), 332 (d) and 326 (d), 2JPP = 13.5 Hz] [13] together with another small singlet assignable to [(Me3Si)2CP]2O (353 ppm) and [(iPrMe2Si)2CP]2O (354 ppm) [12]. Apparently the bulky NMes* groups enhance the rotational barrier(s) of rotameric interconversion of symmetric 5f-sym or 5g-sym and unsymmetric S-shaped 5f-asym or 5g-asym. Initially, the 31P-NMR peak height of symmetric 5g-sym is larger than the added intensities of the AM-pattern from 5g-asym (ratio about 5:2), but after a slight relative increase of the amount of asymmetric species within 2 days, an approximately 5:3 ratio of the two species in solution stays constant for an extended period.
In the context of syntheses of pyridine-bridged bidentate phosphaalkene and iminophosphane ligands such as compound 9, the N-pyridylimido-bridged bisphosphaalkene 10 (an isomer of 9) was detected and structurally characterised [20]. Compound 10 adopts a non-planar S-shaped structure of the heteropentadiene moiety (Schemes 5 and 7). In contrast to 5c, 5e and 8a, PC functions of pyridylimino derivative 10 are directed approximately orthogonal to the CNP2 plane. According to its AM-pattern (δ31P = 340.4 and 328.9 ppm) in solution 31P-NMR at –60 °C, 10 also adopts an unsymmetric structure in solution. Equilibration of the two phosphorus nuclei on the 31P-NMR timescale takes place at room temperature (δ31P = 336.1 ppm) [20].
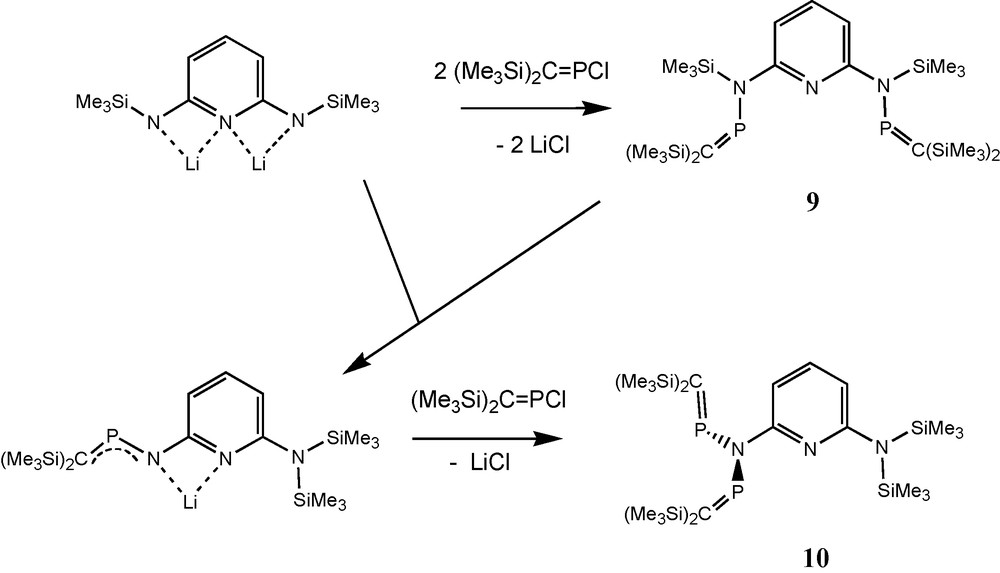
Formation of the N-pyridylimido-bridged bis(phosphaalkene) 10.
The reaction of 10 with nickel tetracarbonyl leads to a product that shows: (i) two 31P-NMR resonances at 96.1 and 66.9 ppm (JPP = ± 8.9 Hz), i.e. far upfield from ligand 10; (ii) two 13C resonances of the coordinated PC functions (75.7 ppm [JPC = ± 108.3 Hz] and 45.6 ppm [JPC = ± 103.4 Hz]; and (iii) four 13C-NMR signals in the “CO” range (192.2, 192.3, 202.0, 202.9 ppm) suggesting η2-coordination of both PC functions, one of them with an Ni(CO)2 acceptor (that will also be in contact with the pyridine N atom) and the other one with an Ni(CO)3 acceptor [20].
2.1.4 DFT calculations on 2,4-diphospha-3-azapentadienes (iminobisphosphaalkenes)
At the B3LYP/6-31+G* level have been found three minima of very similar energy on the potential energy surface of the parent [(H3Si)2CP]2NCH3 system. They are representing S-shaped, W-shaped, and V-shaped CPNPC moieties (Scheme 8). The particular non-planar distortion of compound 8 will have steric grounds [13].
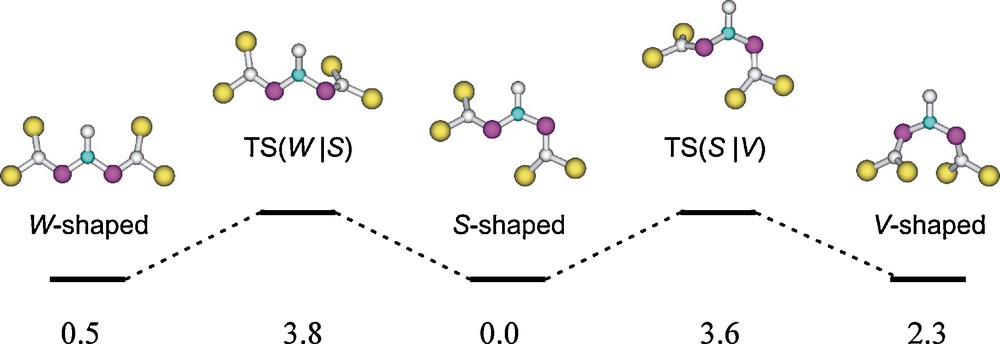
[(H3Si)2CP]2NCH3 rotamers and their connecting transition structures; relative energies (in kcal mol-1) at the B3LYP/6-31+G* level [13].
Concerning silyl group migration (Schemes 3 and 4), the reaction was calculated (Scheme 9; energies in kcal mol−1) for 5a [13].
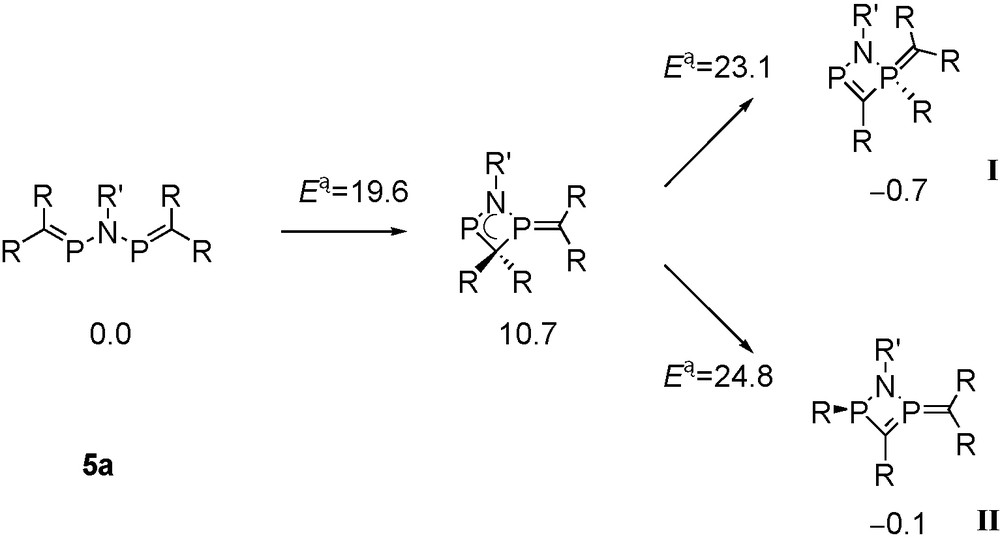
Cyclisation pathway of alkyliminobisphosphaalkenes at the MPW1K/6-311+G**/B3LYP/6-31+G* level of theory (R = SiMe3, R′ = tBu) [13].
2.2 Imido-bridged bisphosphaalkenes
2.2.1 Formation of imido-bridged bisphosphaalkene complexes
2,4-Diphospha-3-azapentadienyl anions were unknown until recently, when reactions of the N-silylimino-bridged bisphosphaalkene 5e became synthetically accessible. Experiments on the reactivity of 5e with AuCl(THT) and with [RhCl(COD)]2 allowed the observation of SiN bond cleavage under very mild conditions by chlorotrimethylsilane elimination furnishing the dinuclear complexes {Au[(iPrMe2Si)2CP]2N}2 (11) and Rh2Cl{[(iPrMe2Si)2CP]2N}(COD)2 (12), which, according to X-ray crystal structure determinations, contain 2,4-diphospha-3-azapentadienyl anions as P,P-coordinated bidentate ligands (Scheme 10) [19].
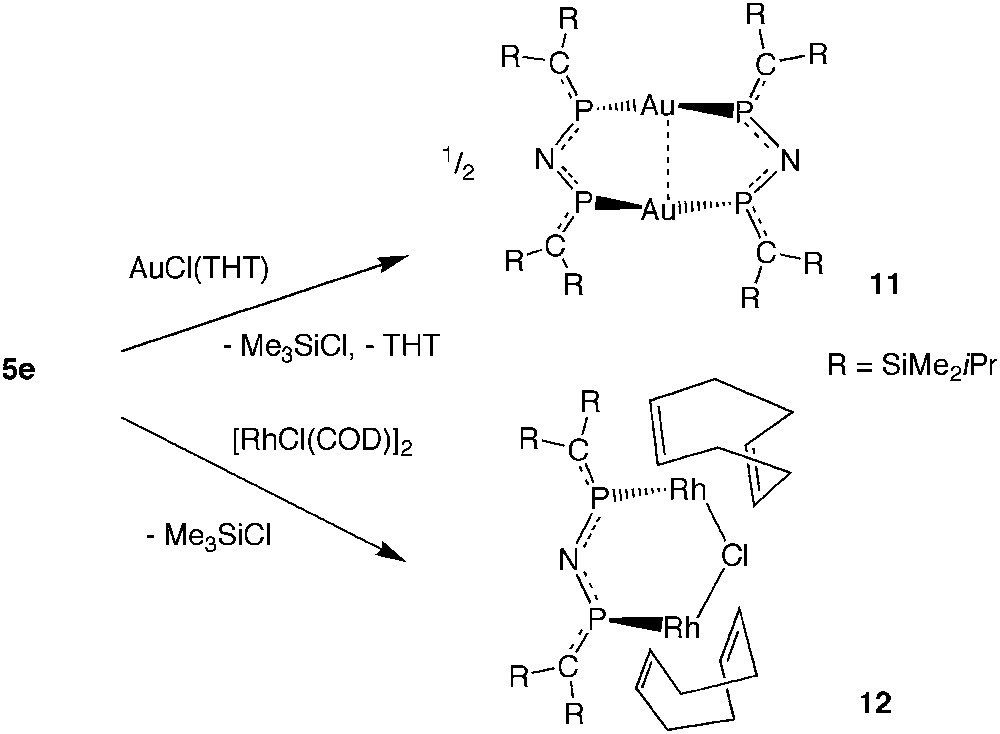
Reactions of 5e with AuI and RhI chloro complexes leading by N-Si cleavage to metal complexes of an imidobis(phosphaalkene) anion.
Yellow 11 [d(Au–Au) 3.02 Å] is related to the insoluble white bis(diphenylphosphanyl)amide gold complex [Au(Ph2P)2N]2 made first by Schmidbaur et al. and Laguna et al. [22,23] and also regarded as a dimer.
The dinuclear Rh(I) complex 12 contains as bridging ligands one chloride anion and one sterically crowded [(iPrMe2Si)2CP]2N− anion, which clearly prevents reactions of 12 with another equivalent of ligand 5e. Each of the COD ligands chelates an Rh atom as in the starting material [RhCl(COD)]2 [24]. The PN distances in 11 and 12 are about 10 pm shorter than those in the uncharged ligand 5e. Since the PC distances are unaffected or even slightly shorter in the complexes, compared with 5e, [19] the large increase of PN bond strength suggests the presence of a highly delocalized heteropentadienide 6π system.
2.2.2 DFT calculations on imido-bridged bisphosphaalkenes
DFT calculations on 2,4-diphospha-3-azapentadienes (type C structures, E = N) [19,25] reveal that, while the (CP)2NSi moieties in compounds [(R3Si)2CP]2N(SiR3) are non-planar even with the small R = H substituent, the (CP)2NSi moiety in the corresponding imide anion [(R3Si)2CP]2N− is planar in the case of R = H. This planarity, together with the short PN distances, indicates that the stabilization of the anion by delocalisation exceeds that of the amine. Indeed, the second order perturbational analysis of the Fock matrix in the NBO basis provides 37.0 kcal mol−1 stabilization energy between the nitrogen lone pair and each π(PC)* orbital, exceeding the stabilization energy for [(H3Si)2CP]2NH (19.7 kcal mol−1) significantly. [(Me3Si)2CP]2N− has an S-shaped and an energetically nearly identical, slightly non-planar W-shaped [still with 33.0 kcal mol−1 n ≥ π(PC)* interaction energy] structure.
The energetic consequences of the aurophilic interaction were estimated using the isodesmic reactions (2) and (3) in Scheme 11. The energy of the reaction (2) is remarkably large (27–30 kcal mol−1 at different levels of theory), comparable with the Au–P binding energy (30–34 kcal mol−1) [19], which was determined as the reaction energy of reaction (3). It is important to note that this unusually large interaction energy [26] is only partly attributable to the aurophilicity because the extended π-conjugation in the dimeric complex (see the MO representing π-delocalisation in Fig. 2) also contributes to the overall stabilization of this reaction.
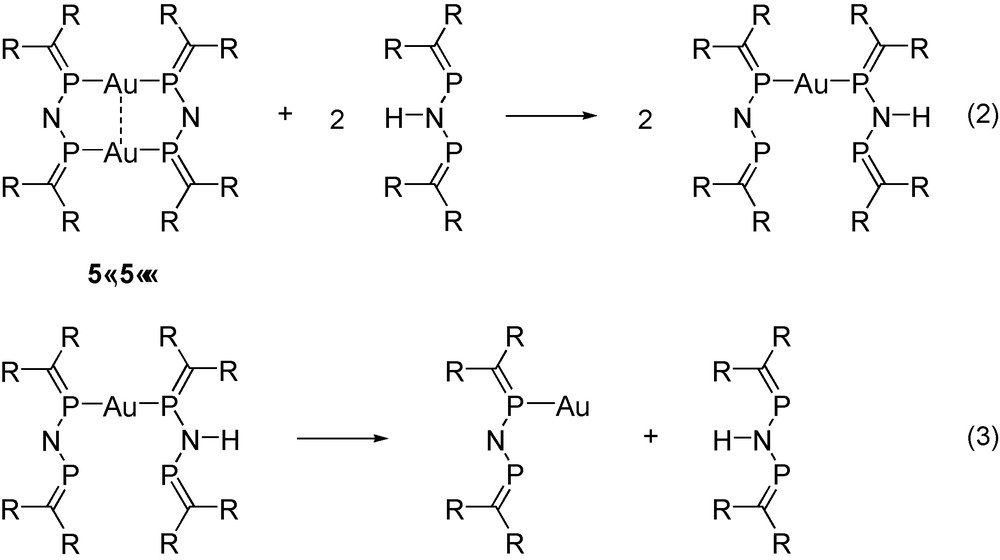
Isodesmic reactions (see text).
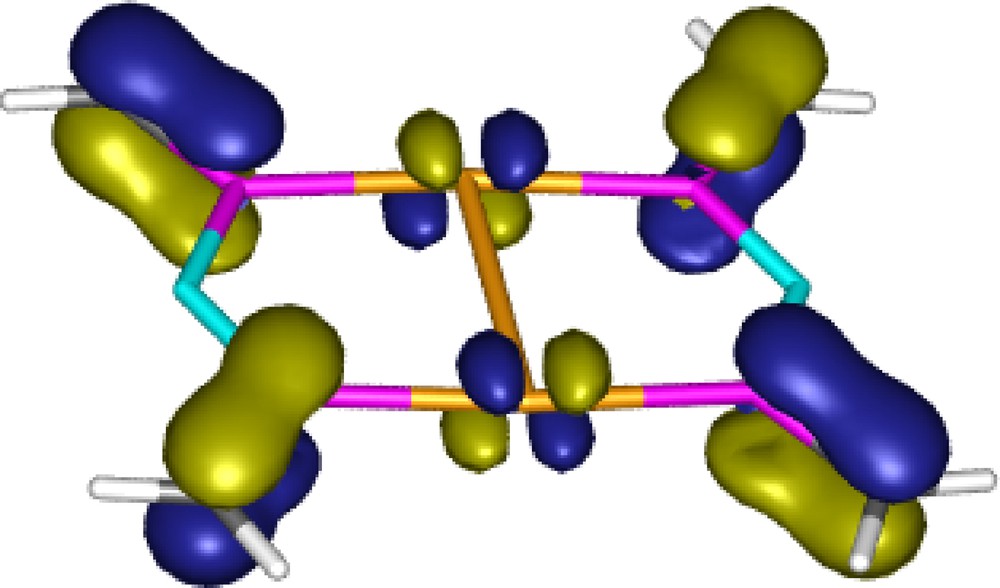
The HOMO-3 of the gold complex 11 [19].
2.3 Oxygen-bridged bisphosphaalkenes
2.3.1 Synthetic aspects
The P–O–P unit is undoubtedly the most common structural motif in phosphorus chemistry. In naturally occurring and anthropogenic phosphorus compounds, PVOPV systems with tetracoordinated phosphorus (such as P4O10 and oligo- or polyphosphates) are the most abundant. Higher coordination numbers (e.g. PVOPV with pentacoordinated phosphorus, e.g. P4O18) [27] and lower coordination numbers (PIIIOPIII, e.g. P4O6 and phosphinous anhydrides) [28] have been investigated to a much lesser extent. POP-systems involving two-coordinated phosphorus, however, are new. When studying reactions with P-halogenophosphaalkenes (Me3Si)2CPX (X = F, Cl, I) for a number of synthetic purposes, we tentatively assigned a minor 31P NMR signal at about +352 (±1) ppm in the reaction mixtures to [(Me3Si)2CP]2O (“POP”) (13a) [12] as a possible hydrolysis product. In the context of current work on related PNP-ligands [(RMe2Si)2CP]2NR′ (R = Me, iPr, Ph; R′ = tBu, Me3Si) [13,19] that exhibit, as does “POP”, 31P NMR resonances in the range of +350 to +360 ppm, it became necessary to undertake a synthetic study of the as yet unexplored unsaturated POP-system (2,4-diphospha-3-oxapentadienes = oxobisphosphaalkenes), in order to enable the unambiguous 31P NMR spectroscopic discrimination of transient PNP compounds from POP hydrolysis products.
Controlled hydrolysis of P-chlorophosphaalkenes is accompanied by formation of alkylphosphinic acids 14 (about 10–15%), but with the help of metal oxides and using more reactive iodophosphaalkenes, pure samples of oxobisphosphaalkenes are available [12] (Scheme 12).
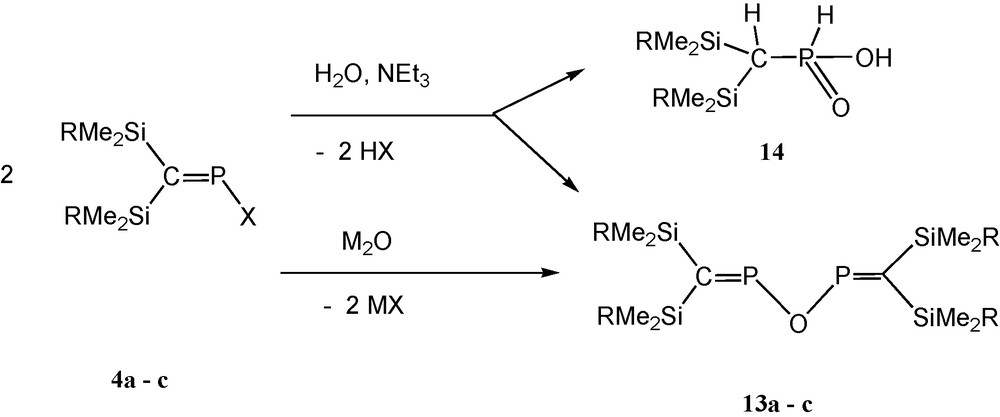
Formation of oxabis(phosphaalkenes) (M = Li, Ag; R = Me, iPr, Ph) [12].
The hydrolysis reaction of P-chlorophosphaalkenes 4 was also investigated by density functional calculations. All the calculations were performed on the model compound (H3Si)2CPCl (4′). As the hydrolysis usually starts with the substitution of the halogen by the OH group, we assumed the formation of (H3Si)2CPOH (disilylmethylene phosphinous acid) in the first step. The activation barrier of this substitution reaction involving a single water molecule (Scheme 13) is quite large. It is clearly seen in the transition structure that the direct transfer of the proton from the O to the Cl atom is energetically demanding because of the formation of the strained four-membered ring. It is known, however, that the barrier of hydrolysis reactions is lowered in the presence of three or four water molecules, which enable the proton transfer via a hydrogen bonded network in the transition structure [12]. Whereas this reaction step is somewhat endothermic, the presence of the base (which then reacts with HCl) drives the formation of the product.

Energy (E, kcal mol−1, in italics) and Gibbs free energy (G, kcal mol-1, in bold) of the hydrolysis products of (H3Si)2CPCl along the reaction coordinate. Left: formation of (H3Si)2CPOH; right: formation of (H3Si)2CH-P(OH)2 [12].
The addition of the second molecule of water at the double bond of (H3Si)2CPOH produces the disilylmethylphosphonous acid (H3Si)2CH-P(OH)2, which is a tautomer of the disilylmethylphosphinic acid 14′. The reaction is exothermic and the activation energy of this step (26 kcal mol−1) is larger than that of the first step (16 kcal mol−1). The experimental finding that 14 is formed only in small amounts besides 13, is in good accordance with the substantial activation barrier of this reaction step. The formation of the bis(phosphavinyl)ether 13′ in a condensation reaction is a well-known reaction that needs no further evaluation.
2.3.2 Structure and bonding
Among the B3LYP/6−31+G* optimized 13a and 13a′ structures (Fig. 3) the 13a isomer is more stable by 1.2 kcal mol−1. In both structures the CPOPC linkage forms a W-shape with C2 symmetry. While the corresponding structure with SiH3 groups is planar, the trimethylsilyl substituted 13a is nearly planar, and in 13a′ the deviation from planarity is substantial, presumably for a better steric accommodation of the bulky trimethylsilyl groups. The bond lengths and bond angles are in the usual ranges.
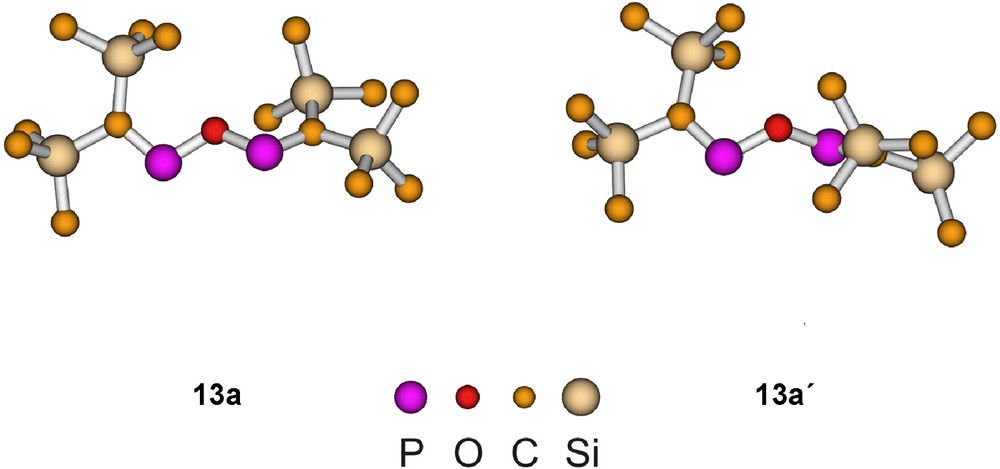
Calculated structures of conformers of [(Me3Si)2CP]2O (13a). Bond lengths and bond angles: 13a: PO 1.697, PC 1.665; POP 120.5, CPO 107.5; 13a′: PO 1.691, PC 1.666; POP 127.9, CPO 107.4.
The stabilising effect of the conjugation for the SiH3 substituted model compounds was estimated by the isodesmic reactions below (see ref. [11a] for the S analogue) (Schemes 14).
Isodesmic reactions of chalcogen-bridged bisphosphaalkenes [11,12].
The computed stabilisation energies are collected in Table 1. Reaction (4) is nearly thermoneutral for each chalcogen, showing that the stabilisation energy in the two CPOH fragments is nearly equal to that in the CPOPC moiety. Data for reaction (5) estimate the interaction between the CP and chalcogen fragments, which is 30.2 kcal mol−1 for oxygen, i.e. a remarkably high value of 15.1 kcal mol−1 for each of the CPO units. In the case of 13a, the isodesmic reaction energy is somewhat reduced to 27.0 kcal mol−1 (13.5 kcal mol−1 for each CPO unit). The slight decrease can be attributed to the non-planarity of the molecule, resulting in a weaker interaction between the lone pair of the oxygen and the π*(CP) orbitals. According to the second order perturbational analysis of the Fock matrix in the NBO basis (which estimates the energy of donor–acceptor interactions between natural orbitals) a value of 9.4 kcal mol−1 was obtained for the interaction between the oxygen lone pair and each antibonding π*(CP) orbital, which is comparable to that obtained using reaction (3) for 13a – see above.
Stabilisation energies of the reactions (4) and (5): E = O, S, Se (kcal mol-1) [11,12].
| O | S | Se | |
| (4) | −1.8 | 0.1 | −1.5 |
| (5) | 30.2 | 17.6 | 20.7 |
The conjugation effect can also be illustrated by the molecular orbitals of [(H3Si)2CP]2O (Fig. 4), which clearly show the characteristic nodal properties of five-membered conjugated systems. The energies of the isodesmic reactions (3) and (4) give similar results for the sulfur and selenium analogues, but the stabilization for the oxygen analogue is higher than for the heavier chalcogens.
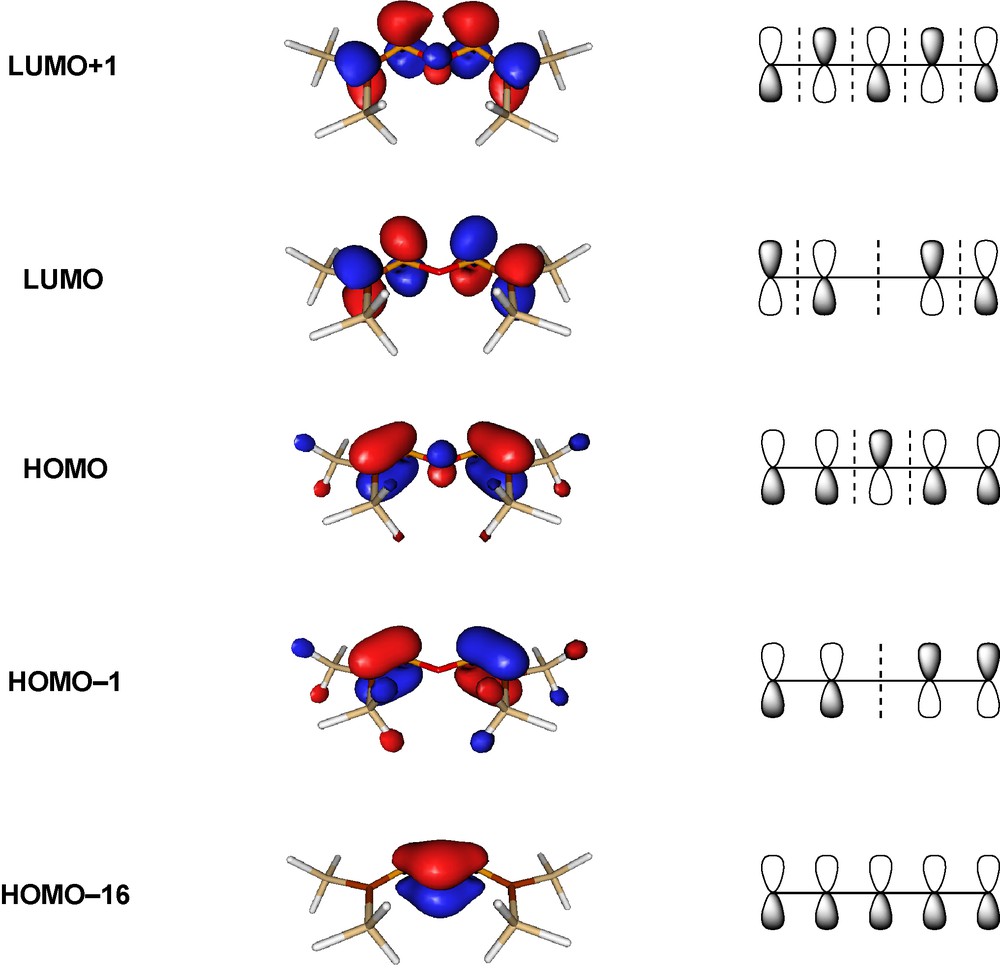
Canonic molecular orbitals of an oxabisphosphaalkene (HF/6-31G*//B3LYP/6−31+G*) [12].
2.3.3 Cycloaddition reactions
13a reacts with two equivalents of cyclopentadiene in dichloromethane at room temperature within 24 h to form the bis-[2 + 4] cycloadduct 15, accompanied by the phosphinous acid 14a because of impurities in the starting material (Schemes 12 and 15). 15 is identified by two singlet signals in the {1H}-31P-NMR-spectrum indicating the presence of new oxygen-bridged bis(2-phospha-norbornene) derivatives as pairs, either: (i) endo,endo and exo,exo isomers of diastereochemically pure RS/SR or RR/SS; or (ii) as RS/SR and RR/SS configurated products, existing either as endo,endo or as exo,exo-isomers [10]. Since in the case of [2 + 4] cycloadditions of related thio- and selenophosphaalkenes (Me3Si)2CPEPR2 (R = tBu, iPr) with cyclopentadiene, rearrangements of the endo-isomers, as initially formed, into more stable exo-isomers occurred within 24 h [10,12], we assume that both diastereomers of the phosphinous anhydride 15 exist as the exo,exo-isomers [10], as depicted in Scheme 15.
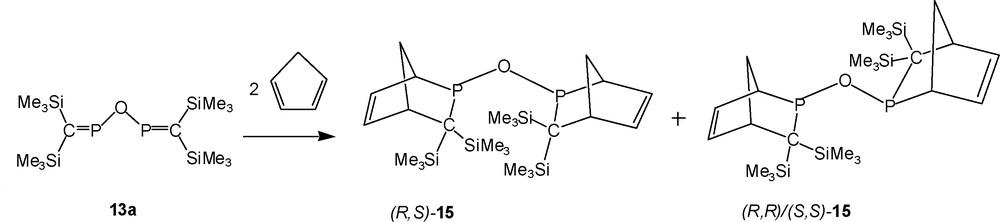
Proposed structures of products from 13a with cyclopentadiene.
The formation of the 1:2 adduct 16 from the reaction of 13a with two equivalents of tetrachloro-o-benzoquinone (TOB) [12] proceeds diastereoselectively (Scheme 16). In the recrystallisation of 16 the phosphinic acid 14a (from the impure starting material) is removed.

Reaction of 13a with tetrachloro-o-benzoquinone (TOB).
The proposed identity and stereochemistry of the oxo-bridged bis(2-phospha-2,5-dioxa-3,4-benzophospholene) derivative 16 were confirmed by X-ray crystallography. Compound 16 crystallizes as a racemic mixture of (RR) and (SS) enantiomers with approximate C2-symmetry.
The C1P1O1P2C2 moiety of 16 exhibits a distorted W-shaped arrangement, whereby the five atoms are approximately coplanar. From this central unit the heterocycles extend in opposite directions, allowing the phosphorus atoms to minimise their lone pair interactions in a “gauche”-type fashion. The angle P–O–P is 118.81(6)°, significantly larger than in the analogous selenium-bridged compound 17 [87.86(3)°] [11]. The two molecules are otherwise very similar, but the structures are not isotypic; the selenium derivative crystallizes as a chloroform solvate [11].
2.4 The heavier congeners [(RMe2Si)2CP]2E (E = PR, P−, As−, S, Se, Te,)
2.4.1 Phosphanediyl-, phosphido-, and arsenido-bridged bisphosphaalkenes
The reaction of tbutylbis(trimethylsilyl)phosphane with two equivalents of the P-chlorophosphaalkene 4a provides the 1,3-bisalkylidenetriphosphane 18 [7], which can act as a bidentate ligand towards the Mo(CO)4 acceptor moiety [1,7] leading to the chelate complex 19.
A charged equivalent of 18 is present in the lithium salt Li(THF)x{[(Me3Si)2CP]2P} 20 [8]. This 2,3,4-triphosphapentadienide anion represented in 1996 the first case of a π-donor heteroatom-bridged bis-phosphaalkene anion allowing electronic communication between the π-systems (PC bonds) of two neighbouring phosphaalkene moieties. Like the uncharged ligand 18, 20 can act as small bite angle chelate ligand towards metal carbonyl acceptors (Scheme 17).
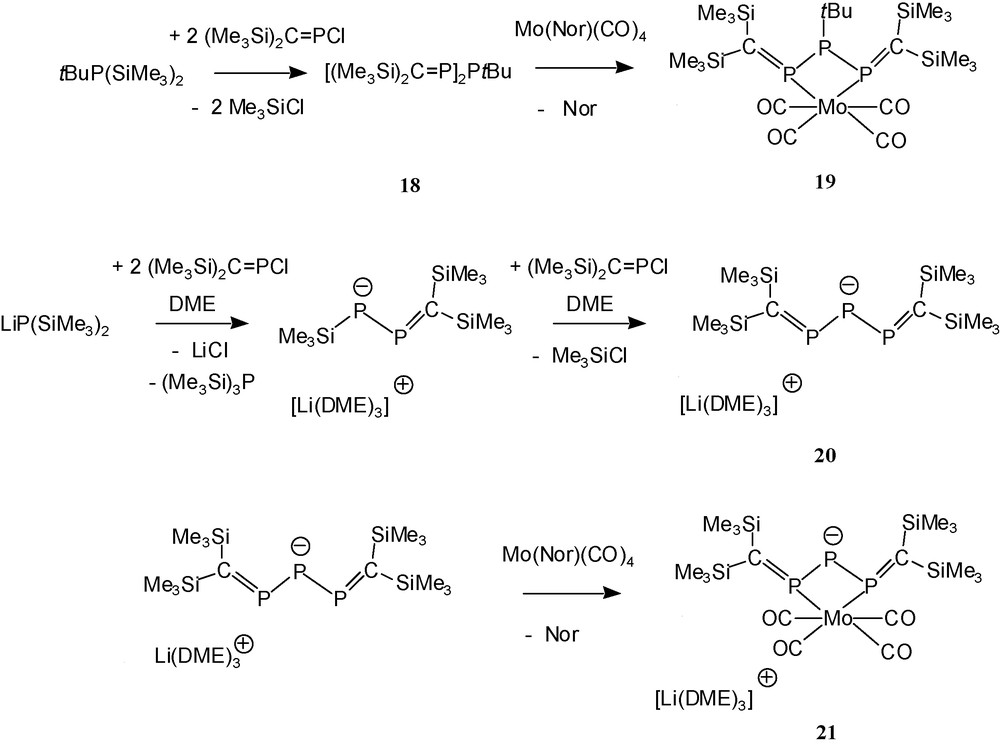
Phosphorus-bridged bis(phosphaalkenes) and their chelate complexes.
The structure determination of the ion pair-like compound 20 reveals some PC/PP π-delocalisation and a remarkably small central bond angle PPP. In the related arsenidobis(phosphaalkene) lithium salt Li(THF)x{[(Me3Si)2CP]2As} 22 [9,29], the bond angle PAsP is even smaller, suggesting attractive 1,3-P…P interactions in both anions. Comparison of the spectroscopic data of chelate complexes 19 and 21 reveals that – as expected – the anionic ligand 20 donates more electron density to the transition metal than the uncharged ligand 18.
2.4.2 Sulfur-, selenium- and tellurium-bridged bisphosphaalkenes
Sulfur-, selenium- and tellurium-bridged bisphosphaalkenes are synthetically available from (RMe2Si)2CPCl (4a–c) with bis(trialkylsilyl)chalcogenides (by chlorosilane elimination, similar as the synthesis of compound 18) or with in situ prepared lithium chalcogenides (by lithium chloride precipitation [11]). Single crystalline samples were obtained from [(PhMe2Si)2CP]2S (23c) and from [(RMe2Si)2CP]2Se (R = Me: 24a [11], iPr: 24b). In the solid, these molecules exist in slightly distorted W-shaped conformations (see Scheme 5). The angles PSP and PSeP in 23 and 24 are larger than angles PPP and PAsP in the isoelectronic anions 20 and 22.
In case of the telluride [(Me3Si)2CP]2Te (25a), the synthesis is – even at –40 °C – accompanied by immediate precipitation of tellurium allowing only the 31P-NMR-spectroscopic observation of the desired product in the black suspension, and within 30 minutes 90% of 25a has lost tellurium to furnish the known bicyclobutane isomer of the elusive diphosphabutadiene derivative [(Me3Si)2CP]2 (Scheme 18) [30].
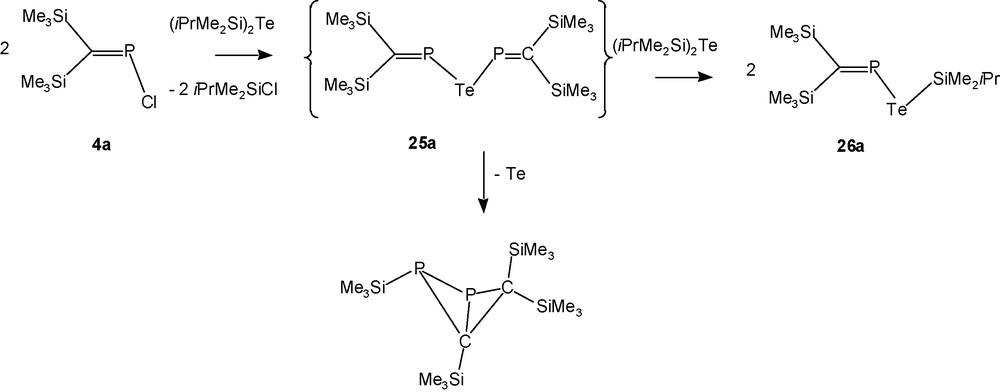
Reaction of 4a with (iPrMe2Si)2Te.
Reaction of the bulkier P-chlorophosphaalkene 4b, however, with (iPrMe2Si)2Te provides solutions containing [(iPrMe2Si)2CP]2Te (25b) in fair yields. Telluride 25b can be kept in solution in the cold for an extended time, but removal of the solvent leads to impure samples through loss of tellurium.
With two equivalents of 1,3-dienes or -heterodienes (such as tetrachloro-o-benzoquinone [TOB]) the sulfide [(Me3Si)2CP]2S (23a) and the homologous selenide 24a furnish S- and Se-bridged bisphosphanes through straightforward [2 + 4] cycloaddition reactions [13], as for 13a. With three equivalents of TOB, PIII–E–PV–units are formed (compounds 27, 28; Schemes 19 and 20).
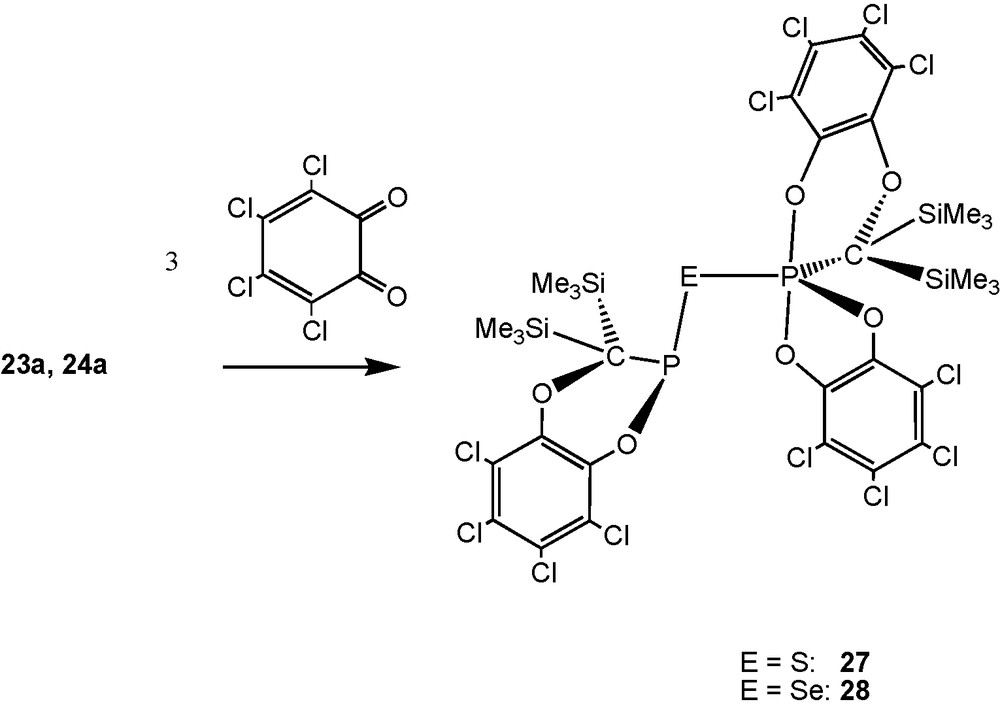
Products from cycloaddtion of [(Me3Si)2CP]2E (E = S, Se) with three equivalents of TOB.
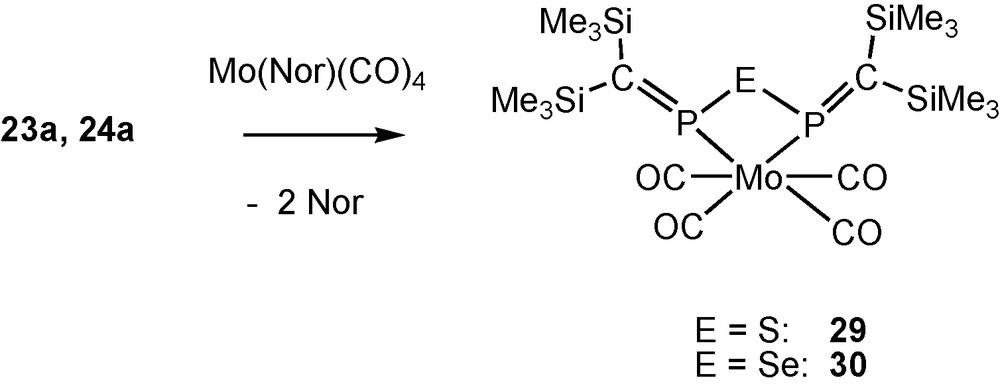
Coordination reactions of 23a and 24a.
From norbornadiene complexes of group 6 metal tetracabonyls and ligands 23a and 24a, P,P′-coordinated chelate complexes such as 29 and 30 (closely related to complex 19) are formed as expected.
With elemental sulfur and selenium, however, the heteropentadienes [(Me3Si)2CP]2E 23a, 24a react in a surprising way that is dissimilar from that of comparable monofunctional phosphaalkenes, delivering bicylic compounds with norbornane-type P2Se5-related [(Me3Si)2CP]E3 backbones (E = S, Se).
[(Me3Si)2C]2P2S5 (33, PV,PV) (Scheme 21) was obtained crystalline as the final product of the addition of sulfur to 23a (Scheme 22).
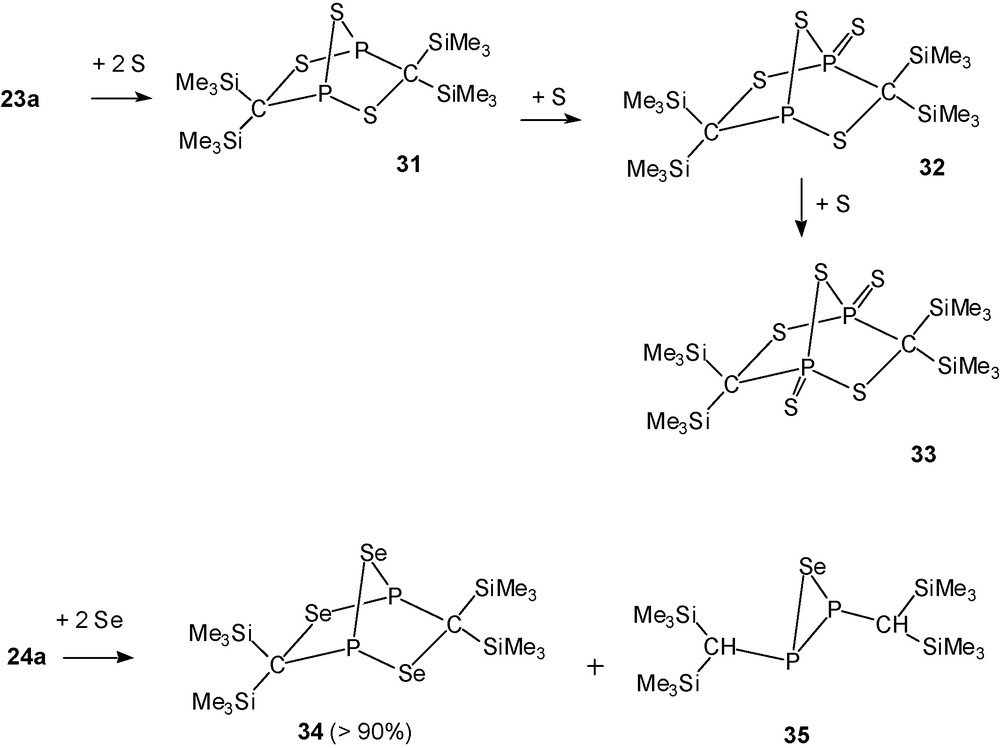
Sulfur addition to 23a and selenium addition to 24a.
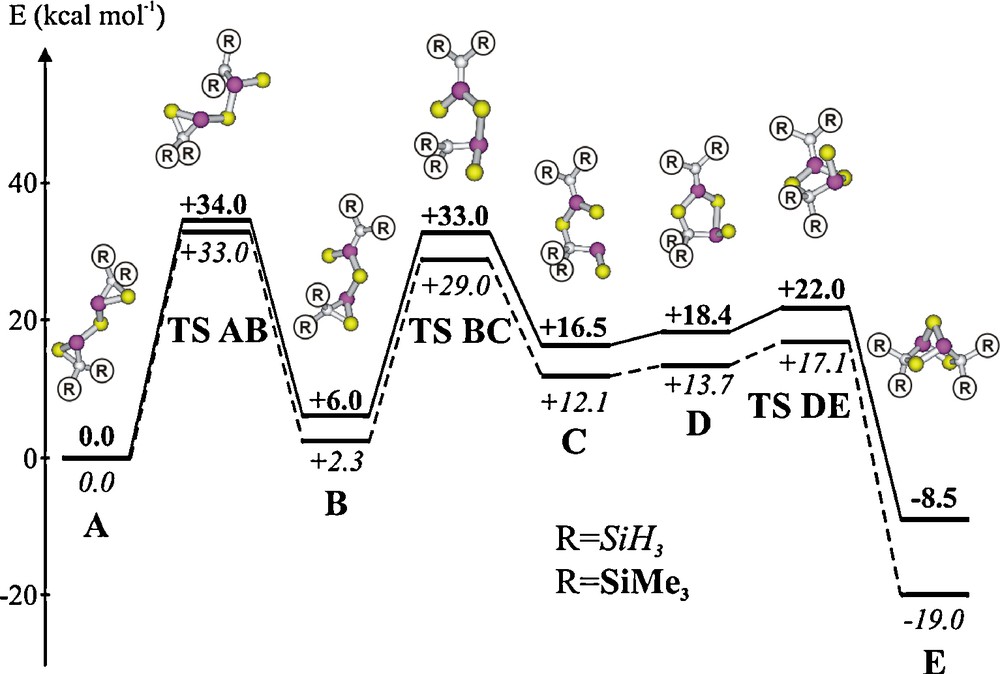
Calculated pathway of isomerisation of a bis(thiophosphiranyl)sulfide A (energies of R = SiH3 in italics and R = SiMe3 in bold, in kcal mol-1) to the related heteronorbornane E.
In a similar way the reaction of 24a with two equivalents of Se provides in high yield [(Me3Si)2C]2P2Se3 (34, PIII, PIII) as a stable yellow solid that is not further oxidised by selenium. Crude 34 is accompanied by minute amounts of P,P′-di[bis(trimethylsilyl)methyl]selenadiphosphirane (35) [11].
The formation of the heteronorbornane-like C2P2E3 cages 31–34 is unexpected because related thia- and selenaphosphiranes do not dimerise under comparable conditions. Quantum chemical calculations on the rearrangements of a bis-phosphirane type model compound A into the norbornane-type isomer E reveal that the most favourable pathway involves a type phosphirane/phosphorane intermediate B [11].
2.4.3 Discussion of the heavier congeners [(RMe2Si)2CP]2E
Containing a W-shaped central SePEPSe moiety that is close to planarity, the selenium-bridged bis-phosphaalkene [(Me3Si)2CP]2Se (24a) [11] is structurally related to Nieckes’ 2,3,4-triphosphapentadienide anion 20 {[(Me3Si)2CP]2P}− [8], which represents a π-donor heteroatom-bridged bis-phosphaalkene anion that allows electronic communication between the π-systems (PC bonds) of two neighbouring phosphaalkene moieties. Recently, it was reported that in the related arsenide anion {[(Me3Si)2CP]2As}− (isoelectronic with 20) the angle PAsP shrinks to 85.8°; quantum chemical calculations implied C–P–As π-delocalisation of the negative charge, including island homoaromaticity for the AsP2 moiety [8,9]. Crystallographic data of 23c and 24a, however, do not provide evidence of a significant ground state π-conjugation in the CPEPC (E = S, Se) system. The reactivity of 23a and 24a is towards sulfur and selenium is markedly different from that of monofunctional phosphaalkenes [11].
3 Phosphanylaminophosphaalkenes (RMe2Si)2CPN(R)PR′2
In this section, initial results of the characterisation of the novel complexes M[(Me3Si)2CP(Cl)N(1-Ada)PPh2]2 (1-Ada = 1-adamantyl; M = Pd, Pt) are presented, which are unprecedented in phosphaalkene coordination chemistry [14].
3.1 Ligand synthesis
Synthetic experiments on the type D (Scheme 1) ligands showed that (Me3Si)2CPCl fortuitously reacted with lithium iminophosphanides Li[Ph2PNR], [31] prepared in situ from the aminophosphanes Ph2PN(H)1-Ada [32] and Ph2PN(H)tBu with LDA, [33] to give the P-(phosphanylamino)phosphaalkenes (Me3Si)2CPN(1-Ada)PPh2 (36) and (Me3Si)2CPN(tBu)PPh2 (37), [14,17] whereas the reaction of the 1-aza-2-phosphaallyl anion [(Me3Si)2C=PNtBu]− [16,17] with R2PCl led preferentially by PP bond formation to the undesired phosphanylphosphorane isomers (Me3Si)2CP(NtBu)PR2 [17]. The molecular structure of solid 36 was determined by X-ray crystallography [14].
3.2 Platinum and palladium dichloride complexes
After adding a solution of the ligand 36 in an aprotic solvent to an equimolar amount of the PtCl2 cyclooctadiene (COD) complex suspended in dichloromethane, 31P-NMR spectra of the orange solution indicate the complete consumption of 36 [AX-pattern, δ = + 373.5, +37.7 ppm, 2JPP = ±12 Hz] and the formation, depending on the reaction conditions, of up to three new species exhibiting AX-patterns that all appear in the chemical shift range of +60 to +15 ppm, showing couplings 2JPP and 1JPtP. Sets of further weak 31P-NMR signals are assigned to an A2M2 pattern [δ = +71, +42; 2JPP = ±21 Hz], and tentatively to higher order multiplets, partially hidden by platinum satellites of the stronger signals. An X-ray structure determination revealed that the compound with the A2M2-like pattern is a very unusual 1:2 chelate complex of PtCl2 with ligand 36 [14]. When two equivalents of ligand 36 were used to favour the formation of the 1:2 complex, ligand 36 and the coordinated species giving rise to AX-patterns were consumed within several days according to 31P-NMR, that showed, inter alia, overlapping signals of multiplets including the A2M2 pattern of 38a as a major product.
The X-ray crystallographic structure determination confirmed that the solid 38a is a complex of the composition [PtCl2(36)2] (as a 1:1 pentane solvate) with tetra-coordinated platinum surrounded by two trans-oriented P,P′-chelating ligands in a square-planar fashion, but is uniquely different from usual cationic platinum bis-chelates of the type {[PtL4]2+(X−)2}; there are covalent bonds between the phosphorus atoms of the hypothetical cationic moiety [Pt(1)2]2+ and the two chlorine atoms. The related 1:1 reaction of ligand 1 with PdCl2(COD) provided a few crystals of the solid pentane-solvated palladium complex 39, which is isotypic to 38a (Scheme 23) [14].
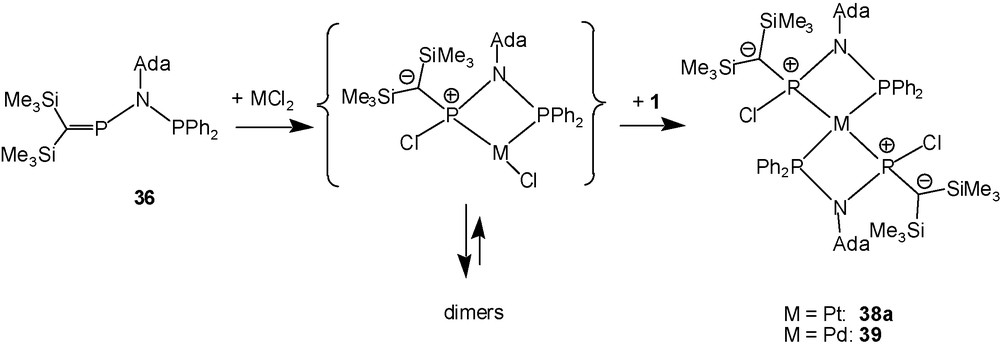
Reaction of 36 with Pt and Pd dichlorides.
The addition of two chloride ions to the phosphaalkene phosphorus atoms of the hypothetical cationic moiety trans-[M(36)2]2+ (M = Pd, Pt) leads formally to two four-coordinated stereogenic phosphorus atoms P(C)(N)(Cl)(M) in the resulting molecular complexes. Complexes 38a and 39 consist of centrosymmetric molecules with trans-orientation of the two types of phosphorus atoms around palladium and platinum; the chlorine atoms are anti-oriented, one above and below the square plane (R,S). The alternative (R)- or (S)-configurations at phosphorus, together with the intrinsic possibility of cis- or trans-P,P’-chelate ligand orientations around square planar platinum or palladium, mean that up to four stereoisomers 38a–38d (or 39a–39d) are possible (Scheme 24).
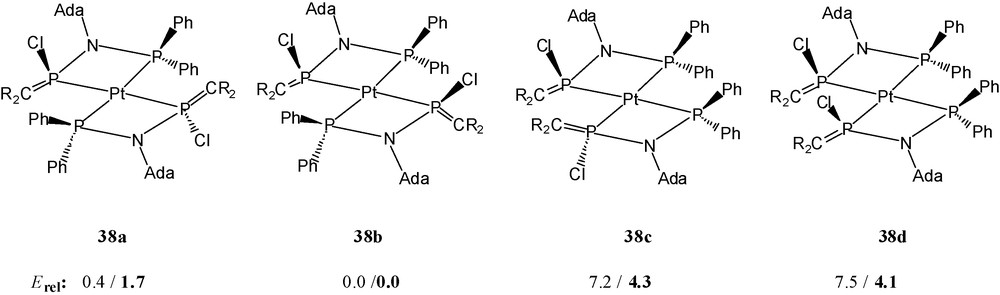
Relative energies of complexes related to isomers 38a–38d (R = Me3Si) for ligand 36’, for 36” in boldface [(H3Si)2CPN(Me)PR’2, R’: Me (36’), Ph (36”)], in kcal mol-1 [14].
Variation of the reaction conditions and of the solvent mixtures for crystallisation allowed the isolation of a few single crystals of two further isomers 38b and 38c (Scheme 24) of the platinum complex 38. Solid 38b and 38c exist as racemic mixtures of (R,R) and (S,S) enantiomers.
Bond distances and angles of 38b and 38c show no significant differences to those of 38a. In all three isomers, each bidentate ligand 36 unit has acquired one chlorine atom bonded to a (formerly) phosphaalkene phosphorus atom. This phosphorus atom acts as a σ-donor towards platinum and as a σ-acceptor towards the chlorine atom, i.e. the phosphaalkene phosphorus atom has inserted in a carbene-like fashion into the PtCl bond. In order to understand the remarkable electrophilicity of the P-(phosphanylamino)phosphaalkene ligands, model calculations based on density functional theory have been performed [14]. On the basis of these results, the known strong electron acceptor behaviour of the π*(CP) unit [6] is remarkably increased by the silyl groups (the LUMO energy drops). The effect is corroborated by considering phenyl groups at the σ3, λ3-P atom instead of methyl groups. Further enhancement of the electrophilicity is achieved for the entire ligand by the metal complexation [14].
The coordination of ligand 37 to platinum dichloride in dichloromethane solution competes with a reaction that involves elimination of chlorotrimethylsilane, furnishing as a yellow solid the cyclic C-metallophosphaalkene 40, which exists according to NMR as an isomeric mixture of Cl-bridged dimers. Crystallisation of this material from acetonitrile led to a monomeric acetonitrile complex 41 (Scheme 25). Complex 41 is expected to be formed from a P-chloroylide complex that may undergo β-elimination of Me3SiCl, leading to a kind of silyl(phosphanyl)carbene intermediate. From this species, P→C migration of platinum can lead to the unusual C-metalated cyclic phosphaalkene that crystallises as acetonitrile complex 41 [21b,32].

Chlorosilane elimination following the reaction of ligand 37 with PtCl2.
In the course of the attempts to isolate the palladium complex 39 and related species based on ligands 36 and 37, we fortuitously isolated a unique tetranuclear hexachloropalladium complex 42 consisting of two planar Pd2Cl3 moieties connected by two of the bidendate anionic hydrogenated phosphane/phosphido ligands [Ph2PN(tBu)P(−)CH2SiMe3]− [21b,32]. Each phosphido function brigdes two Pd atoms from the two different planar Pd2Cl3 moieties, and the PPh2 groups play their usual role as terminal ligands. The anion (Scheme 26, below/right) is related to the C-coordinated anionic ligand in the platinum complex 41 by addition of two hydrogen atoms to the “carbene” carbon atom (of the “carbene” intermediate, see Scheme 25).
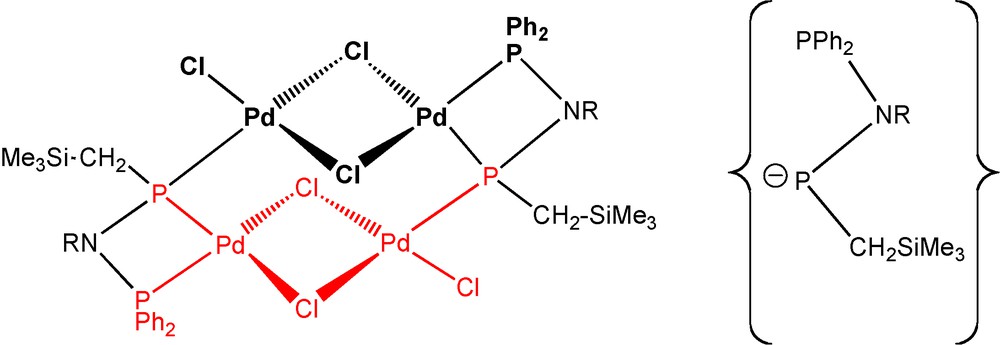
Left: topolgy of complex 42; right: an anionic ligand in 42; R = tBu [21b,32].
3.3 Heavier congeners: sulfur- amd selenium-bridged “hybrid ligands” (Me3Si)2CPEPR2 (E = S, Se)
In a study on the reactivity of P-phosphanylphosphaalkenes (Me3Si)2CPPR2 towards chalcogens, type D molecules (Me3Si)2CPEPR2 (R = iPr, tBu; E = S: 43; Se: 44) were recognized to be products of the monoinsertion of sulfur and selenium into the PP bonds [10]. Subsequent chalcogen addition leads by PIII→PV oxidation of 43 and 44 to dithio- and diselenophosphinato-phosphaalkenes (Me3Si)2CPEP(E)R2. This oxidation is followed by S or Se addition to the PC double bond, providing P-dithio- and diselenophosphinato-thia- or -selenaphosphiranes as final products. Since pure 43 and 44 could not be isolated, the coordination with M(CO)4 moieties was used as a trapping reaction. This led also to the crystallographic characterisation of a molybdenum complex 45 of the “hybrid” ligand (Me3Si)2CPSeP(iPr)(tBu) (44a) [10] (Scheme 27).
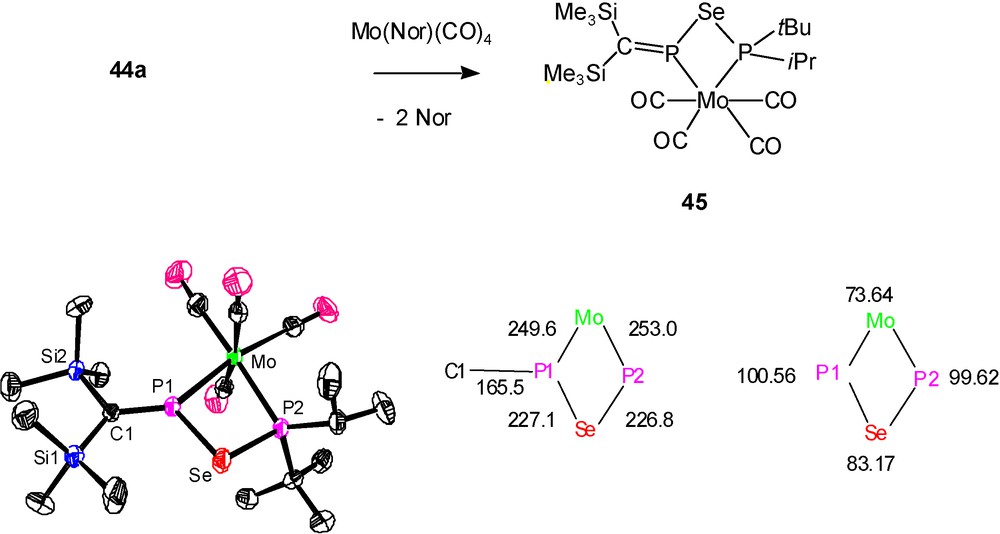
Formation (above) and structural features (below) of complex 45: molecular structure (left), distances (central) and angles (right) [10b].
4 Conclusion
Following previous work on heavier heteroatom-bridged bifunctional phosphaalkenes [(RMe2Si)2CP]2E (E = S, Se, Te, PR, P−, As−) and (RMe2Si)2CPEPR’2 (E = S, Se, Te), nitrogen- and oxygen-bridged bidentate phosphaalkene ligands [(RMe2Si)2CP]2E (E = O, NR′, N−; R = Me, iPr, Ph) and (RMe2Si)2CPN(R′)PPh2 are now accessible by salt elimination methods from the corresponding P-halogenophosphaalkenes (RMe2Si)2C=PX with lithium imidophosphaalkenes and imidophosphanes. Alkyliminobisphosphaalkenes without bulky C-iPrMe2Si substituents undergo intramolecular rearrangements leading to mixtures of four-membered heterocycles. The stable N-trimethylsilylimino derivative [(iPrMe2Si)2CP]2NSiMe3 reacts with AuI and RhI chloro complexes with NSi bond cleavage furnishing binuclear complexes of the 6π-delocalised imidobisphosphaalkene anion [(iPrMe2Si)2CP]2N−. The most exceptional property of P-phosphanylaminophosphaalkene ligands (RMe2Si)2CPN(R′)PPh2 is the chlorotropic formation of molecular PdII and PtII metallochloroylid complexes. Several stereoisomers of the 2:1 complex 38 were characterised by X-ray crystallography. Chlorosilane elimination from an intermediate P-platina-P-chloroylid 1:1 complex (Scheme 25) can explain the formation of the unusual C-platina phosphaalkene complex 41 and of the tetranuclear palladium complex 42, suggesting a considerable synthetic potential of the chloroylid complexes derived from P-phosphanylaminophosphaalkene ligands.
Acknowledgment
The COST CM0802 action and the DFG (MO290) are acknowledged for financial support. We thank Professor Ludger Ernst and Dr. Ulrich Papke (both TU Braunschweig) for support with heteronuclear NMR (L.E.) and with mass spectrometry (U.P.), and Professor Peter G. Jones for X-ray crystallography and his help to finalize the manuscript.