

1 Introduction
In 1981, a new concept of bulky protecting groups was introduced for the kinetic stabilization of highly reactive unsaturated bonds between the main group elements heavier than the second row elements [1]. Since then, many experimental efforts have been directed toward a variety of unsaturated compounds of the heavy elements [2], including the Si=P compounds, i.e., phosphasilenes [3]. In 1984, Bickelhaupt et al. reported the first stable compound containing a Si=P double bond [4a]. Since then, various phosphasilenes have been prepared by taking advantage of the steric protection with appropriate bulky substituents [4–7]. For example, the first X-ray structural analysis of phosphasilene was reported by Niecke et al. in 1993 [5a]. A series of phosphasilenes having bulky silyl groups at the phosphorus atom have been studied by Driess et al. [6a–d]. The “half”-parent phosphasilene with a PH group and its metallated compounds were also reported [6e,f]. However, despite the unconventional polar heavy double bonds, the electronic properties of the phosphasilenes have been only slightly explored; weak absorptions probably due to the n–π* transition have been reported only in rare cases [4c, 6c]. Very recently, a “push-pull” phosphasilene was synthesized by Sekiguchi and Escudié groups [7], the EPR studies of its anion radical species was reported.
Last year, we reported the first experimental evidence for the π-conjugation between the Si=P double bond and the carbon π-electron system linked to the silicon atom [8]. Organic π-conjugated architectures comprised of multiple bonds of the heavier main group elements have attracted much attention because of their potentially useful properties and unique functions [9]. For example, a variety of oligomers and polymers encapsulating P=E (E = C, P) [10] and Si=Si [11] bonds have been investigated, which would open the way to new elemento-organic hybrid materials. In 2007, Scheschkewitz's group [12] and we [13] independently reported the synthesis and characterization of the disilene analogues of the oligo(p-phenylenevinylene)s (Si-OPVs) (Scheme 1). In our case, the newly developed fused-ring bulky 1,1,3,3,5,5,7,7-octaethyl-s-hydrindacen-4-yl groups, abbreviated as “Eind” groups (“E” for ethyl), effectively protect the reactive Si=Si fragments, thus providing the highly coplanar arrangement of the Si-OPV dimer framework. Notably, our 1,4-bis(disilenyl)benzene derivative showed a unique fluorescence at room temperature based on the efficient π-delocalization over the skeleton.
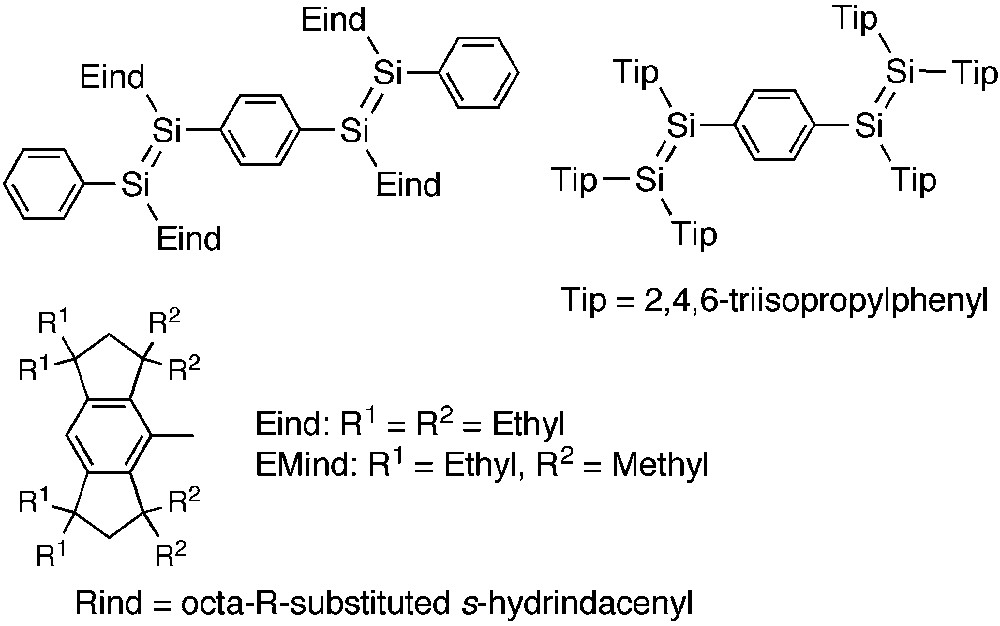
Si-OPV dimers and Rind groups.
After the initial findings of the effectiveness of the Eind groups in the Si-OPVs, we concentrated on the development of π-conjugated systems containing heavy unsaturated bonds by taking advantage of the bulky “Rind” groups based on a fused-ring octa-R-substituted s-hydrindacene skeleton [14], where R denotes the initial of the substituents. For example, 1,1,7,7-tetraethyl-3,3,5,5-tetramethyl-s-hydrindacene-4-yl group, abbreviated as an “EMind” group (“M” for methyl), is sometimes useful as the less bulky group. In this report, we review our recent results for a series of π-conjugated phosphasilenes and their gold(I) complexes.
2 Preparation of π-conjugated phosphasilenes
At the beginning of this study, as shown in Scheme 2, we examined the one-step coupling reaction between the Eind-dichlorophosphine 3 and Eind-dilithiosilane 2a, which was formed by the reaction of the dibromosilane 1a with lithium naphthalenide (LiNaph) in THF at low temperature [7,15]. This reaction, however, led to a mixture of the Eind-stabilized diphosphene 4 [16], phosphasilene 5a, and disilene 6 [13]. We have isolated 4 as orange crystals, 5a as yellow crystals, and 6 as orange crystals in 20, 33, and 30% yields, respectively, using silica gel chromatography in a glove box filled with argon gas. However, this synthetic approach seemed to be not suitable for the large-scale synthesis of the phosphasilenes.

Synthesis of 4, 5a, and 6.
The selective synthesis of phosphasilenes has been achieved by the stepwise construction of the Si=P double bonds, starting from the Eind-phosphine 7 as shown in Scheme 3. The coupling reaction between the lithium phosphide (8) and 1a and subsequent removal of HBr from the resulting phosphasilane 9a with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) afforded the Eind-stabilized phosphasilene 5a in 65% yield. Similarly, we synthesized a series of Eind-stabilized phosphasilenes 5b–g with various aryl substituents at the silicon atom: 2-naphthyl (5b), 2-anthryl (5c), 2-fluorenyl (5d) [17], 1-naphthyl (5e) [17], 2-thienyl (5f) [17], and 2,2’-bithienyl (5 g) [17]. Each compound was isolated as yellow to yellow-orange crystals in 15–35% yields. We have also synthesized the 1,4-bis(phosphasilenyl)benzene 12, in which two Si=P bonds are connected by a para-phenylene linker as shown in Scheme 4. Thus, the reaction of 1,4-bis(dibromosilyl)benzene 10 with 2 equiv. of 8 and subsequent treatment with DBU produced 12 as orange crystals in 29% yield.
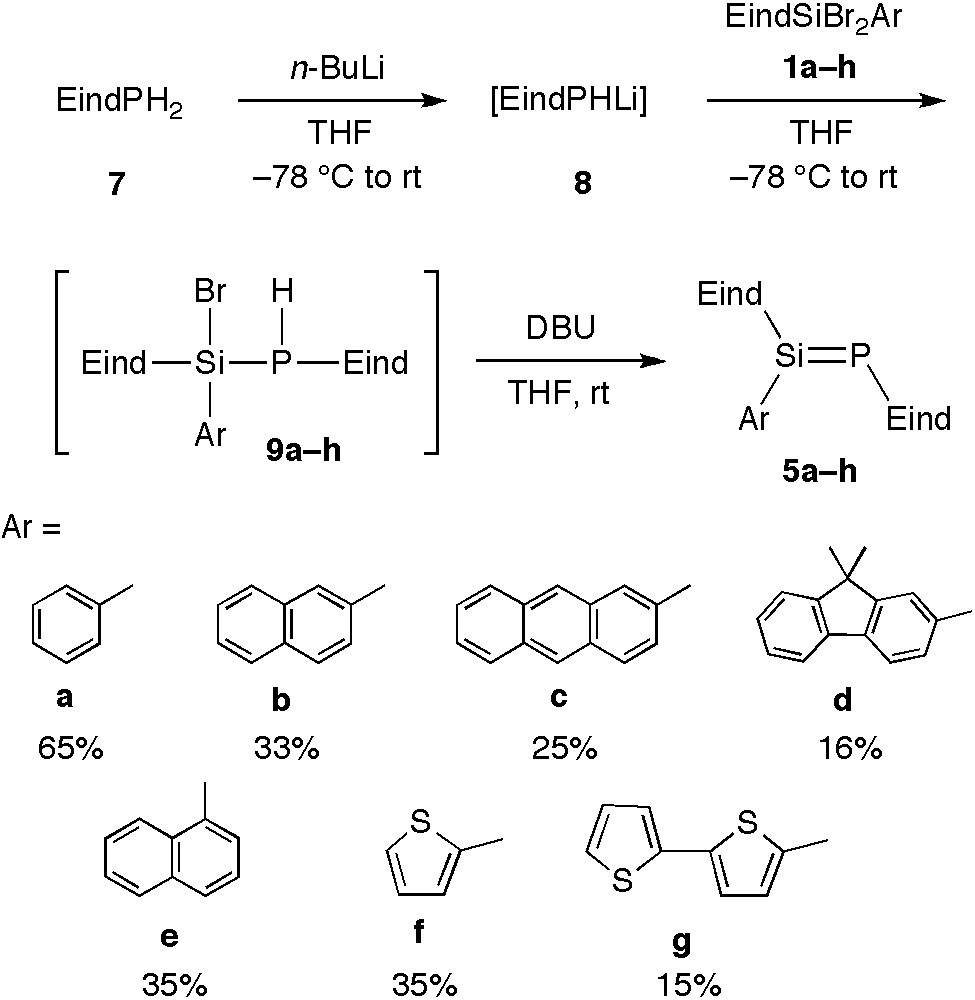
Synthesis of 5a–g.

Synthesis of 12.
The 29Si and 31P NMR spectral data of 5a–g are listed in Table 1. In the 29Si NMR spectra of 5a–d, 5f and 5 g, one signal is observed at δ = 147.7–162.1 ppm as a doublet with a 1JSiP coupling constant of 171–178 Hz. The characteristic 31P NMR signal is found at δ = 77.2–89.7 ppm. Compound 5e exhibits two signals in both the 29Si and 31P NMR spectra, respectively, indicating the existence of two rotational isomers around the Si–Cipso(aryl) bond, the s-cis and s-trans, in solution as shown in Fig. 1. However, all attempts to separate the isomers have been unsuccessful. For compound 12, the solid-state NMR experiments have been utilized due to its poor solubility in organic solvents. Both the 29Si and 31P NMR spectral peaks appear as single signals, indicating a highly symmetrical structure for 12 in the solid-state, which is in agreement with the crystal structure (vide infra).
Selected NMR spectral data for phosphasilenes.
| Compound | δ (Si, ppm) | 1JSiP (Hz) | δ (P, ppm) |
| 5aa | 161.7 | 171 | 89.2 |
| 5ba | 161.9 | 174 | 89.7 |
| 5ca | 162.1 | 174 | 89.2 |
| 5da | 160.0 | 174 | 82.5 |
| 5ea | 149.6 (major) | 183 | 84.0 (major) |
| 147.9 (minor) | 180 | 84.4 (minor) | |
| 5fa | 147.7 | 178 | 77.2 |
| 5ga | 148.6 | 175 | 81.5 |
| 12b | 160.9 | 182 | 109.1 |
| 97.9c |
a In benzene-d6 at 25 °C.
b In the solid state.
c In THF-d8 at 65 °C.
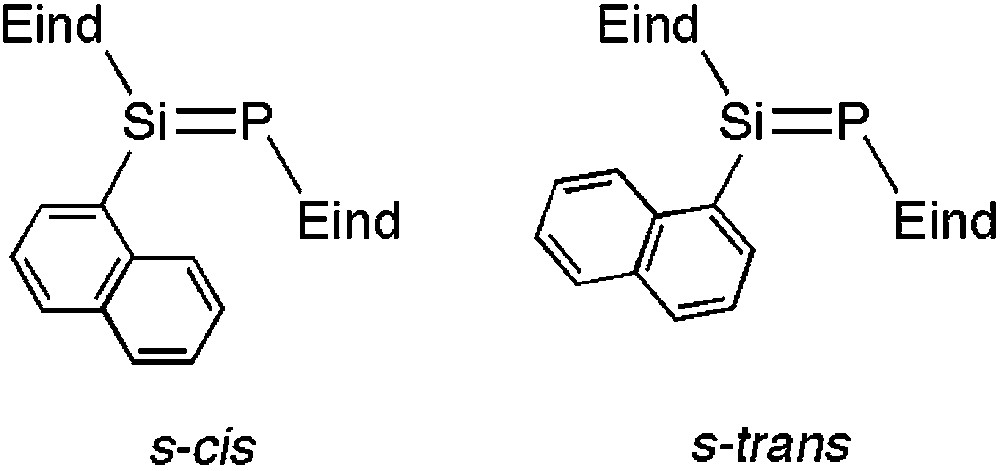
Rotational isomers of 5e.
The resulting phosphasilenes are stable in air for months in the solid state and for a week as a dilute solution, demonstrating the efficient protection ability of the Eind groups. Each compound has a high thermal stability; 5a–g melted at > 200 °C without decomposition under an argon atmosphere in a sealed tube. The dimer 12 has decomposition points of > 340 °C under an argon atmosphere in a sealed tube.
3 X-ray structural aspects
The molecular structures of 5a–c and 12 have been determined by X-ray crystallography; their structural parameters are summarized in Table 2, and two of them, 5a and 12, are reproduced in Figs. 2 and 3. The aryl and the arylene groups are entirely coplanar with the Si=P double bond(s), having the P–Si–C–C torsion angles of 3.04(13) (5a), 1.1(4) (5b), 2.0(2) (5c), and 0.8(2)° (12), respectively, which is well suited for the 2pπ–3pπ conjugation. This coplanarity is apparently attained by the rigid Eind groups, oriented trans and perpendicular with respect to the Si=P bond, as observed in the disilene case [13]. The space-filling model of 12 shows that the peripheral ethyl groups effectively protect the Si=P moieties (Fig. 4). The ethyl side chains on the rigid s-hydrindacene skeletons interlock with one another above and below the Si=P moiety to enforce the planar geometry. The resulting cavity surrounded by the ethyl groups can fix the framework aryl groups in a coplanar conformation with the P=Si plane, which is ideal for the extension of the π-conjugation.
Selected X-ray crystal structural data for phosphasilenes.
| Compound | Bond lengths (Å) | Torsion angle (°) | |||
| Si=P | Si–C(Eind) | Si–C(aryl) | P–C | P–Si–C–C | |
| 5a | 2.0917(5) | 1.9006(12) | 1.8633(13) | 1.8706(12) | 3.04(13) |
| 5b | 2.0958(16) | 1.901(4) | 1.855(4) | 1.876(4) | 1.1(4) |
| 5c | 2.0915(8) | 1.899(2) | 1.860(2) | 1.872(2) | 2.0(2) |
| 12 | 2.0980(9) | 1.902(3) | 1.857(3) | 1.871(3) | 0.8(2) |
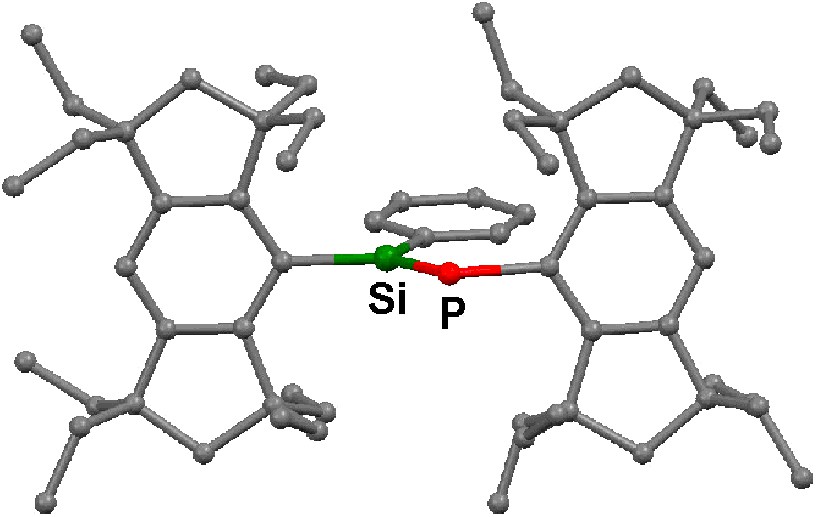
Molecular structure of 5a. Hydrogen atoms are omitted for the clarity.
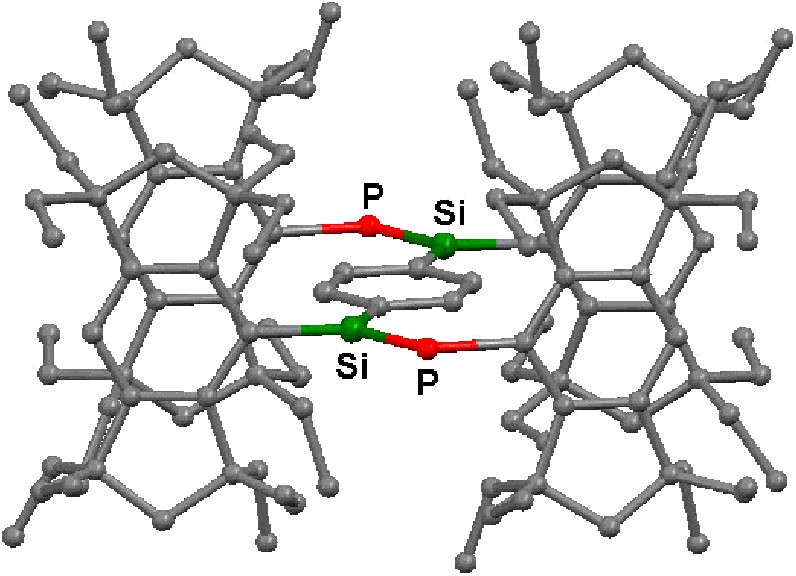
Molecular structure of 12. Hydrogen atoms are omitted for the clarity.
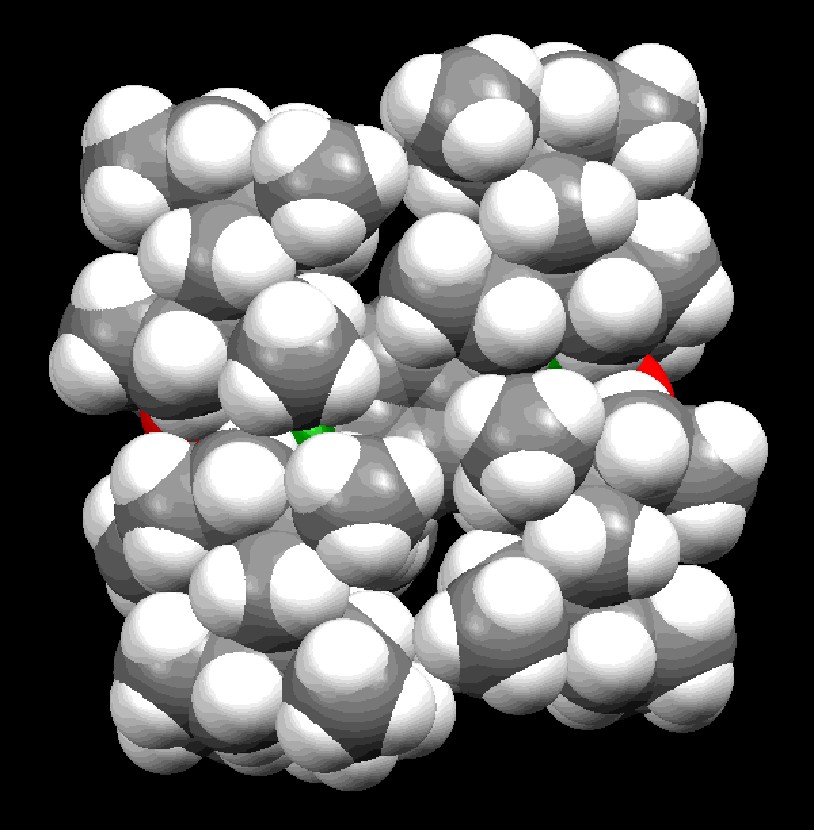
Space-filling model of 12.
The Si=P bond lengths of 5a–c and 12 (ca. 2.09–2.10 Å) are in the range of those for typical phosphasilenes [5–7]. The Si–Cipso(Eind) bond lengths (ca. 1.90 Å) are somewhat longer than the Si–Cipso(aryl) bond lengths (ca. 1.86 Å), presumably due to the small steric repulsion between the ethyl groups of the Eind groups. The P–C bond lengths (ca. 1.87–1.88 Å) are normal. Each silicon atom in 5a–c and 12 adopts an essentially planar geometry; the sum of the bond angles around the Si atom are ca. 360°.
For 5b and 5c, the arrangement between the Si=P bond and the polycyclic aromatic substituents about the intervening Si–Cipso(aryl) bond is described as an s-cis conformational geometry [18] in the crystals.
4 Photophysical and electrochemical properties
Phosphasilenes 5a–g and 12 exhibit an intense absorption band in the electronic absorption spectrum. The data are summarized in Table 3. The UV-visible absorption spectra of 5a–c and 12 are shown in Fig. 5 [8]. The phenyl-substituted phosphasilene 5a shows an absorption peak (λmax) at 385 nm (ɛ = 1.16 × 104), which is about 50 nm bathochromically shifted and two-orders stronger relative to the λmax and ɛ of the previously reported phosphasilenes [4c,6c]. The introduction of the 2-naphthyl (5b) and the 2-anthryl (5c) groups to the silicon atom causes further λmax red-shifts to 405 nm (ɛ = 1.06 × 104) and 430 nm (sh, ɛ = 1.27 × 104), respectively. Moreover, the λmax of 12 is observed at 449 nm (ɛ = 2.20 × 104), which is 64 nm red-shifted from 5a, indicating the extension of the π-conjugation over the 1,4-bis(phosphasilenyl)benzene skeleton.
Electronic absorption data for phosphasilenes in THF.
| Compound | λmax (nm) | νmax (cm−1) | ɛ (cm−1 M−1) |
| 5a | 385 | 26,000 | 11,600 |
| 5b | 405 | 24,700 | 10,600 |
| 5c | 430 (sh) | 23,300 | 12,700 |
| 5d | 403 | 24,800 | 15,500 |
| 5e | 404 | 24,800 | 9,200 |
| 5f | 394 | 25,400 | 9,500 |
| 5g | 425 | 23,500 | 10,600 |
| 12 | 449 | 22,300 | 22,000 |
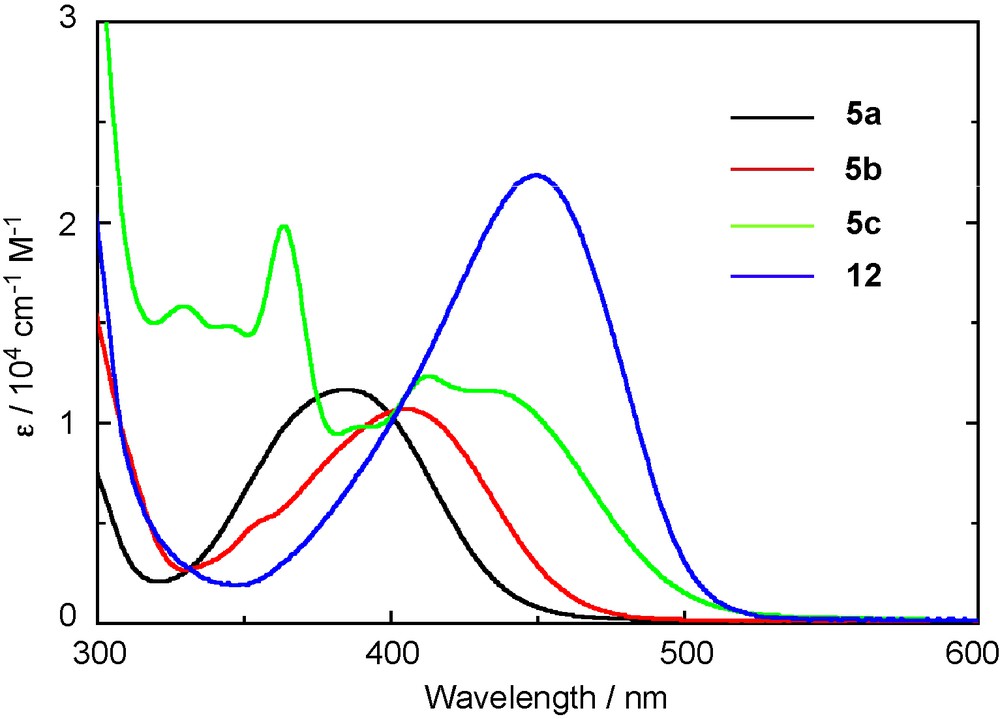
UV-vis absorption spectra of 5a–c and 12 in THF.
While 5a does not show any fluorescence, the π-extended 12 shows a weak, but distinct emission in the solid state, as shown in Fig. 6 [8]. The emission maximum of 12 is observed at 592 nm. The large Stokes (5380 cm−1) shift may be attributed to the structural change from the coplanar structure in the ground state to the twisted structure in the excited state.
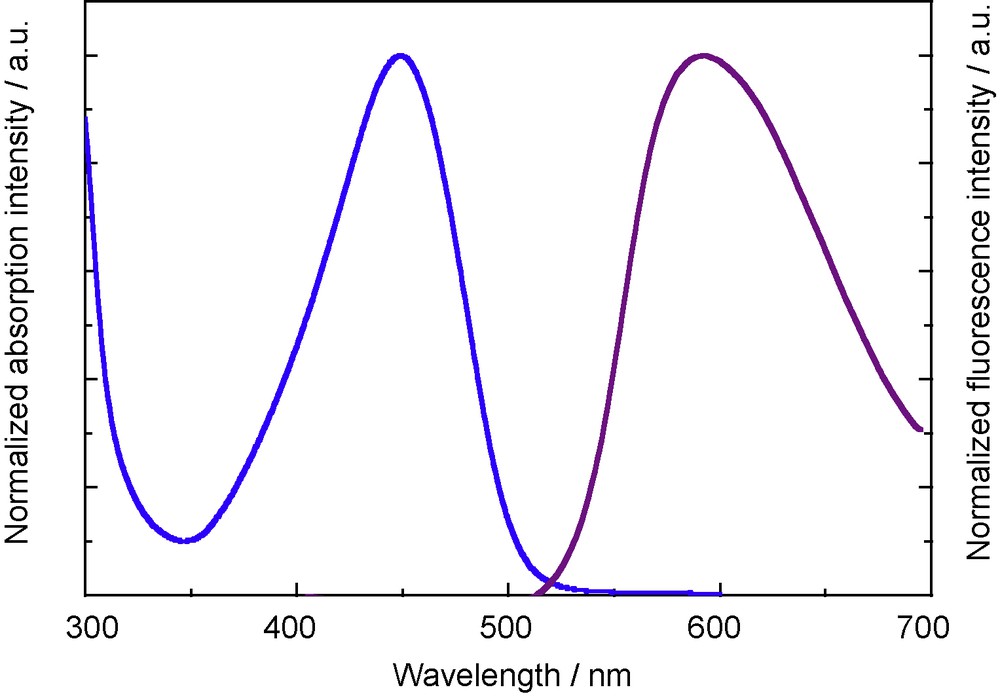
Solid-state luminescence spectrum of 12 excited at 360 nm (purple). UV-vis absorption spectrum of 12 in THF (blue).
Although there have been a number of reports regarding the electrochemical properties of the diphosphenes [19], those of phosphasilenes have rarely been explored [7]. We performed the electrochemical studies of 5a and 12 by cyclic voltammetry [8]. The irreversible oxidation waves at +0.94 V (5a) and +0.97 V (12) as well as the quasi-reversible and the irreversible reduction waves at –2.55 V (5a) and –2.12 V (12) were observed for the cyclic voltammograms, respectively (Fig. 7). These experimental results exhibit good qualitative agreement with the DFT calculations (vide infra).
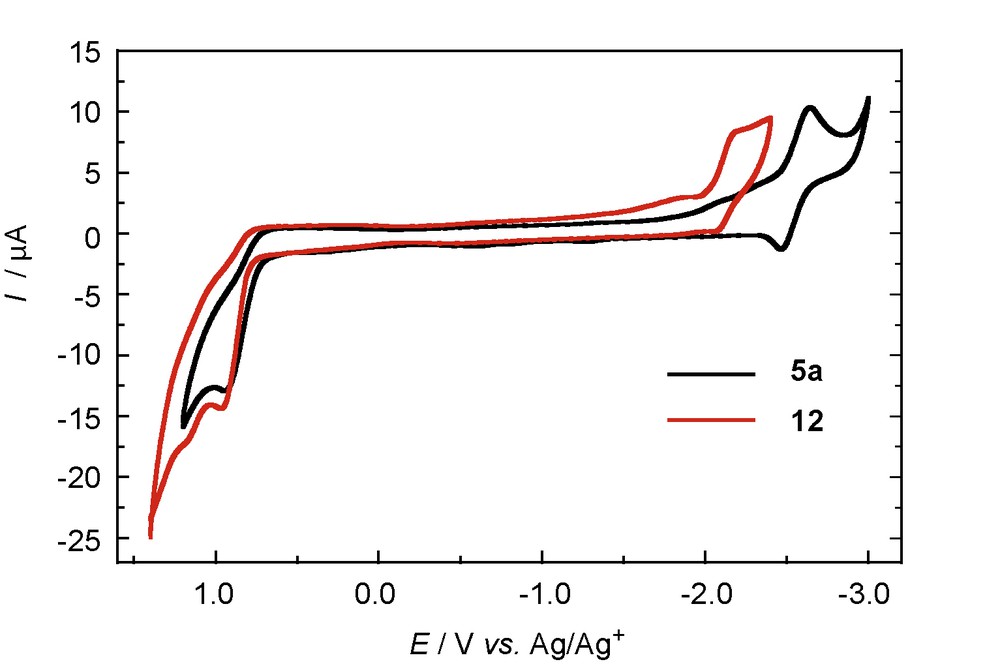
Cyclic voltammograms of 5a (black) and 12 (red).
5 Theoretical studies of π-conjugated phosphasilenes
To delineate the nature of the chemical bonding in the phosphasilenes, DFT computations at the B3LYP/6-31G** level were carried out for 5a and 12. The optimized structures well reproduced the X-ray crystal structures.
The three pertinent molecular orbitals (MOs) of 5a are depicted in Fig. 8 [8]. While the HOMO of 5a is primarily represented by the 3pπ(Si–P) orbital, the LUMO involves the substantial contribution of the 3pπ*(Si–P)–2pπ*(phenyl) conjugation. The lower-lying HOMO-1 involves the 3n–2pπ conjugation due to the lone pair at the phosphorus atom and the π-orbital on the Eind benzene ring; the maximum orbital interaction of these two orbitals is attained by the perpendicularly-fixed Eind group.
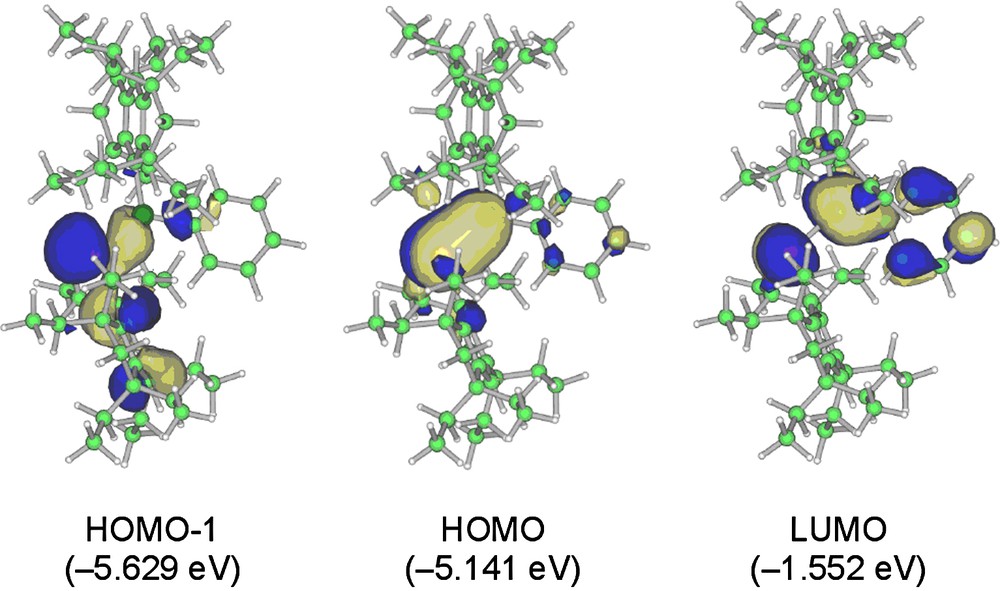
Selected molecular orbitals of 5a (top view).
Fig. 9 shows the selected MOs of the π-extended 12. Calculations for 12 have demonstrated that the LUMO level is lowered [–1.552 (5a) and –2.108 (12) eV], thus keeping the HOMO levels nearly constant [–5.141 (5a) and –5.137 (12) eV], in agreement with the orbital shapes for 5a and the previously mentioned electrochemical experimental results. The significant 3n–2pπ conjugation are also found in the HOMO–2 and HOMO–3 of 12.
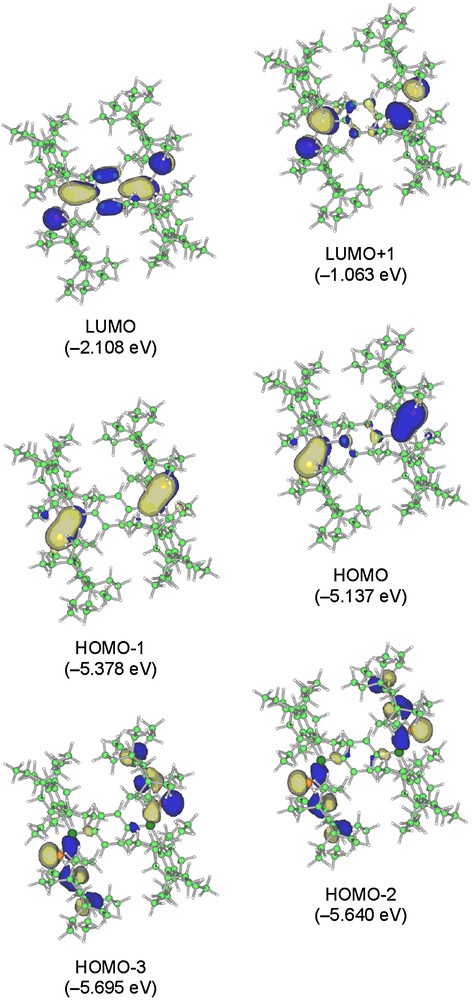
Selected molecular orbitals of 12 (top view).
The absorption wavelengths were calculated for the ground-state geometries using the TD-B3LYP/6-31G** method. The calculated wavelengths of 364 (5a) and 456 (12) nm are comparable to the observed values. The absorption at 364 nm of 5a is assignable to the mixed version of the HOMO→LUMO (π–π*) and HOMO-1→LUMO ((n + π of Eind benzene)–π*) transitions with almost equal weighting. In contrast, the absorption at 456 nm of 12 is mainly assigned to the HOMO→LUMO (π–π*) transition.
6 Gold(I) complexes of π-conjugated phosphasilenes
Although the P-zinciophosphasilene [6e] and P-plumbyleniophosphasilene [6f] have been reported to be obtained by the metallation of the corresponding hydrogen-substituted phosphasilene, the transition metal complexes bearing neutral phosphasilene ligands had been unexplored until we succeeded quite recently in preparing some gold(I) complexes [20]. Thus, as a part of the studies aimed at developing new conjugated hybrid materials consisting of both the main group elements and transition metals, we have focused on the synthesis of the gold(I) complexes of π-conjugated phosphasilenes stabilized by the bulky Rind groups.
As shown in Scheme 5, the reaction of 5a with AuCl(tht) (tht = tetrahydrothiophene) led to the quantitative formation of the phosphasilene gold(I) chloride complex 13 as a yellow powder. We have also designed a new phosphasilene 14 with the less bulky 1,1,7,7-tetraethyl-3,3,5,5-tetramethyl-s-hydrindacene-4-yl (EMind) groups. The corresponding AuCl complex 15 was prepared in 82% yield.
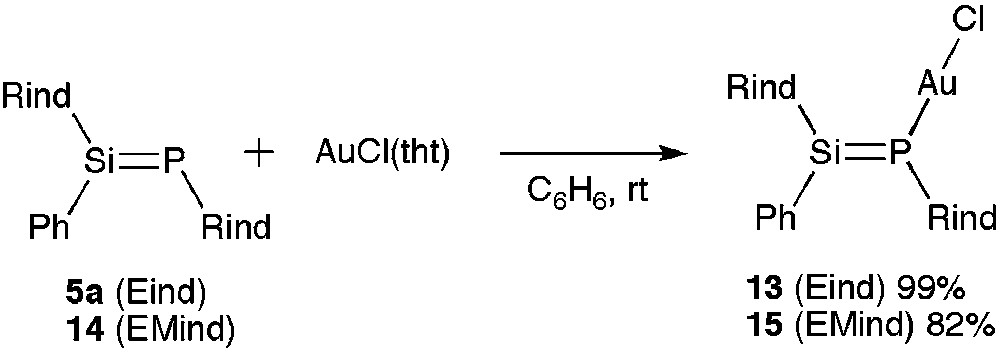
Synthesis of 13 and 15.
The molecular structure of 13 has been determined by X-ray crystallography (Fig. 10). The Au atom has a linear two-coordinate geometry with the P–Au–Cl angle of 178.83(5)°. The Au–P and Au–Cl distances are 2.2295(9) and 2.2739(10) Å, respectively. The Si=P bond length of 13 is 2.0924(12) Å, which is similar to that of 5a [2.0917(5) Å]. The phenyl group is slightly twisted to the Si=P bond; the P–Si–C–C torsion angle is 13.83(15)°.
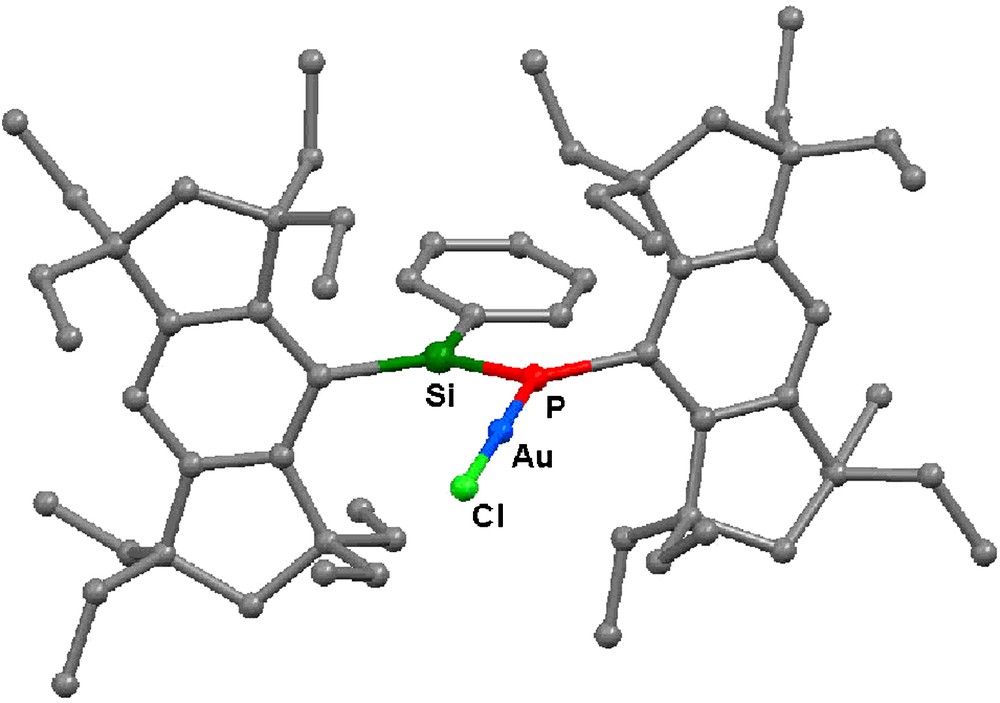
Molecular structure of 13. Hydrogen atoms are omitted for the clarity.
The 29Si NMR signal of 13 (δ = 144.4 ppm) is shifted upfield relative to 5a (δ = 161.7 ppm) with the decreasing 1JSiP coupling constant (39 Hz for 13; 171 Hz for 5a). The 31P NMR signal of 13 (δ = 43.2 ppm) is also shifted to a higher field relative to 5a (δ = 89.2 ppm). The complex 13 has an absorption maximum at 395 nm, which is 10 nm red-shifted from 5a, with a higher molar extinction coefficient (ɛ = 1.75 × 104). These data suggest that the degree of the π-conjugation has been increased by the Au(I) ion complexation.
7 Conclusions
We have described our recent results concerning the π-conjugated stable phosphasilenes based on the Rind groups in this report. The phosphasilenes with a variety of aryl groups have been clearly characterized by NMR spectroscopy and X-ray structural analyses. All of the phosphasilenes demonstrate the strong π–π* absorptions resulting from the π-conjugation between the Si=P bond and the carbon π-electron system. The fused-ring bulky Rind groups successfully protect the reactive polar Si=P bonds, making the highly coplanar arrangement of the π-framework. The unique electronic properties of the π-conjugated phosphasilenes have been elucidated by UV-visible absorption spectroscopy as well as DFT calculations.
The gold(I) complexes of the π-conjugated phosphasilenes have also been isolated to demonstrate the extension of π-conjugation through the gold atom. Elucidation of the chemistry of the phosphasilene metal complexes has just begun and further investigations are needed in order to understand their properties and reactivities.
Acknowledgment
We thank the Ministry of Education, Culture, Sports, Science, and Technology of Japan for the Grant-in-Aid for Specially Promoted Research (No. 19002008). The calculations and experiments discussed in this article have been performed with many collaborators, whose names appear in the references. The authors wish to express their thanks to all of them for their kind collaboration, particularly Professor Kazuyoshi Tanaka and Dr Hiroyuki Fueno (Kyoto University), Dr Daisuke Hashizume, Dr Takeo Fukunaga (RIKEN), Dr Vincent Lemau de Talancé (Université de Rennes 1), and Ms Qian Luo (Peking University). We also thank Dr Yayoi Hongo and Mr Takashi Nakamura (RIKEN) for their kind help with the mass spectrometry and solid-state NMR spectroscopy. Numerical calculations were partly performed at the Supercomputer Laboratory, Institute for Chemical Research, Kyoto University. Finally, the authors express their sincere thanks to Pr Hayato Tsuji (The University of Tokyo), Dr Yongming Li (The Chinese Academy of Sciences), Dr Aiko Fukazawa, and Pr Shigehiro Yamaguchi (Nagoya University) for their initial contribution to this study.
Vous devez vous connecter pour continuer.
S'authentifier