

1 Introduction
One of the major aims of chemical biology [1], the young and developing scientific field between chemistry and biology, is to find matches between the biological and chemical space [2]. The chemical space comprises (small) molecules, some of which show complementary features to certain points of the biological space constituted by the structure of binding sites of biomacromolecules (mainly but not only proteins). Good matches may result in efficient agonists/antagonists of receptors or activators/inhibitors of enzymes. Such interactions contribute to the basic understanding of the way of biological action of the macromolecule, and may ultimately be utilised in drug design and discovery.
In the context of this survey, the biological space is represented by glycogen phosphorylase (GP), the main regulatory enzyme of glycogen metabolism. GP, catalysing the rate-determining step of glycogen degradation in the liver by phosphorolysis, is directly responsible for the regulation of blood glucose levels. Thus, the enzyme has become a validated target in combatting non-insulin-dependent, or type 2, diabetes mellitus (NIDDM or T2DM), and its inhibitors are considered as potential antidiabetic agents. The biochemical and pharmacological background of this research has been amply summarized in several reviews of the past decade, therefore, the reader is kindly referred to those papers [3–8].
Diverse classes of compounds [4,9–12] can be found among inhibitors of GP binding to one (or in specific cases more) of the so far discovered binding sites of the enzyme (Fig. 1). The most populated class of compounds is that of glucose derivatives, first proposed and investigated [4,13,14] by Fleet, Johnson, and Oikonomakos,1 which bind primarily to the active site of GP. This article highlights the most important “historical” moments of GP inhibitor design among glucose analogues, and the main emphasis is put on developments of the past couple of years, not, or not fully, included in the last comprehensive reviews [11,12]. Although the design of compounds was heavily based on and supported by results of crystallographic investigations of enzyme–inhibitor complexes and molecular dockings, the syntheses and structure–activity relationships of the inhibitors are pointed out in this overview.
2 Early glucose analogue inhibitors of glycogen phosphorylase
The weak binding of d-glucose anomers 1 and 2 to the catalytic site of GP to act as the physiological regulator of the enzyme [15] raised the possibility to design glucose derivatives with much higher affinity to the active site. Enzymatic tests of a large series of α- and β-d-glucopyranosides, 1-thio-d-glucopyranosides, N-acyl-β-d-glucopyranosylamines and related compounds [13] revealed 1-deoxy-d-gluco-heptulopyranose 2-phosphate (3) and N-acetyl-β-d-glucopyranosylamine (4) as the first glucose derivatives with an inhibitor constant (Ki) in the low micromolar range. Anhydro-heptonamides 5 and 6 were less effective, however, a formal combination of 6 with an anomeric substituent similar to that of 4 gave again a low micromolar inhibitor 7. Ring closure of 7 to glucopyranosylidene-spiro-hydantoin 8 strengthened the binding by a factor of ∼5. The spiro-epimeric hydantoin 9 proved much less efficient, indicating that the presence of a β-d-anomeric NH was very important to make a good inhibitor. This was rationalized by crystallographic investigation of the enzyme–inhibitor complex [16] to show the presence of a specific H-bridge between NH and His377 next to the catalytic site also present in N-acyl-β-d-glucopyranosylamine type inhibitors (Fig. 2a for an illustration). The synthetic problems with the stereoselective preparation of the properly configured spiro-hydantion 8 [17–19] were essentially overcome by the highly stereoselective synthesis of spiro-thiohydantoin 10 [20] which proved equipotent with 8 (Fig. 3).

Outline of binding of glucose analogues at the active site of glycogen phosphorylase (GP) highlighting (a) important H-bonds between N-acyl-β-d-glucopyranosylamine type inhibitors and His377 and (b) binding modes of N-acyl-N’-β-d-glucopyranosyl ureas as observed by X-ray crystallography.
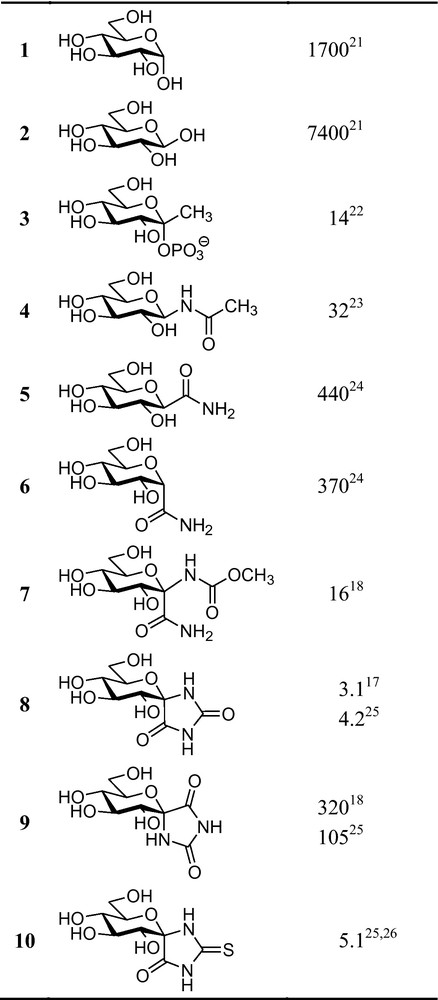
Inhibition of glycogen phosphorylase (GP) by d-glucose and the most efficient inhibitors of early glucose analogue derivatives (Ki [μM] against RMGPb) [21–26].
3 Glucose derivatives tested recently as inhibitors of glycogen phosphorylase
3.1 N-Acyl-β-d-glucopyranosylamines and related compounds
Following the success of the first N-acyl-β-d-glucopyranosylamine type inhibitors like 4, several modifications of the acyl group were carried out. A widely applied general method for the preparation of such compounds starts with the reaction of per-O-acetylated β-d-glucopyranosyl azide 11 with triaryl- or trialkyl phosphanes (PMe3 proved the most advantageous [27]) and the intermediate phosphinimine is then reacted with a carboxylic acid or acid chloride or anhydride to get protected amides 14 (Scheme 1; for an exhaustive review see [28]). Reduction of 11 to 15 followed by acylation can be an alternative synthetic route. Subsequent deprotection yields test compounds of type 14 (R = H), and several recent examples as inhibitors of rabbit muscle GP b (RMGPb) are shown in Fig. 4.

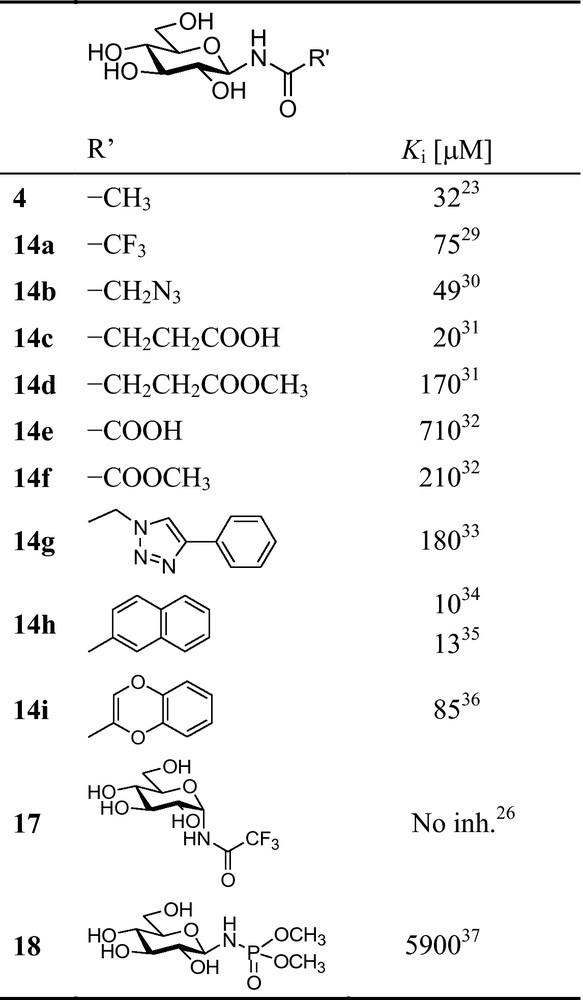
Inhibition of rabbit muscle glycogen phosphorylase b (RMGPb) by N-acyl-β-d-glucopyranosylamines and related compounds. Illustrative examples of the most efficient members of larger series of compounds detailed in the referred papers [29–37].
Substitution in the methyl group of N-acetyl-β-d-glucopyranosylamine makes the inhibition weaker (Fig. 4, compare 4 and 14a,b). The α-anomeric trifluoroacetamide 17 proved configurationally stable (for a discussion on the stability of N-acyl-glycosylamine anomers, see ref. [27]) but showed no inhibition. From a larger collection of monoamides of dicarboxylic acids, 14c showed similar inhibition to that of 4, while its methylester 14d proved significantly weaker. In the series of oxamic acid derivatives, the efficiencies of acid 14e and ester 14f were reversed, both being much less effective than 4. Introduction of a large side chain as in 14g made a weak inhibitor. Among aromatic amides, the 2-naphthoyl derivative 14h proved the most efficient, and in this series, the position occupied by the aromatic moiety becomes also important (Fig. 5 also). Necessity of the intact homoaromatic system is indicated by 1,4-benzodioxane carboxamide 14i. Changing the acyl part to a dimethoxyphosphoryl residue (18) resulted in a practical loss of inhibition.

Comparison of inhibition of rabbit muscle glycogen phosphorylase b (RMGPb) (Ki [μM]) by N-acyl-β-d-glucopyranosylamines, N-substituted-N’-β-d-glucopyranosyl ureas and related compounds. Against rat liver glycogen phosphorylase (GP) [48–51].
Syntheses of analogues 19 of spiro-hydantoins 8-10 were envisaged by photocyclization of acyl urea derivatives 20 outlined in Scheme 2a. To this end, reported cyclizations of 3-oxoalkyl glycosides [38,39] 23 resulting in stereoselective formation of spiro-acetals 24 (Scheme 2b) served as analogies. Thus, a photoexcitation of 20 might have resulted in intermediate 22 which, upon intramolecular hydrogen abstraction to give 21 and subsequent radical combination could have given the target compounds 19.
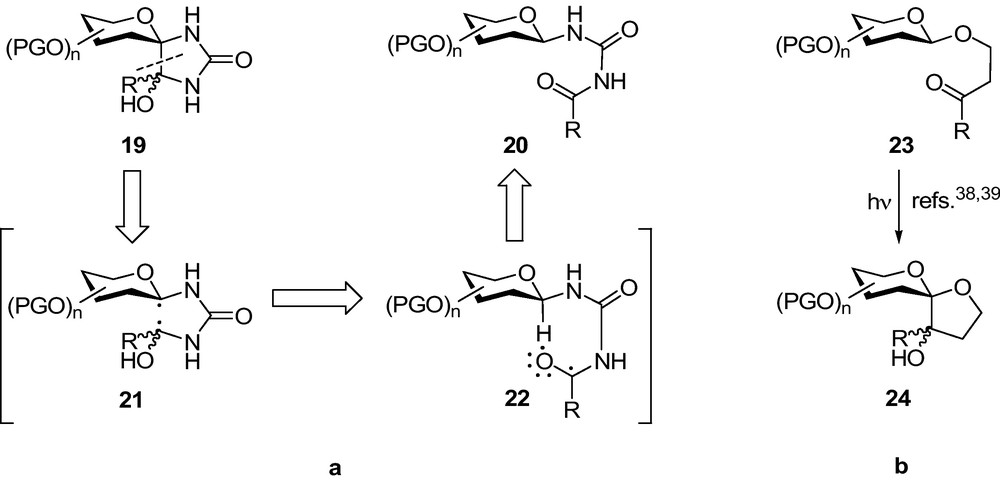
To test this hypothesis, N-acyl-N’-β-d-glucopyranosyl ureas of type 27 (Scheme 3) were needed. Only two examples of this class of compounds were known in the literature [40] which were obtained by a modification of the original synthesis. Azide 11 was transformed to urea 26 by Pintér et al.’s method [41] and then acylation was carried out to give 27 (R = Ac, R’ = Me or Ph). Irradiation of 27 under various conditions brought about a Norrish I type cleavage of the R’CO moieities leading back to 26 instead of the expected Norrish II type cyclization [42]. Quite unexpectedly, the deprotected compounds 27 (R = H, R’ = Me Ki = 305 μM; R’ = Ph Ki = 4.6 μM) proved efficient inhibitors of GP [43] and the benzoyl derivative had similar potency to those of spiro-hydantoins 8 and 10. Initiated by this serendipitous finding, synthetic and enzymatic studies were started to get insight in structure–activity relationships of β-d-glucopyranosyl derivatives attached to aromatic rings by linkers of 3-6 atoms analogous to amide groups.

N-Aryl-N’-β-d-glucopyranosyl ureas 13 were obtained (Scheme 1) either via acid catalysed hydration of carbodiimide 12 obtained from azide 11 by a Staudinger type transformation, or by reacting glucosylamine 15 with isocyanates, or by in situ conversion of 15 into glucosylisocyanate 16 [44] followed by amine addition. Removal of the protecting groups was straightforward under Zemplén conditions. Further compounds of the protected N-acyl-N’-β-d-glucopyranosyl urea series 27 (Scheme 3) were obtained in reactions of glucosylamine 15 with acyl-isocyanates or from glucosylisocyanate 16 upon treatment with arenecarboxamides. During these syntheses, anomerization was observed in almost every cases thereby diminishing the yield of the target compounds [45]. Furthermore, deprotection of acyl ureas 27 was always accompanied by the cleavage of the R’CO group, both under base or acid catalysed transesterification conditions. These side reactions could be circumvented by the addition of unprotected β-d-glucopyranosylamine obtained in situ from β-d-glucopyranosylammonium carbamate [46] (25) to various acyl-isocyanates to give directly the unprotected 27 ureas [45]. Biurets 28 [47] and 29 [42] were prepared in reactions of urea 26 with phenyl and 2-naphthoyl isocyanates, respectively.
Most important results of the enzyme kinetic studies are collected in Fig. 5. Comparison of entries 1, 2, 4, 13, and 14 shows that the inhibition is strongest for the acyl urea type compounds (entry 4). Introduction of a tetrahedral element into the linker makes weaker inhibitors (compare entries 2–3, 4–6). Replacement of one NHCO by a more rigid bond (entries 4, 7, 9) seems less detrimental, although the inhibition is weakened, showing the necessity of a polar part capable for participation in H-bonds as well. Entries 7 and 8 indicate again that higher flexibility due to a rotatable element of the linker is not advantageous (of course, the absence of the H-bond donor amide moiety from the anomeric carbon must also contribute to the weaker binding). Constitutional isomers of the NHCONHCO moiety (entries 10–12) also make significantly less efficient inhibitors. Comparison of columns A–C demonstrate the importance of the size and orientation of the aromatic appendage the 2-naphthyl derivatives exhibiting the strongest binding. Accordingly, N-2-naphthoyl-N’-β-d-glucopyranosyl urea (entry 4C) was the first nanomolar glucose analogue inhibitor of GP. Protein crystallography showed acyl ureas of entries 4A and 4C to bind also to the new allosteric site of the enzyme [43].
X-Ray crystallographic studies of GP–N-acyl-N’-β-d-glucopyranosyl urea complexes revealed that, contrary to the N-acyl-β-d-glucopyranosylamines, there is no H-bond between the β-anomeric NH and His377 (Fig. 2b) [43]. As the acyl ureas are much more inhibitory than the corresponding glucosylamines (Fig. 5, entries 1 and 4), the stronger binding must be due to extended interactions of the urea and especially the aromatic parts of the molecules in the β-channel2 of the enzyme. This observation was utilized in further inhibitor design discussed in Section 3.4.
Very recently, a new series of aldehyde 4-(β-d-glucopyranosyl)-thiosemicarbazones [52,53] was prepared from per-O-acetylated β-d-glucopyranosylisothiocyanate (Fig. 6) and several of them showed micromolar inhibition.
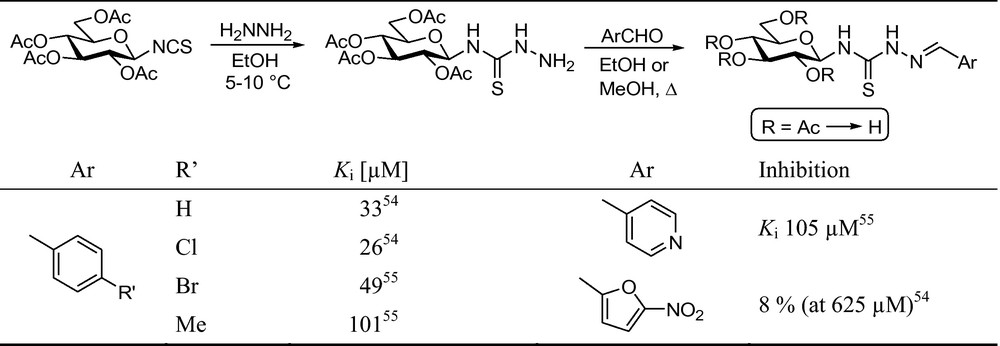
Synthesis of aldehyde 4-(β-d-glucopyranosyl)thiosemicarbazones and their enzymatic evaluation against rabbit muscle glycogen phosphorylase b (RMGPb) [54,55].
3.2 N-β-d-glucopyranosyl heterocycles
The problems encountered in the synthesis of N-acyl-β-d-glucopyranosyl ureas necessitated a quest for more stable compounds. To this end, bioisosteric replacement of NHCO moieties in acyl ureas and related compounds was envisaged. As the first example of such studies, the NHCO unit of N-acyl-β-d-glucopyranosylamines was changed to 1,2,3-triazole because some literature examples indicated similarities [56] of these two moieties. Three series of 1-d-glucopyranosyl-4-substituted-1,2,3-triazoles [51] were prepared by copper(I) catalysed azide-alkyne cycloaddition (CuAAC) [57] outlined in Scheme 4. From β-d-glucopyranosyl azide 11 conditions 1a, frequently applied in the literature, proved to be a straightforward way to the per-O-acetylated 1-β-d-glucopyranosyl-4-substituted-1,2,3-triazoles in 58–96% yields. Transformations of the α-azide 30 required higher catalyst loads (conditions 1b) and the yields for the corresponding per-O-acetylated 1-α-d-glucopyranosyl-4-substituted-1,2,3-triazoles were lower (36–72%). The aqueous conditions were unsatisfactory for the reactions of (hept-2-ulopyranosylazide)onamide 31 for which conditions 1c were found the best to give 75–87% of the corresponding O-protected glucosyl triazoles with 51–73% conversion of the starting 31 in one day. Removal of the protecting groups was effected by the Zemplén protocol to give triazoles 32-34 in generally very good yields.
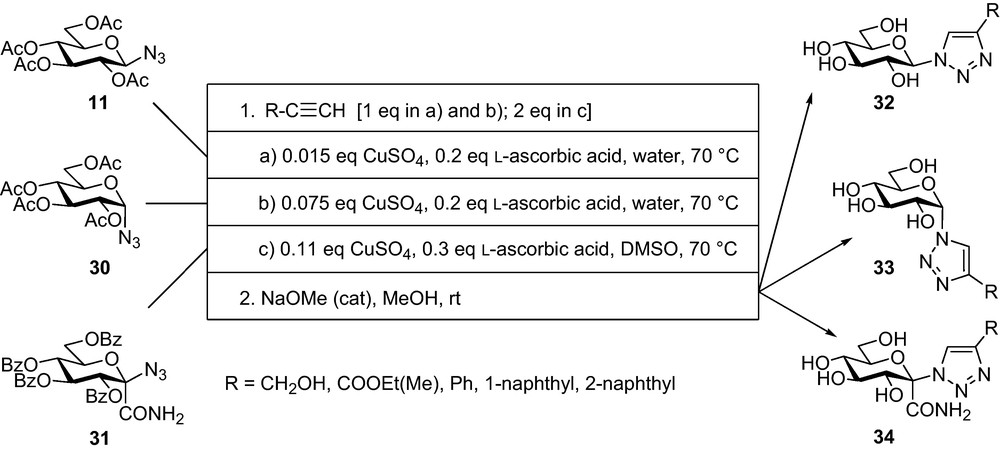
From these 1,2,3-triazoles, only compounds 32 showed significant inhibition (e.g. R = CH2OH Ki = 26 μM [51] or 14 μM; [35]). Inhibitor constants for other members of this series can be found in Fig. 5, entry 15 to show acceptable similarity with those of glucosyl amides in entry 1. Comparative crystallographic studies of the amide and triazole series revealed that pairs of the compounds with the same aglycon bound to the enzyme in essentially the same way in most cases [35]. Thereby, the bioisosteric relationship for NHCO-1,2,3-triazole was proven for the GP case as well.
Investigations of some N-β-d-glucopyranosyl derivatives of pyrimidine and purine heterocycles (“glucosyl nucleosides”) showed these compounds to have inhibitory effect towards GP, and the best inhibitors are collected in Fig. 7.
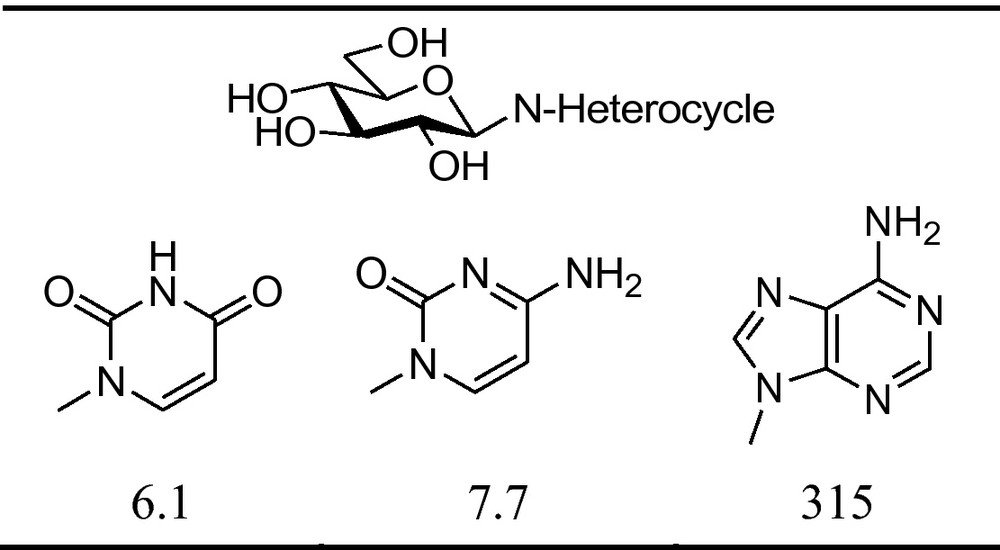
Inhibitory effect of β-d- glucopyranosyl nucleosides against rabbit muscle glycogen phosphorylase b (RMGPb) [58] (Ki [μM]) [58].
3.3 C-β-d-glucopyranosyl derivatives
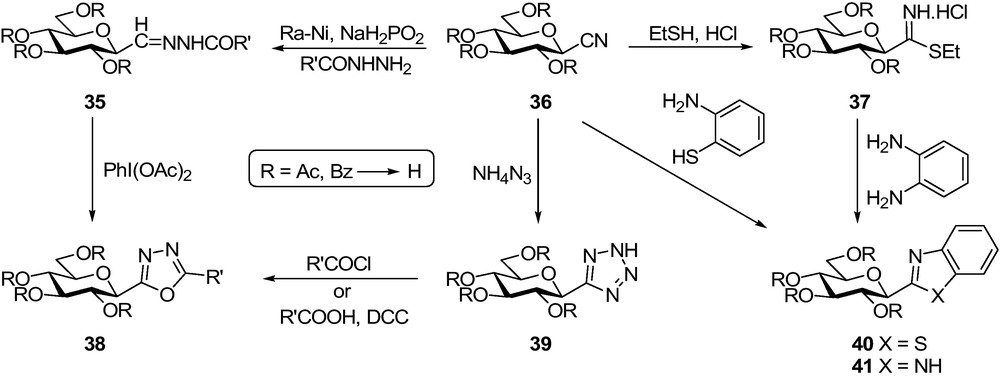
The first C-β-d-glucopyranosyl heterocycles tested as inhibitors of GP were methyl-1,3,4-oxadiazole 38, tetrazole 39, benzothiazole 40, and benzimidazole 41 (Scheme 5, R = H in each) [59]. Common starting material for the syntheses of these compounds was the per-O-acetylated or -benzoylated 2,6-anhydro-aldononitrile (β-d-glucopyranosyl cyanide) 36. 1,3-Dipolar cycloaddition of protected 36 with azide ion gave 5-β-d-glucopyranosyl tetrazole 39 which was transformed into 2-β-d-glucopyranosyl-5-substituted-1,3,4-oxadiazoles 38 via an N-acyl-nitrilimine intermediate obtained by acylation of 39 [59,60]. Oxadiazoles 38 could also be prepared by oxidation [60] of 2,6-anhydro-aldose acylhydrazones [61] 35, and the two pathways proved comparable with respect to yields and operational difficulties. Nitrile 36 was ring-closed to benzothiazole 40 with 2-aminothiophenol. The analogous reaction with 1,2-diaminobenzene was unsuccessful, therefore, benzimidazole 41 was obtained via thioimidate 37. Deprotection was carried out by the Zemplén method.
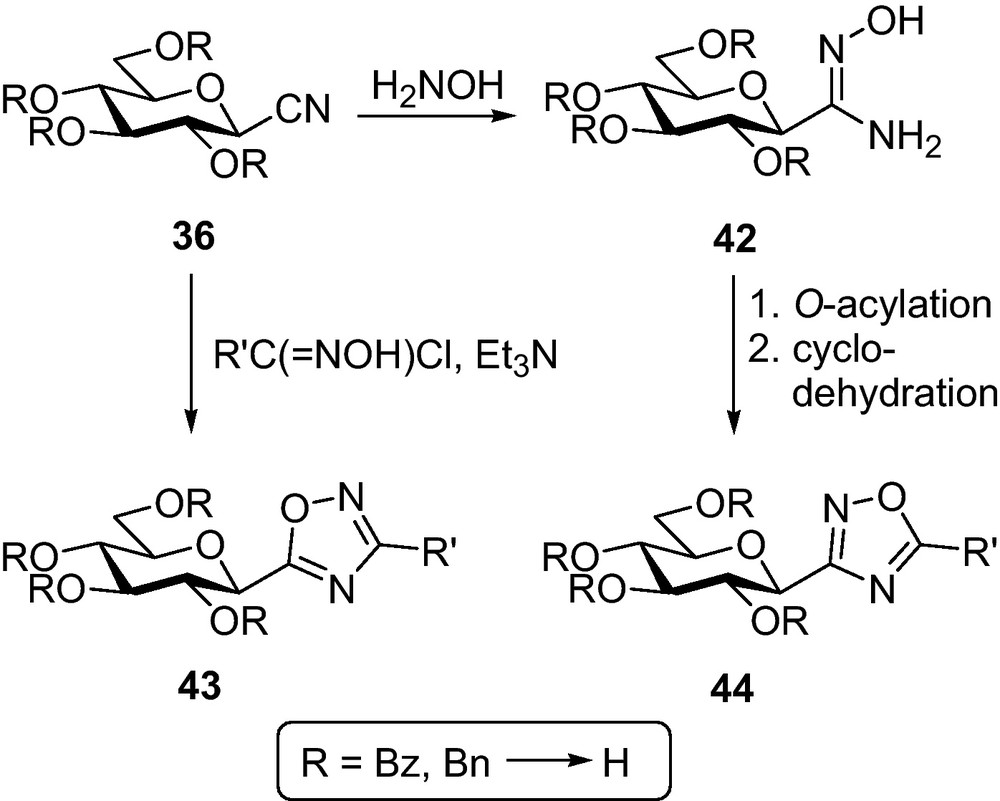
Per-O-benzoylated or -benzylated nitriles 36 were also transformed into two other series of 1,2,4-oxadiazoles (Scheme 6). 1,3-dipolar cycloaddition with nitrile-oxides generated in situ furnished 5-β-d-Glucopyranosyl-3-substituted-1,2,4-oxadiazoles 43 [60,62]. Addition of hydroxylamine to 36 produced amidoxime 42 which upon O-acylation with either carboxylic acids or acid chlorides followed by cyclodehydration gave 3-β-d-glucopyranosyl-5-substituted-1,2,4-oxadiazoles 44 [63]. The protecting groups were removed by standard methods.
Results of enzyme kinetic studies are presented in Fig. 8. β-d-Glucopyranosyl cyanide 36 is a somewhat better inhibitor than anhydro-aldonamide 5, while tetrazole 39 and amidoxime 42 are inactive. Benzimidazole 41 binds stronger than benzothiazole 40, and this can be attributed to the H-bond between the NH of the heterocycle and His377 which is necessarily absent for 40. X-ray crystallography has shown 41 also to be present at the new allosteric site and the new “benzimidazole site” has been discovered by investigating this compound (Fig. 1) [64]. From the three oxadiazole series (38, 43, 44), compounds 43 are the most active. The tendency of strengthening the inhibition by a large and properly oriented aromatic substituent can be observed in the oxadiazoles, too: compounds with a 2-naphthyl appendage (43d, 44d) are the best inhibitors. Although all three oxadiazoles could be considered as bioisosteric replacements [65,66] of NHCO, these results suggest that in the case of GP, 5-β-d-glucopyranosyl-3-substituted-1,2,4-oxadiazoles 43 are the best choice.

C-β-d-glucopyranosyl heterocycles and their precursors as inhibitors of rabbit muscle glycogen phosphorylase b (RMGPb) (Ki [μM]).
β-d-Glucopyranosyl hydroquinone derivative 46 in its O-acetyl protected form was prepared by aromatic electrophilic substitution in 1,4-dimethoxybenze using penta-O-acetyl-β-d-glucopyranose as a source of glucosylium ion. Subsequent oxidation gave protected benzoquinone 47 which was reduced to 45 [67]. The deprotected compounds were moderately inhibitory against GP (Fig. 9). Cyclopropane 48 was obtained from per-O-benzoylated nitrile 36 by EtMgBr–Ti(OiPr)4 followed by Zemplén deprotection [68]. This compound had no inhibition of GP.

β-d-Glucopyranosyl carbocycles as inhibitors of rabbit muscle glycogen phosphorylase b (RMGPb) (Ki [μM]).
3.4 Glucopyranosylidene-spiro-heterocycles
Studies on N-acyl-β-d-glucopyranosylamines and N-acyl-N’-β-d-glucopyranosyl ureas allowed one to conclude that it is possible to make very efficient inhibitors even in the absence of a H-bond to His377, provided that interactions in the β-channel are strong enough. Combining these facts with the spirobicyclic structure of hydantoins, a novel design principle for efficient glucose-based inhibitors of GP could be set up [69,70]:
- • such molecules should have a rigid spirobicyclic scaffold in which a (preferably five-membered hetero) cycle is attached to the anomeric carbon of d-glucopyranose;
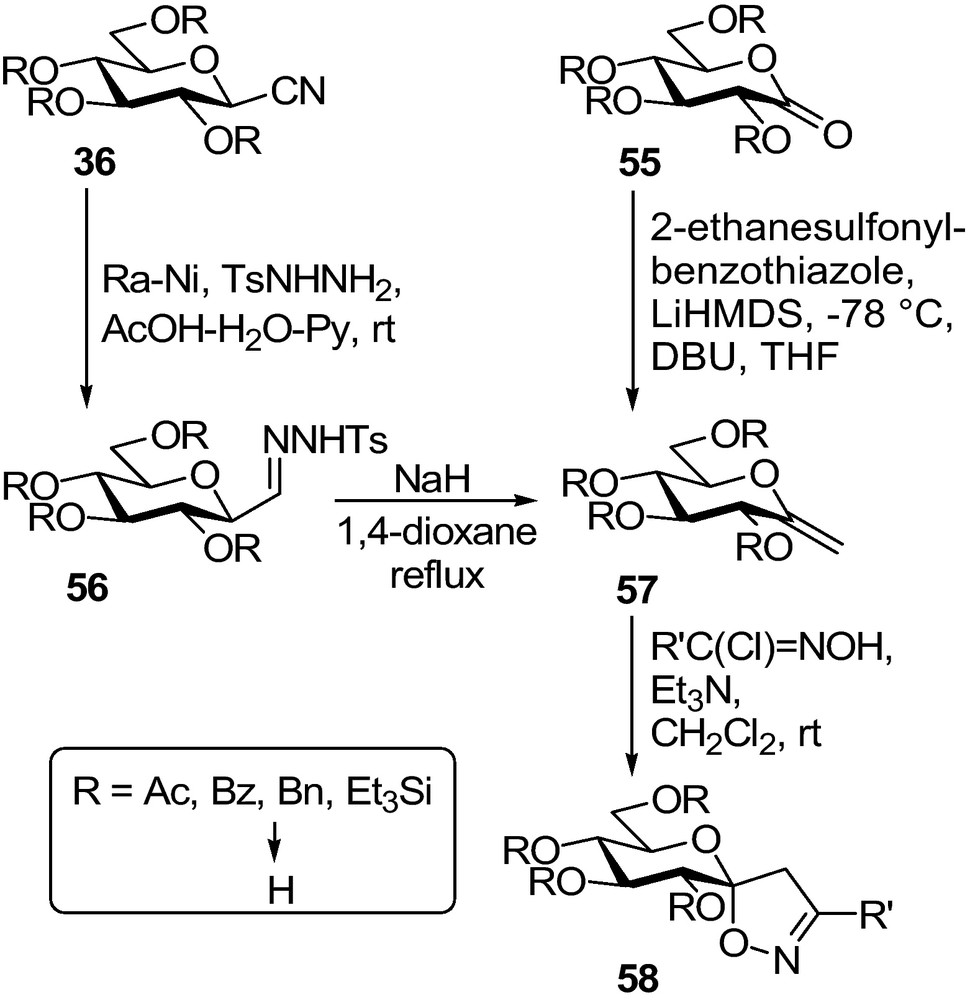
- • this cycle, although it may, should not necessarily be a H-bond donor towards His 377;
- • a suitably oriented, large aromatic appendage must be present on this cycle to fit into the β-channel.
This principle was first verified by spiro-oxathiazolines 53, the synthesis of which followed well elaborated pathways [71] (Scheme 7): per-O-acetylated 1-thio-β-d-glucopyranose 49 was reacted with in situ generated nitrile-oxides to give hydroximothioates 50 which underwent a ring-closure upon oxidation by NBS to yield the target compounds 53 after Zemplén deprotection. Synthesis of the analogous spiro-oxadiazoline 54 was also attempted. To this end, glucosyl azide 11 was transformed in a Staudinger type reaction into N-β-d-glucopyranosyl amidoxime 51. Oxidative treatment of 51 gave oxadiazole 52 probably via 54. The driving force for the tautomeric ring opening must be the aromatization of the heterocycle.
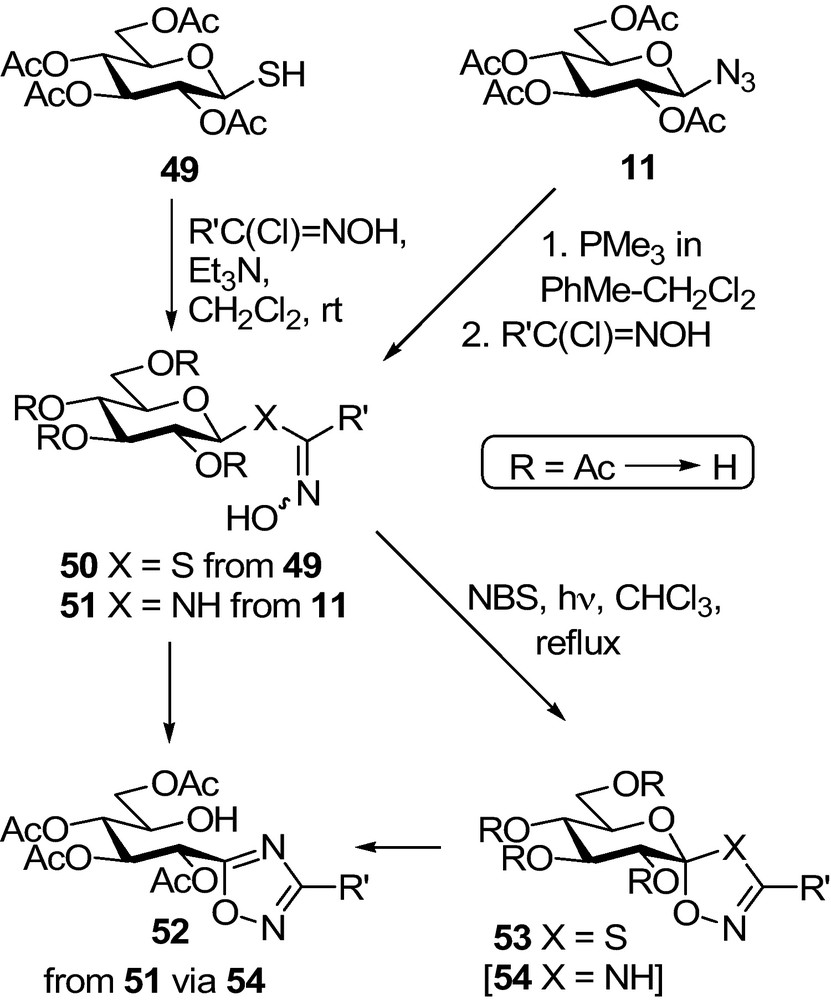
A series of glucopyranosylidene-spiro-isoxazolines 58 was prepared by 1,3-dipolar cycloaddition of nitrile-oxides to exo-glycals 57 (Scheme 8) [62]. The exomethylene sugars were made by Julia olefination of per-O-benzylated or -silylated lactone 55. Protecting group exchange to get the per-O-acetylated 57 was necessary because upon hydrogenolytic debenzylation of 58, the isoxazoline ring also opened up due to a cleavage of the N–O bond. O-deacetylation of 58 could be achieved by the Zemplén protocol. Another way to 57 was reported by transforming per-O-acylated nitriles 36 to 2,6-anhydro-aldose tosylhydrazones followed by a Bamford-Stevens type carbene generation to yield the target exo-glycals [72,73].
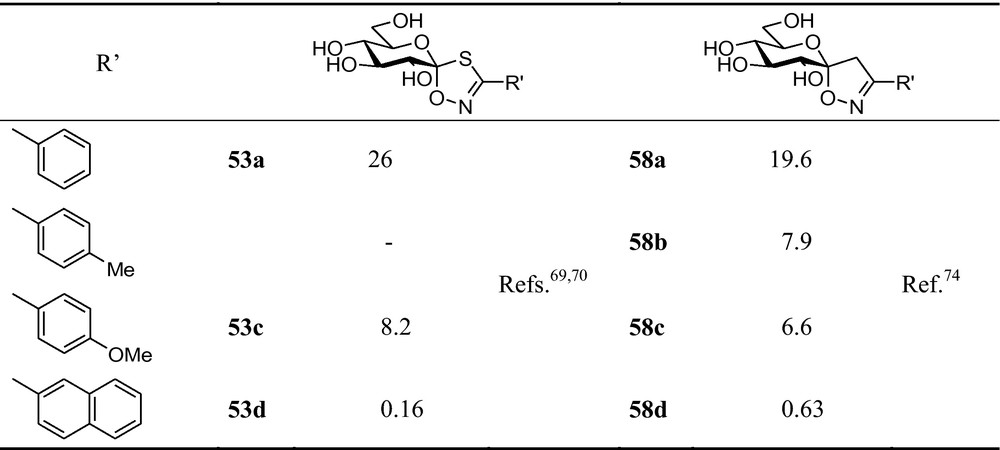
Enzyme kinetic investigation of these spirocycles (Fig. 10) indicated low micromolar inhibition of GP by the phenyl substituted derivatives 53a and 58a. Substitution in the para-position of the aromatic ring gave somewhat better inhibitors (53c, 58b,c). The 2-naphthyl derivatives (53d, 58d) were nanomolar inhibitors, thereby fully validating the design principles.
Inhibition of rabbit muscle glycogen phosphorylase b (RMGPb) (Ki [μM]) by glucopyranosylidene-spiro-heterocycles [74].
3.5 Miscellaneous compounds
Several O-, S-, and N-glucosides (Fig. 11, 59-63) of β-d-gluco-hept-2-ulopyranosonamide were prepared by nucleophilic substitutions of the corresponding glycosyl bromide [75]. These compounds can be regarded as anomerically extended variants of amide 6 for which a β-anomeric carbamate moiety (7) significantly improved the inhibitory efficiency. On the other hand, the new substitution patterns of 59-63 weakened the inhibition.

Inhibition of rabbit muscle glycogen phosphorylase b (RMGPb) by various monosaccharide derivatives [78].
Sulfonamide 64 prepared recently by two different methods [76,77] had no inhibition against RMGPb.
Very recently, multivalent molecules have been designed and proposed for inhibition of GP [79]. Compound 65 (Fig. 12) was prepared by acylation of amidoxime 42 with trimesic acid chloride. To get compound 67 containing a spacer, 42 was acylated with 4-pentynoic acid followed by CuAAC with 1,3,5-tris(azidomethyl)benzene. These compounds have three glucose units, each potentially capable to bind to an active site of GP. It was found that the homotrivalent derivatives 65 and 67 had slightly better inhibitory activity than the corresponding monovalent compounds 44b and 66, respectively. Homobivalent compounds 68 were made by CuAAC from N-ω-azidoalkanoyl-β-d-glucopyranosylamines and 1,7-octadiyne, but had no effect on the enzyme [80].

Probing multivalency for the inhibition of rabbit muscle glycogen phosphorylase (RMGP).
Potentially heterobivalent compounds were designed by tethering pentacyclic triterpenes and d-glucose derivatives [80]: C-28 propargyl esters of oleanolic, ursolic, or maslinic acids were coupled by CuAAC with β-d-glucopyranosyl azide and N-ω-azidoalkanoyl-β-d-glucopyranosylamines to give compounds 69 and 70, respectively. Derivatives with both per-O-acetylated and unprotected sugar parts were tested against GP and the best inhibitors are shown in Fig. 12. Micromolar inhibitors could be identified among both protected and unprotected glucose derivatives, and also the triterpene part and, in some cases, the linker length had a bearing on the efficiency of the compounds.
Oleanolic acid and d-glucose were also conjugated via C-6 ethers and glucuronic esters in several ways [81]. Most efficient compounds are 71b and 72a (Fig. 13) interestingly with an unprotected and a protected sugar unit, respectively. Based on molecular docking, 71b was proposed to bind at the allosteric site of GP. Per-O-benzylated precursor sugars 73 and 74 containing a propargyl group also exhibited inhibition of GP, the latter in the low micromolar range.

Triterpene–glucose conjugates and protected monosaccharide derivatives [81] as inhibitors of rabbit muscle glycogen phosphorylase a (RMGPa) (IC50 [μM]).
4 Conclusion
Extensive synthetic efforts supported by crystallographic studies on enzyme–inhibitor complexes have resulted in several new types of glucose analogue inhibitors of GP. Among them, N-acyl-N’-β-d-glucopyranosyl ureas, glucopyranosylidene-spiro-oxathiazolines and -isoxazolines represent novel scaffolds which, in the presence of suitable substituents, exhibit nanomolar efficiency. Further increase in the binding strength of glucose analogues may be expected from a better exploitation of interactions of the molecules in the β-channel of the enzyme. This will need a strong collaboration between synthetic and computational chemists, as well as crystallographers and biochemists. Nevertheless, due to the extremely high flexibility of the catalytic site of GP, synthesis and enzyme kinetic study of a large number of compounds will be inevitable.
5 Note added in proof
While this manuscript was under review, an interesting paper appeared on enzyme kinetic and crystallographic investigations of a series of 3-deoxy-3-fluoro-β-d-glucopyranosyl pyrimidine derivatives [82].
Acknowledgement
This work was supported by the Hungarian Scientific Research Fund (OTKA CK 77712) and an ongoing bilateral project of the Hungarian Academy of Sciences and Centre national de la recherche scientifique (PICS 4576). Sincere thanks are due to coworkers in the author's laboratory (V. Nagy, M. Tóth, K. Czifrák, Zs. Hadady, N. Felföldi, E. Berzsényi, É. Bokor, B. Kónya, S. Kun, L. Kóder, C. Hüse, K. Telepó) participating in the syntheses of the compounds, as well as P. Gergely and T. Docsa (Debrecen) for the kinetic studies. Cooperations with J.-P. Praly and S. Vidal (Lyon), H.-B. Sun (Nanjing), D. Postel (Amiens) in the field of organic synthesis, N.-G. Oikonomakos and E.-D. Chrysina (Athens, protein crystallography, enzyme kinetics), I. Tvaroška (Bratislava, computational chemistry) continuously gave new impetus and ideas for the work. These collaborations were supported by the Hungarian Academy of Sciences, the National Office for Research and Technology and the relevant funding organizations of the partner countries.
1 Passed away on Aug 31, 2008.

2 The β-channel or β-pocket is an empty space next to the catalytic site of GP in the direction of the β-anomeric substituent of bound d-glucose surrounded by amino acid side chains of mixed character.
Vous devez vous connecter pour continuer.
S'authentifier