

1 Introduction
From the writings of Hippocrates (460–356 BC) about the consumption of cress and poems by Horatius (65–68 BC) on the flavour of cabbage to the “Natural History” by Pliny the Elder (23–79 AD) and the “De Materia Medica” pharmacopoeia drawn up by Dioscorides (40–90 AD), the beneficial effects of vegetables of the crucifer family have long been observed. Nowadays, this positive health impact is recognized to be due to sulfur-containing secondary metabolites known as glucosinolates (GLs).
These chemical tags are the most strikingly bio-relevant thiosaccharidic molecules in the order Brassicales, which includes 16 plant families (Table 1) of dicotyledonous angiosperms among which the Brassicaceae (synonymous of Cruciferae) are by far the most important, because they encompass 350 plant genera and ca. 3200 species [1].
The botanical order Brassicales.
| Bataceae | Moringaceae |
| Brassicaceae (cabbages) | Pentadiplandraceae |
| Bretschneideraceae | Phytolaccaceae |
| Capparaceae (capers) | Pittosporaceae |
| Caricaceae (papaya) | Resedaceae (reseda) |
| Euphorbiaceae | Salvadoraceae |
| Gyrostemonaceae | Tovariaceae |
| Limnanthaceae | Tropaeolaceae (Indian cress) |
Associated in plants with an atypical glycohydrolase, myrosinase (E.C.3.2.1.147), GLs behave and, in fact, operate like bio-precursors – in accordance with Robiquet's prediction [2] – to produce electrophilic isothiocyanates, compounds displaying a diversified and generally marked biological activity including, in certain concentrations, high cytotoxicity [3]. Indeed, myrosinase-assisted hydrolytic cleavage of those S-glucopyranosyl thioesters – erroneously called “thioglucosides” – releases a labile aglycon which is converted into isothiocyanate through a Lossen-type rearrangement (Scheme 1).
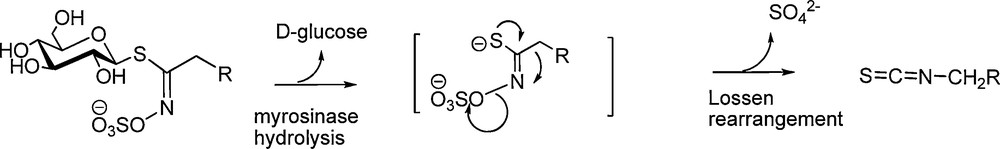
All known GLs (ca 120 molecules) display a remarkable structural homogeneity based on a hydrophilic β-D-glucopyrano unit, a O-sulfated anomeric (Z)-thiohydroximate function connected to a rather hydrophobic side chain whose constitution, depending on plant species, is the sole structural variant (Scheme 2) in which diversified aliphatic, arylaliphatic or heterocyclic arrangements can be found.
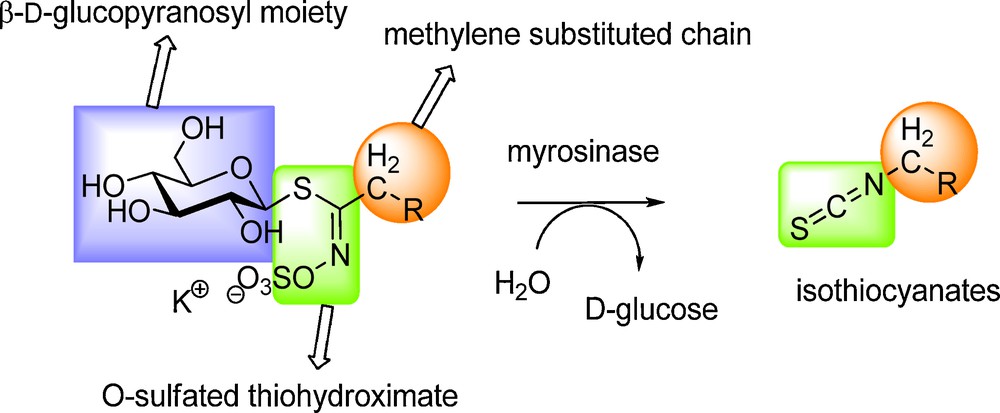
The glucosinolate-myrosinase couple underlies intriguing and complex relationships, not only between plants and animals but also between scientific disciplines such as ecology, agronomy, physiology, biochemistry and, at the molecular level, chemistry. Since several decades, scientists have put many efforts into clarifying this unique enzyme-substrate relationship and the mechanism of action of myrosinase – the only glycohydrolase able to break an anomeric carbon-sulfur bond.
Although they are usually not straightforward operations, extraction of GLs from appropriate vegetable sources and refining by dedicated chromatographic procedures can prove convenient in a number of cases to obtain pure GLs [4]. Nevertheless, the chemical synthesis approach has appeared to be a more general and more efficient way to access GLs – either natural or artificial (mainly targeting the myrosinase inhibition process) – in pure form. An overview of the chemical methods developed to produce tailor-made GLs and diversified analogues is presented.
2 Synthesis of glucosinolates – the methods
From a chemical synthetic point of view, two major approaches – depicted in Scheme 3 – for the elaboration of GLs structures have been developed by a limited number of groups over the past 50 years: these are based on a retrosynthetic scheme where a single specific bond formation affords the GL skeleton: two types of disconnection have been considered, either on the anomeric center (A) or onto the hydroximoyl moiety (B).
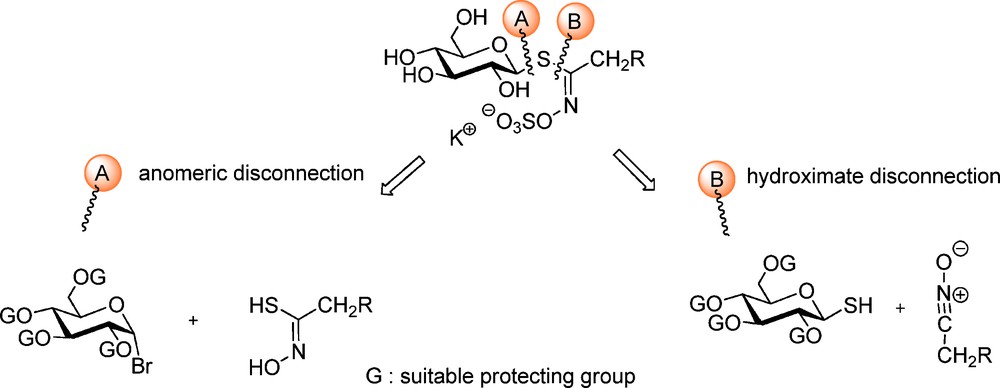
GLs display an uncommon thiofunction indeed – a (Z) O-sulfated thiohydroximate invariably connected through a methylene knuckle to the variable aglycon chain (up to 120 moieties). Several synthetic routes to naturally-occurring different GLs have been developed since the pioneering synthesis of glucotropaeolin (benzyl GL) by Ettlinger and Lundeen [5]. During the 1960–1980 period, syntheses of simple aliphatic and arylaliphatic GLs were mainly performed by three groups: A. Kjaer, the major Brassicale chemistry expert in Denmark [6], M.H. Benn in Canada [7] and A. MacLeod in Great-Britain [8]. In the course of the 1990–2000 decade, indole-type GLs and their glyco-analogues [9,10] and thiofunctionalized GLs [11] were synthesized in our group. Moreover, a major part of this activity has been devoted to the synthetic elaboration of tailor-made artificial glucosinolate-like structures, with a view to exploring the recognition process of myrosinase, estimating the relative importance of topical zones in the active site and searching for enzyme inhibitors.
Miscellaneous synthetic modifications were planned and realized either on the glycosidic moiety or on the aglycon chain. Some of the most significant analogues prepared were: alpha-anomers of some of the naturally-occurring beta-glucosinolates [12], a phosphated analogue of glucotropaeolin [13], deoxyglucosinolates [14], 2-deoxy-2-fluoroglucotropaeolin [15], C-glucosinolates [16], and 5a-carbaglucotropaeolin [17].
2.1 The anomeric disconnection
The anomeric disconnection scheme implies a “glycosidation-type” approach involving a standard electrophilic glucosyl donor and a thiohydroxamic acceptor. The first synthesis of glucotropaeolin by Ettlinger and Lundeen [5] is based on that scheme: phenylacetothiohydroxamic acid (prepared in 33% yield from benzylmagnesium chloride, carbon disulfide and hydroxylamine) was reacted with acetobromoglucose under basic conditions to produce the glucosyl thiohydroximate in reasonable yield (Scheme 4). Subsequent O-sulfation of the hydroximino group using sulfur trioxide pyridine complex gave the peracetylated glucotropaeolate anion, which could be isolated either as potassium or tetramethylammonium salt. Standard de-O-acetylation finally afforded glucotropaeolin after cation exchange purification.
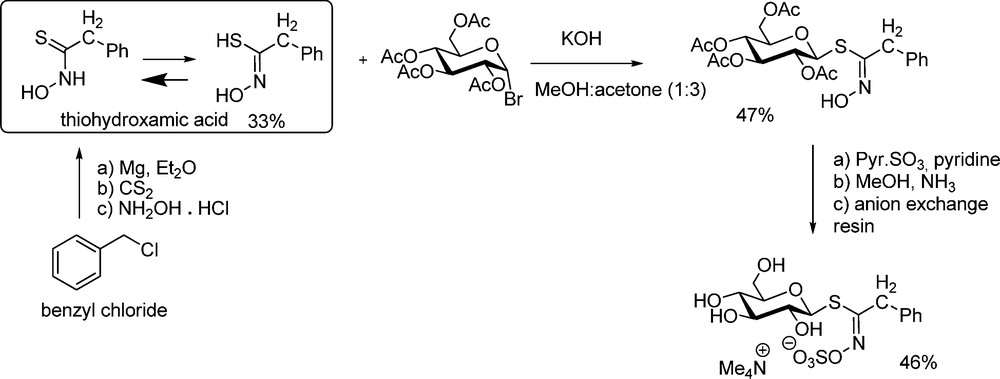
To date, this example remains unique as it has not been further developed (with one isolated exception [18]). This might likely be ascribable to: (a) the low-yield formation of the intermediate alkylthiohydroxamic acid, whose stability can be regarded as rather low [19]; (b) the moderate efficiency of the nucleophilic displacement at the anomeric carbon of acetobromoglucose. It turns out that most of the following GLs syntheses have followed the second disconnection pattern.
2.2 The hydroximate disconnection
The thiohydroximate disconnection scheme is based on the 1,3-addition of a glycosyl mercaptan on a nitrile oxide [20]. Because of their high lability [21], nitrile oxides have to be generated in situ from hydroximoyl chloride precursors through a 1,3-elimination under basic conditions (Scheme 5). The key intermediate in the reaction is in fact the hydroximoyl chloride, which in turn also appears to be quite unstable in most cases. Indeed, the different approaches developed over the years for synthesizing GLs depend on three different ways to access hydroximoyl precursors – from aldoximes, from aliphatic nitronates or from nitrovinyl derivatives.
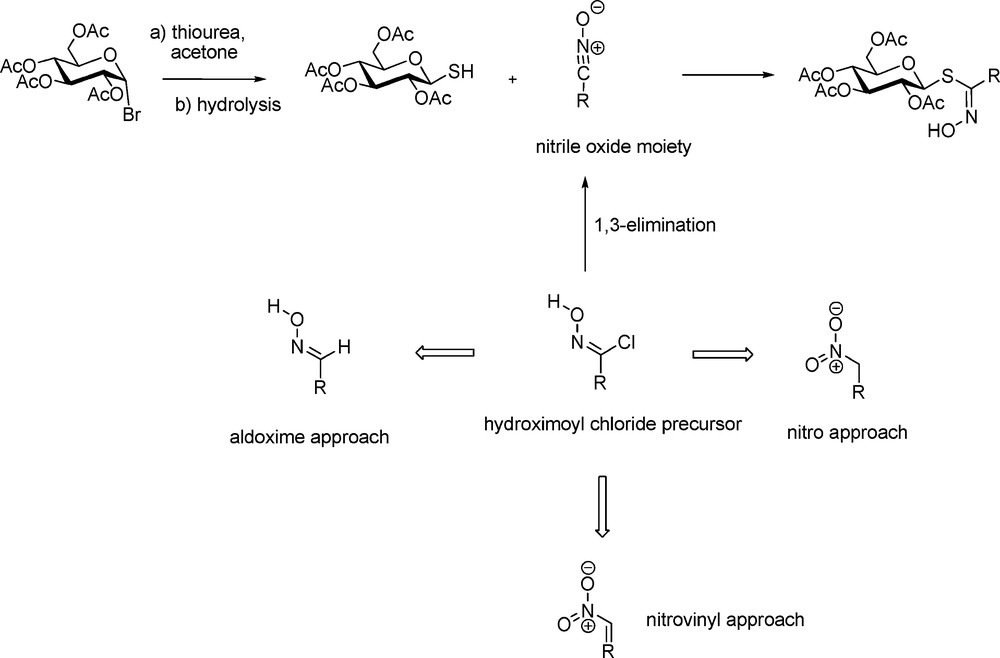
2.2.1 The aldoxime pathway
The pioneering work of M.H. Benn, who first devised efficient methods for the synthesis of several naturally-occurring GLs has to be tremendously acknowledged. In the early 1960s, aliphatic [22] and arylaliphatic [23] GLs were synthesized by Benn using the aldoxime pathway (Scheme 6). Chlorination of aldoximes using either chlorine gas or N-chlorosuccinimide [24] afford transient α-chloronitroso derivatives [25], which are readily rearranged into the corresponding hydroximoyl chlorides. Without further purification, those electrophilic acceptors were reacted in the presence of an organic base with 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranose to produce in good yields the anomeric (Z)-thiohydroximate intermediates with complete stereocontrol [20a].
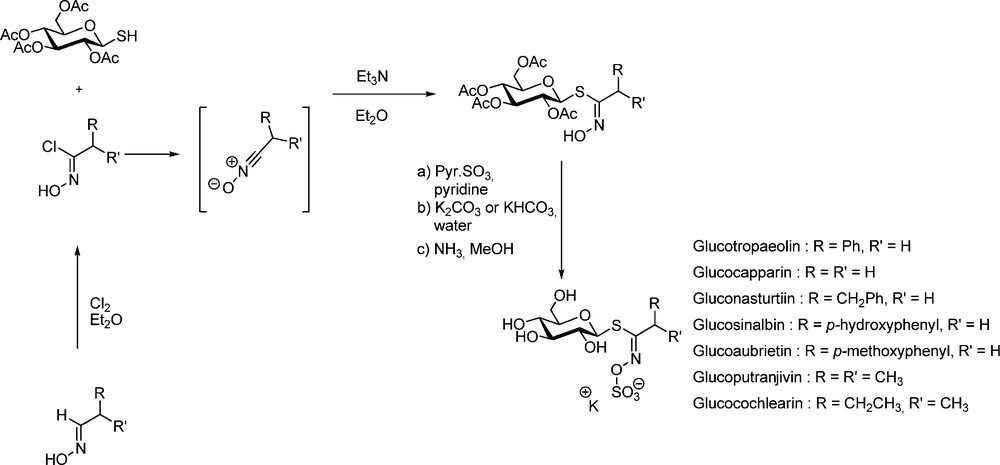
Subsequent O-sulfation with sulfur trioxide pyridine complex, followed by pyridine displacement with KHCO3 and standard de-O-acetylation finally delivered the expected GLs in acceptable overall yields.
2.2.2 The nitronate pathway
The foreground role of Benn and Ettlinger was again illustrated in a joint publication reporting in 1965 the first synthesis of sinigrin [26a]. This latter GL – widespread in the Brassicaceae family, notably in mustards [1,2,27] – bears an allyl aglycon chain, whose alkenyl character prevents bringing an aldoxime chlorination step in the synthesis. In such case, the key-hydroximoyl chloride was obtained via nucleophilic chlorination of an alkenyl nitronate (Scheme 7).
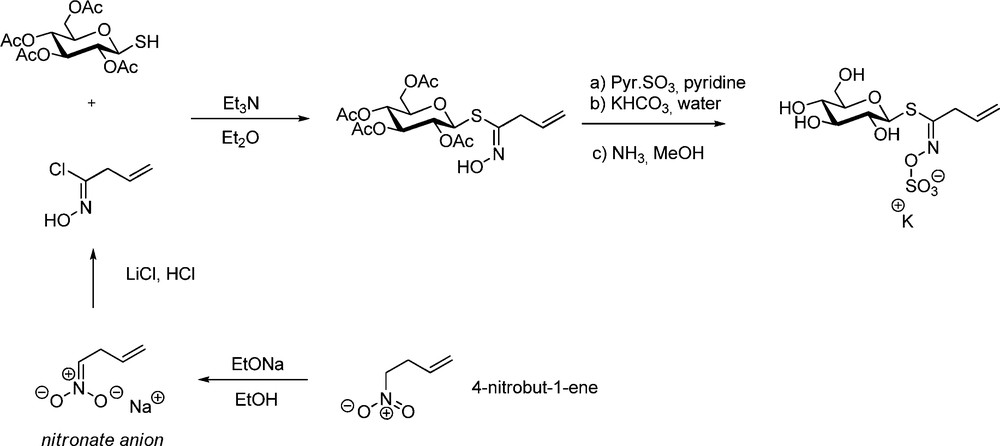
A few years later, A. Kjaer applied the same protocol to the synthesis of gluconapin (but-3-enyl GL) starting from 5-nitropent-1-ene [28]. Later on, a similar sequence – in which SOCl2 was used to generate the hydroximoyl chloride (Scheme 8) – was applied by the same authors to synthesize a diastereomeric mixture of the 2(R and S)-hydroxybut-3-enyl GLs progoitrin and epiprogoitrin [29].
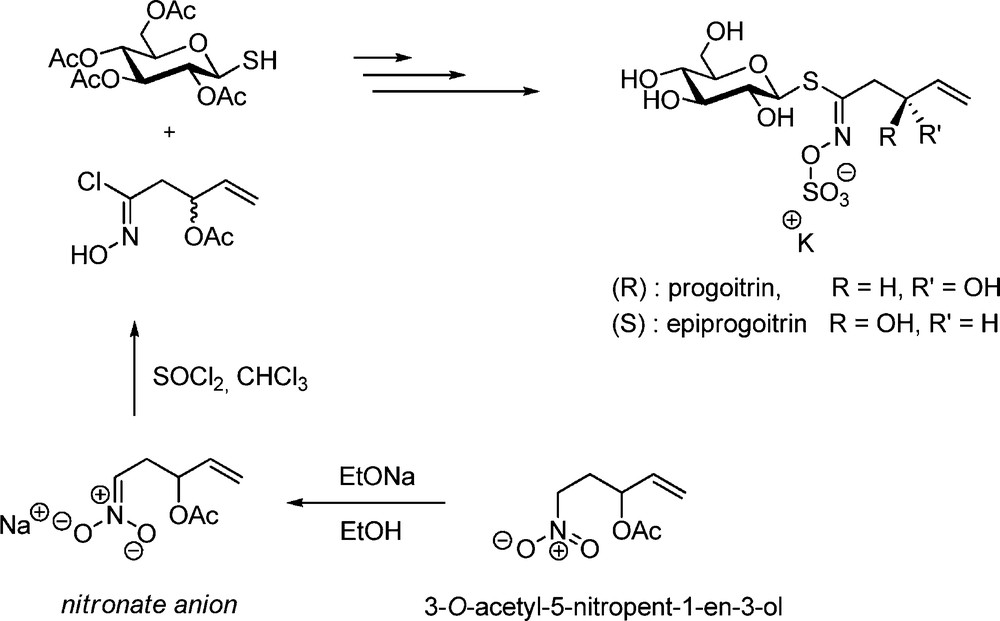
A decade after, a total synthesis of stereopure progoitrin was devised by MacLeod and Rossiter [30]. In a comparative study, the nitronate methodology was also applied to a larger-scale synthesis of gluconasturtiin [8]: MacLeod concluded that the hydroximoyl chloride formation from the nitronate was far less efficient than from the aldoxime.
Nevertheless, the use of electrophilic chlorinating agents was precluded in the case of indole-type or methylsulfanyl-type GLs. Rollin et al. thus followed a nitronate pathway for the first synthesis from 3(2-nitroethyl) indoles of the parent glucobrassicin (indol-3-ylmethyl GL) and its 4- and 5-methoxy derivatives [9]. The same approach was adopted for the synthesis of another important class of GLs – the linear ω-methylsulfanylalkyl GLs [11]: several representatives including glucoibervirin (n = 2), glucoerucin (n = 3) and longer chain representatives were thus prepared. Both synthetic sequences (Scheme 9) introduced two noteworthy technical modifications: (a) sulfation was effected using chlorosulfonic acid in pyridine; (b) final de-O-acetylation under Zemplen conditions was catalyzed by potassium methoxide.
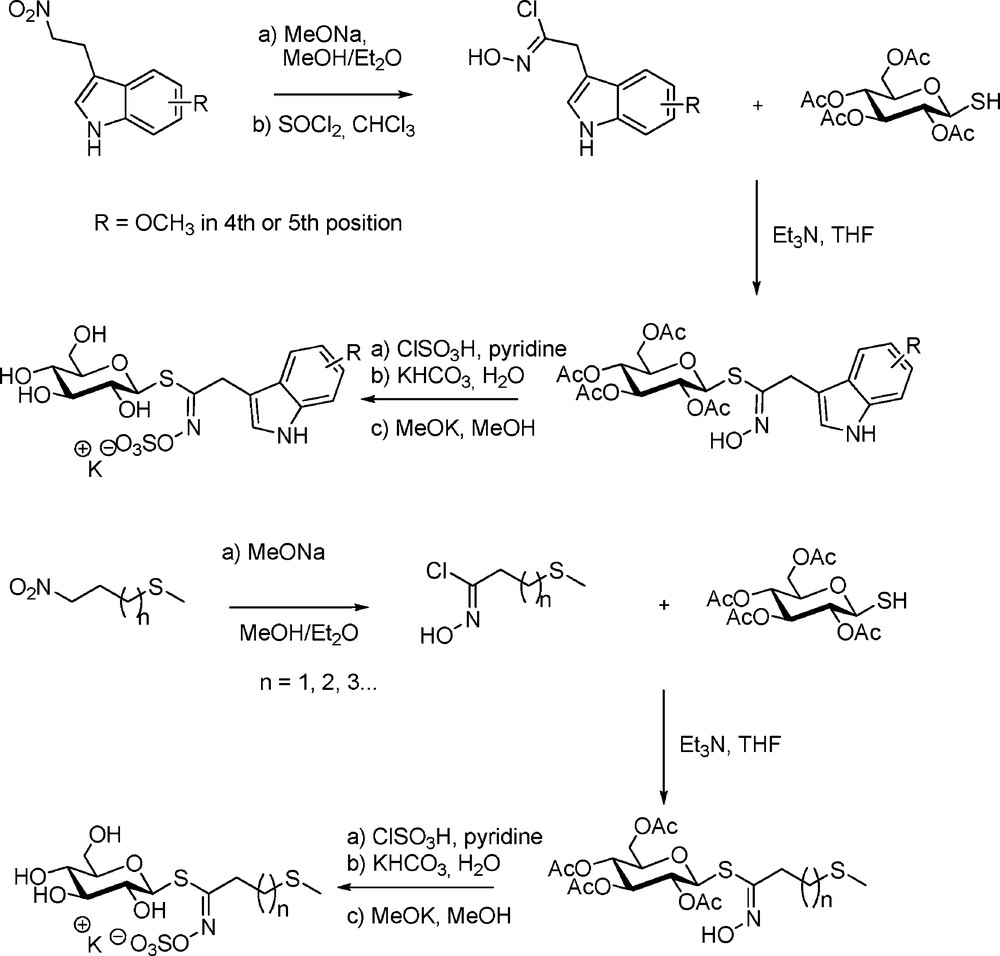
In the light of patents reporting the reaction of thiols on nitroalkanes in basic medium to produce “α-oximino sulfides” [31], the nitronate approach was revisited by Benn with the introduction of stabilized O-silylated nitronates as electrophilic partners in the coupling with the sugar mercaptan [32]. On that basis, a comparative study was performed to prepare glucolepidin (ethyl GL) starting either from propanaldoxime or from nitropropane: the latter method proved superior with a 84% condensation yield, but a ca 2:1 mixture of Z- and E-thiohydroximates was obtained (Scheme 10). This lack of stereoselectivity – which, in fact, allows synthetic access to the unnatural E-stereoform of GLs – is a consequence of (a) the absence of a nitrile oxide intermediate and (b) the rapid O-O migration of the silyl group in silylated nitronates [33].

2.2.3 The nitrovinyl pathway
This more recent methodology was applied after Kulkarni had reported his efficient one-step conversion of nitroalkenes into hydroximoyl chlorides [34]. Reacting readily accessible nitrovinyl derivatives [35] under Lewis acid activation with triethylsilane as the source of the hydride ion led to the formation of substituted acethydroximoyl chlorides with good yields. This is significantly shorter than the previously mentioned nitronate method. Rollin et al. followed this nitrovinyl pathway to synthesize several arylalkyl GLs of the glucosinalbin (p-hydroxybenzyl GL) group [36]. Several O-protected 2-nitrovinyl phenols were readily converted into the corresponding hydroximoyl chlorides, which in turn were efficiently coupled with the thiosugar moiety to afford the GLs (Scheme 11).
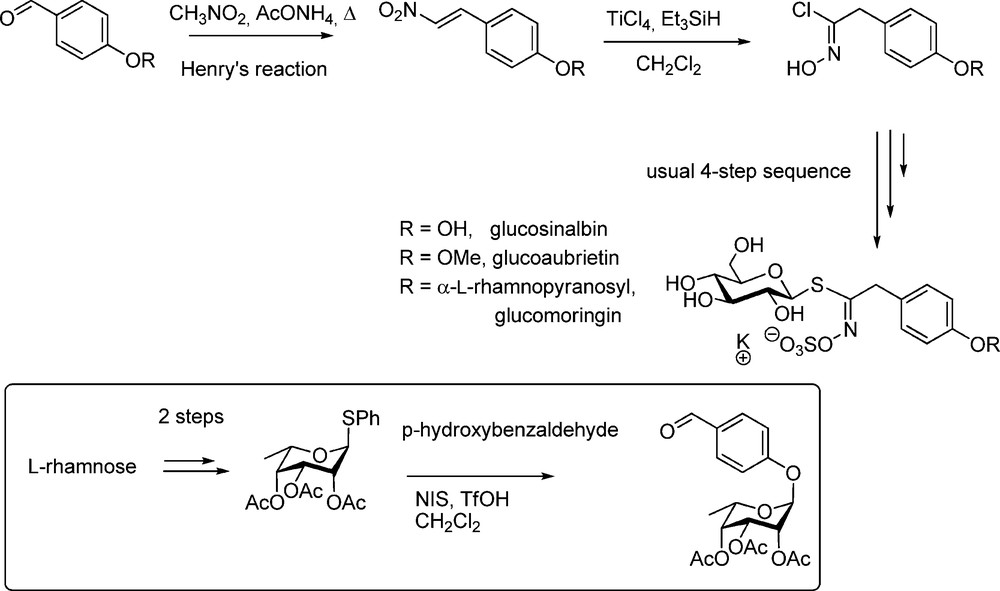
A more striking application of the nitrovinyl method was reported for the first synthesis of the major GL of Moringa sp. (“glucomoringin”), an O-rhamnosylated form of glucosinalbin [37]. The key-intermediate used by Rollin et al. was a per-O-acetylated p-O-rhamnosylated nitrostyrene (Scheme 11) elaborated via a modification of Kunz's method [38].
Also spectacular was the application of the nitrovinyl pathway to the particularly attractive glucobrassicin family [36]. The free NH of indole being poorly compatible with the harsh Kulkarni conditions, diversely N-protected 3-(2-nitrovinyl)indoles [39] were synthesized to be converted into the transient hydroximoyl chlorides. The overall yields of glucobrassicin (Scheme 12) and N-substituted derivatives were significantly superior to that previously obtained using the nitronate pathway [9].
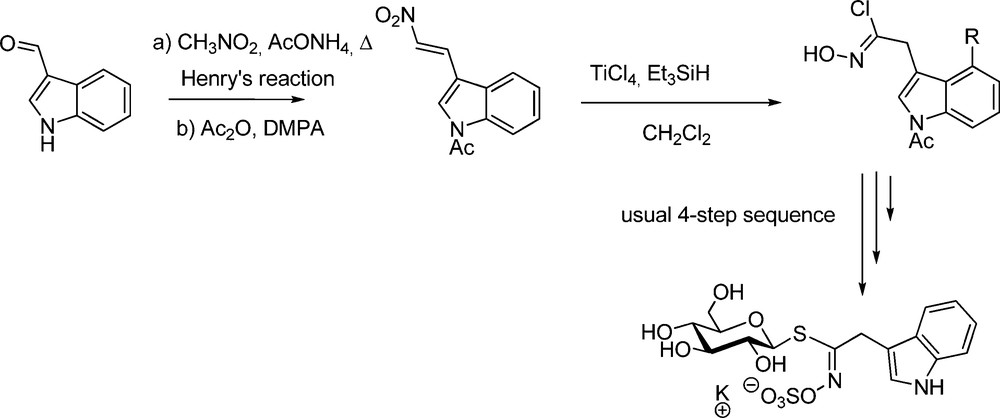
To generate the key-hydroximoyl chlorides, the nitrovinyl pathway thus appears as a real improvement compared to the nitronate pathway, by facilitating the benchwork and shortening the synthetic sequence. Nevertheless, as recently illustrated by a total synthesis of glucoraphasatin (4-methylsulfanyl-3-butenyl GL) [40], the aldoxime pathway still remains a method of choice whenever the functionality of the chain can endure halonium species.
3 Synthesis of glucosinolate analogues
3.1 Isotope-labelled glucosinolates
Isotope-labelling of GLs has been considered an important tool not only to decipher the metabolic pathways of GLs but also to follow their multifaceted biological activity. Extension to isotope-labelling of desulfo-GLs is also useful in unravelling biosynthetic sequences. With a view to studying the in vivo biotransformations of indole-type GLs, a first synthesis of glucobrassicin radio-labelled on the aglycon chain was performed in 1993 (Scheme 13) [41a]: prepared according to the modified nitrovinylation method [42], 5-bromo-3-(2-nitrovinyl)indole was converted into 5-bromoindol-3-ylmethyl GL via the nitronate pathway. Palladium-catalyzed tritiolysis furnished 98% radiopure tritium-labelled glucobrassicin, which was used in breakdown studies of indole GLs [41b].
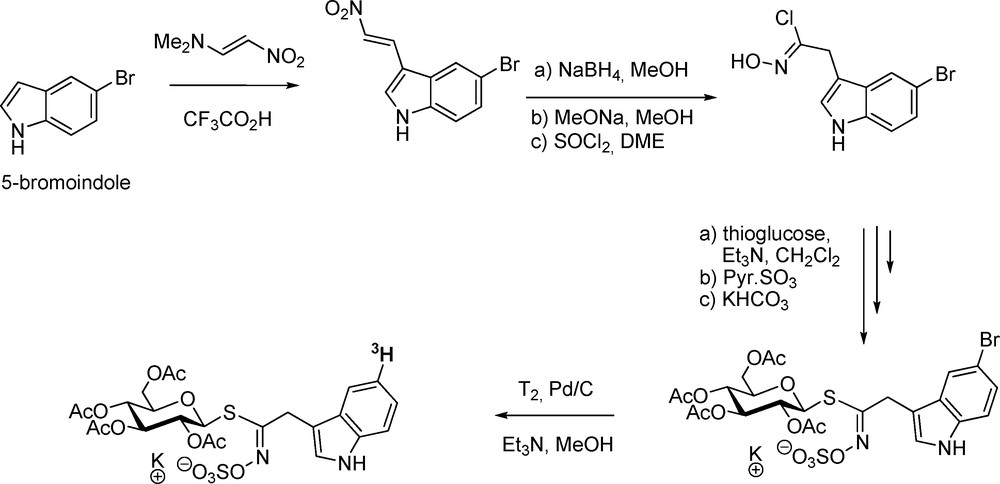
A similar protocol was applied for the preparation of tritium-labelled glucotropaeolin starting from p-bromo-(2-nitrovinyl)benzene [43]. In relation with biosynthetic studies, tritium-labelled desulfo-GLs were also synthesized via tritium addition on alkenyl [19b,20b,43] or alkynyl [44] precursors. Radio-labelling at the thiohydroximate carbon in desulfo-gluconasturtiin (2-phenylethyl GL) was performed using 14C-labelled 3-phenylpropanaldoxime [45].
Labelling with non-radioactive isotopes such as 2H or 13C is critical to elaborate analytical standards to aid accurate and reproducible quantitation of GLs. Rossiter et al. recently reported a synthesis of [2,3-2H2]sinigrin [46] while Robertson and Botting have developed quite a number of syntheses of either deuterated or 13C-labelled GL derivatives. Botting's first paper [47] reported the synthesis of:
- • pentadeuterated desulfo-gluconasturtiin [48] via the aldoxime pathway, starting from [phenyl-2H5]hydrocinnamaldoxime;
- • [2H3]4-methoxy- and [2H3]1-methoxy-desulfo-glucobrassicins via the nitronate pathway, starting from the related isomeric [2H3]methoxy-3-(2-nitroethyl)indoles (Scheme 14).
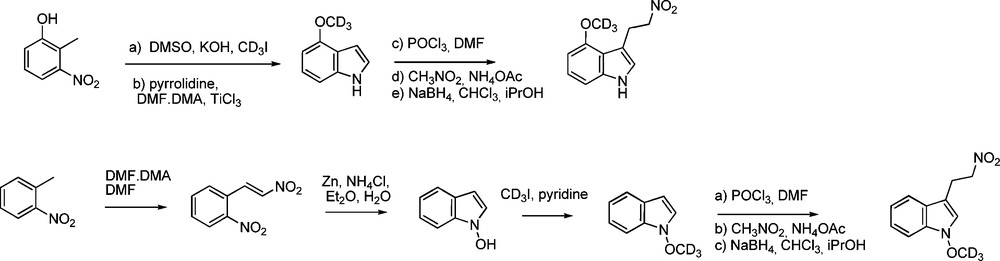
More recently, Robertson and Botting reported deuterium labelling in the gluco moiety [49]: through introducing 2,3,4,6-tetra-O-acetyl-1-thio-β-D-[1-2H, 6-2H2]glucopyranose in the aldoxime pathway, [1-2H, 6-2H2]desulfo-gluconasturtiin was synthesized (Scheme 15) for use as internal standard for MS-based analytical methods.
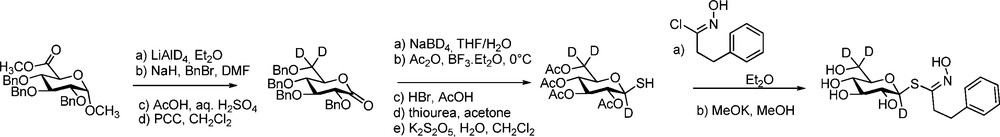
Another breakthrough was made by Botting in GLs isotope-labelling when he reported the synthesis of glucoraphanin (4-methylsulfinyl GL), the bio-precursor of sulforaphane, major anti-carcinogenic compound in broccoli [50]. Starting from 2-(2-bromoethyl)-1,3-dioxolane, 10 steps – involving chlorination of an ω-sulfinyl aldoxime – were required to obtain glucoraphanin in 9% overall yield as a mixture of epimeric sulfoxides. 13C-labelling of position 3 in the side chain by use of K13CN and deuterium labelling of both positions α to the sulfinyl group by use of hexadeuterated DMSO gave access to [10-13C, 11,12-2H5]glucoraphanin (Scheme 16).
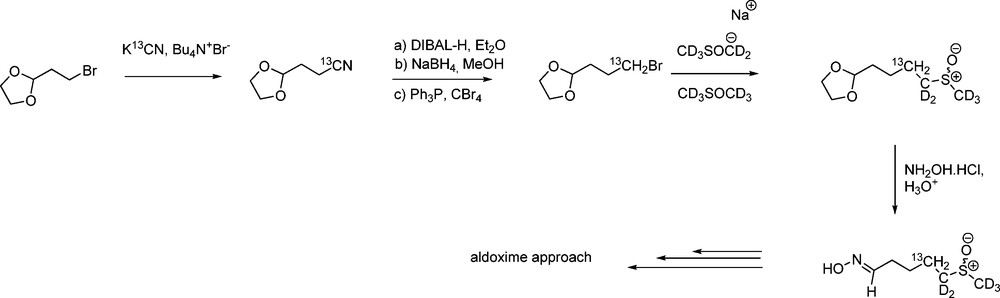
More recently, starting from commercially available [13C6]-D-glucose, Botting et al. have developed a three-step synthesis of hexa-13C-labelled 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranose [51]. This thiol can be employed to synthesize any [gluco-13C6]-GL for use as chemical tags in LCMS analysis or metabolic studies: [gluco-13C6]-gluconasturtiin, sinigrin and glucoerucin (4-methylsulfanyl GL) were thus produced (Scheme 17) either via the aldoxime or the nitronate pathway.

3.2 Glucosinolates with a modified sugar framework
With a view to better understanding this unique “thio”-glycosidase, and developing new chemical tools for biochemical and biological studies, the myrosinase-GL recognition mechanism has been investigated. Synthesized substrate-analogues with tailor-made structures involving modifications at different sites of the GL template have been designed with a classic targeting of the sugar framework.
3.2.1 Deoxyglucosinolates
The compared importance of the hydroxyl groups in the glucopyrano frame was investigated by using synthetic 2-, 3-, 4- and 6-deoxyglucotropaeolins and their glucobrassicin counterparts [52]. The key-reactions consisted in the elaboration of the corresponding per-O-acylated 2-, 3-, 4- and 6-deoxyglucosylmercaptans, to be further coupled with the hydroximoyl chlorides according to Schemes 6 or 9, respectively (Scheme 18).

Through the above study, it could be demonstrated that the lack of hydroxyl in C-2 of the D-gluco moiety is responsible for a strong inhibitory effect on myrosinase [53]. With a view to trapping the glycosyl-enzyme intermediate for a determination of the molecular mechanism, 2-deoxy-2-fluoroglucotropaeolin was similarly synthesized [15] from protected 2-deoxy-2-fluoro-1-thio-β-D-glucopyranose (Scheme 19) and tested as substrate in the stereochemical course and the mechanism of the hydrolysis catalyzed by myrosinase [54].
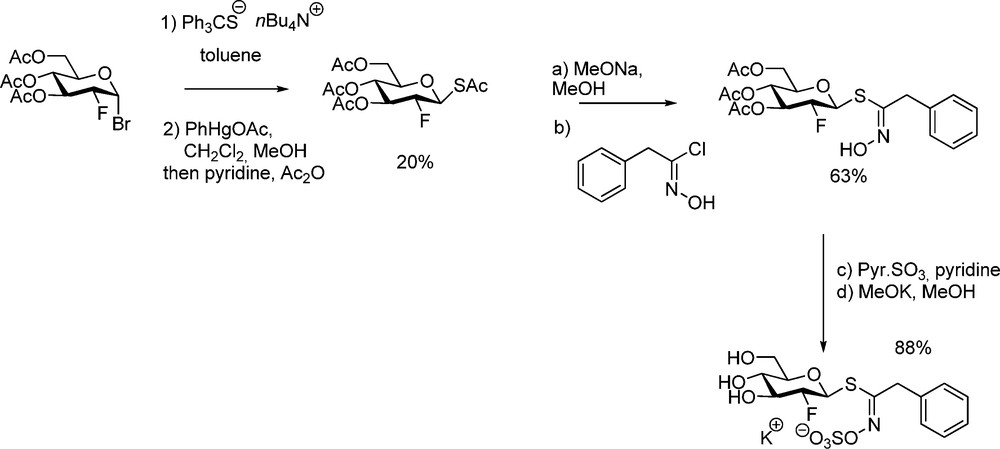
3.2.2 Glycosinolates
An early synthesis of non-gluco analogues of GLs was reported by Uchiyama, who used the Ettlinger [5] methodology to produce galacto- and xylotropaeolins to perform comparative testing of myrosinase [18]. Several decades later, Rollin et al. synthesized a range of eight sugar-variants – including the L-rhamno analogue – of naturally-occurring glucobrassicin [10], using the nitronate pathway (Scheme 20).
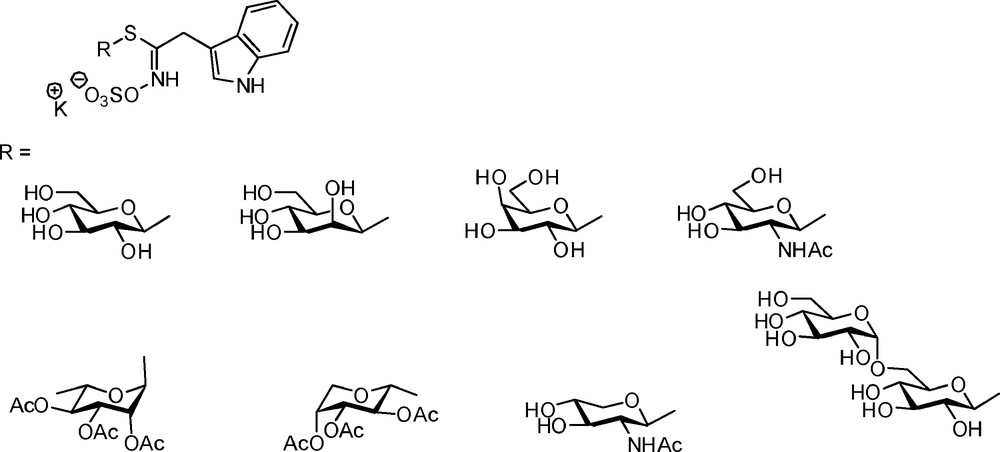
3.3 Glucosinolates modified at the anomeric site
3.3.1 Alpha-glucosinolates
Natural GLs display exclusively a β-D-gluco configuration at the anomeric carbon: it was therefore crucial to develop a synthetic access to the unknown α-anomers. The main difficulty consisted in the elaboration of the required O-protected α-glucopyranosyl mercaptan via a reliable method [55]: further coupling with hydroximoyl chlorides according to Schemes 6 or 9 (Scheme 21) led to the isolation of five α-GLs (Scheme 21) – including the anomers of glucotropaeolin, gluconasturtiin and glucobrassicin [12].
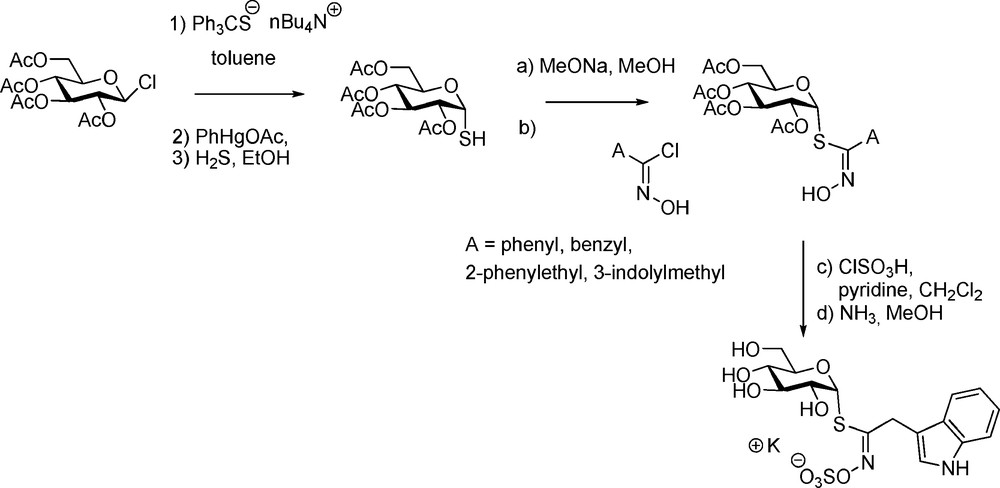
3.3.2 Selenoglucosinolates
Although selenium-containing GLs do not seem to be congeners of GLs in higher plants, Kjaer – in view of biosynthesis studies – achieved a striking synthesis of selenoglucosinolates (Scheme 22) starting from per-O-acetylated isoselenuronium bromide.
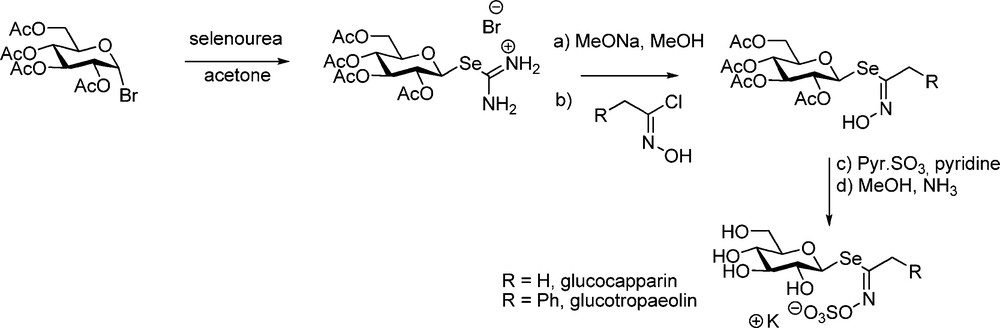
Seleno-analogues of desulfo-gluconapin [56], of glucotropaeolin and glucocapparin [57] were prepared and the last two were shown to be hydrolytically cleaved by myrosinase.
3.3.3 C-glucosinolates
The design and synthesis of non-hydrolysable GL analogues such as C-glucosidic derivatives is of interest for obtaining additional information about the substrate conformation and binding into the active site of myrosinase. Rollin et al. have designed a short synthetic sequence (Scheme 23) to produce C-analogues of glucotropaeolin [58] and glucocapparin [16].
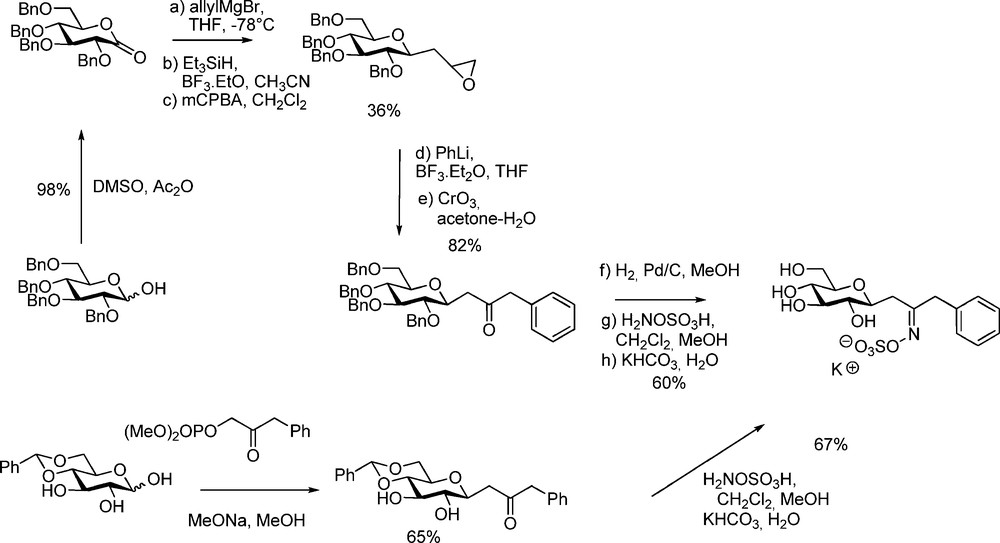
Applying a Horner-Emmons approach on 4,6-protected D-glucopyranose stereoselectively afforded C-glucopyranosyl ketones, which were converted by hydroxylamine-O-sulfonic acid into C-glucosinolates. Those compounds surprisingly did not display inhibitory activity: this is the first example of a glycosylhydrolase of non recognition of the C-analogue of its natural substrate.
3.4 Glucosinolates with a modified thiohydroximate function
The anionic site of GLs being critical in the recognition process by myrosinase, replacement of O-sulfate by another anion appears of prime interest. For that reason, Rollin et al. synthesized a phosphate bio-isostere of glucotropaeolin, which was able to undergo myrosinase hydrolysis [13].
Another synthetic project of Rollin's group was to replace the hydroximino moiety in desulfo-GLs by a hydrazono fragment for diverse purposes [59]. The required electrophilic partners for condensation with the protected 1-thio-β-D-glucopyranose are nitrilimines, which can be readily generated from hydrazonoyl chlorides resulting from Appel chlorination of hydrazides (Scheme 24). Several glucosyl thiohydrazonoates were thus efficiently prepared.

4 Artificial glucosinolates
The development of diversified substrate mimics for myrosinase investigation has found extension in synthesizing miscelleanous thiohydroximate systems which can be designated “artificial glucosinolates”. Most of the structural modifications have been realized either at the aglycon chain level or inside the sugar ring.
4.1 Glucosinolates bearing a non-natural aglycon chain
4.1.1 Synthesis of aryl glucosinolates
Without known exception, the structure of naturally-occurring GLs is characterized by connection of the aglycon chain to the pyrano ring through a methylene knuckle. Removing this fragment in benzyl-type GLs has been performed by several authors.
An early synthesis of phenyl GL (“nor-glucotropaeolin”) was first reported in a patent [60] by Ettlinger, who had switched from his first method [5] to a modified version of the aldoxime pathway: benzhydroximoyl chloride (prepared by tert-butyl hypochlorite chlorination of benzaldoxime) was first O-sulfated to provide a pre-salified form of the electrophilic partner to be coupled with the sodium salt of the thiosugar (Scheme 25).
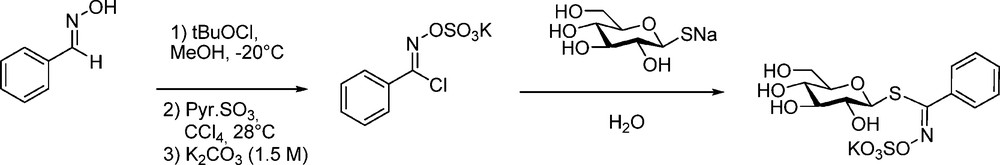
The more simple general procedure previously described (Scheme 6) was further employed by Saito et al. [61] and Rollin et al. [20b] to synthesize a collection of diversely substituted desulfo-nor-glucotropaeolins as customized internal standards for HPLC analysis of GLs. This was further applied by Praly et al. [62] and Somsak et al. [63] for the elaboration of glycogen phosphorylase inhibitors.
Using the same standard procedure, a synthesis of pentadeuterium-labelled nor-glucotropaeolin was reported: sequential conversion of commercially available [2,3,4,5,6-2H5] benzoic acid to [2,3,4,5,6-2H5] benzhydroximoyl chloride gave access to [2,3,4,5,6-2H5] phenyl GL, a useful internal standard for LC/MS techniques [64].
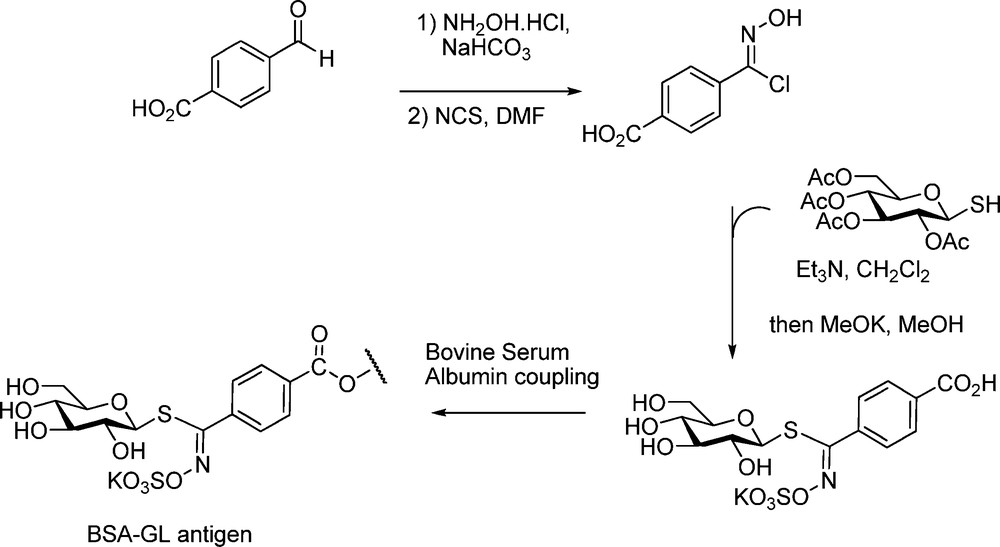
An application to the determination of GLs by enzyme-linked immunosorbent assay (ELISA) was developed [65] via synthesizing the p-carboxyphenyl analogue of nor-glucotropaeolin (Scheme 26); further coupling of this hapten to BSA allowed production of anti-GLs polyclonal antibodies.
4.1.2 Synthesis of alkyl and arylalkyl glucosinolates
The aglycon length of the known natural alkyl GLs does not exceed a C6 chain, whereas minor ω-methylsulfanylalkyl GLs can reach a C10 chain length. For the analytical purpose of setting up a regular homologous series of compounds, GLs or desulfo-GLs with increasing lipophilic character were synthesized via the aldoxime pathway:
- • GLs bearing C3-C5, C7 and C9 chains for a structure-activity study to investigate the interaction of GLs with leaf surfaces [66]; an interesting ω-carboxyalkyl GLs was also reported in the same paper;
- • desulfo-GLs bearing C1–C17 chains to examine the thermotropic and lyotropic liquid crystalline properties [67].
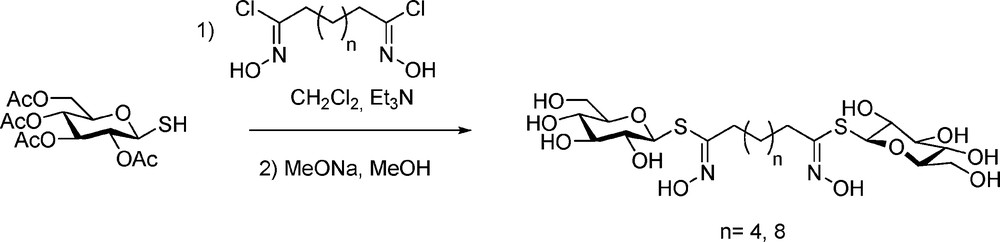
More innovative was the synthetic elaboration of bis-desulfo-GLs, designed as bolaamphilic structures [68] with mono- or disaccharidic moieties connected by a C6 or a C10 linear spacer (Scheme 27):
Artificial arylalkyl GLs – namely glucotropaeolins bearing substituents on the phenyl nucleus – were synthesized for various purposes [44,69]; additional arylalkyl analogues with more “heavy” aryl moieties like naphthyl, biphenylyl or benzhydryl were also constructed for a novel RP-HPLC assay to monitor myrosinase-dependent hydrolysis reactions [66,70].
Also worth mentioning is the synthesis of a useful [43] closely related artificial GL: 2-phenylethenyl GL, which can be regarded as a dehydrogluconasturtiin [20b].
4.2 In-the-ring modified glucosinolates
Replacement of the endo-oxygen in the glucopyrano ring allows delivery of non-hydrolyzable substrates – new tools to investigate the substrate conformation and binding inside the myrosinase pocket.
4.2.1 Synthesis of 5-thioglucosinolates
The central part of the synthesis (Scheme 28) consisted in the elaboration of the corresponding 2,3,4,6-tetra-O-acetyl-1,5-dithio-β-D-glucopyranose, to be further coupled with hydroximoyl chlorides (Schemes 6 or 9). Prepared from 5-thio-D-glucose according to a slightly modified Cerny methodology [71], this hemi-dithioacetal is the first representative of a new class of thiosugars.
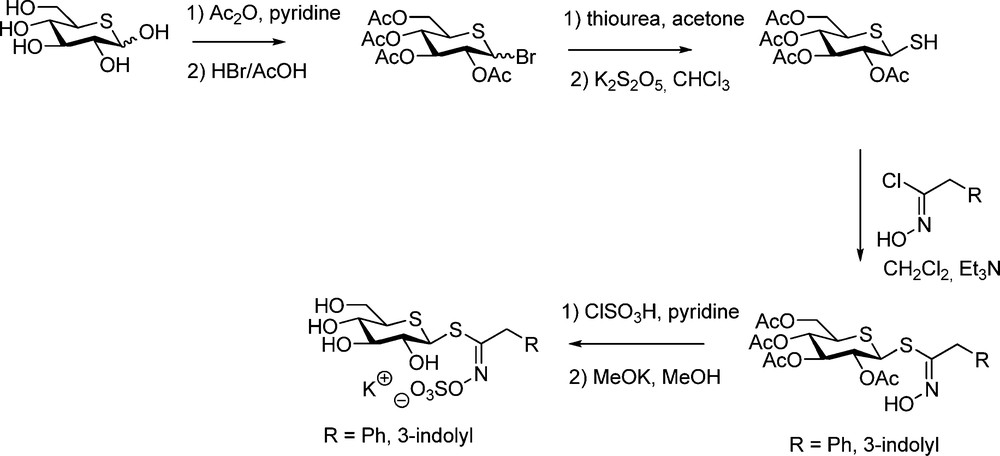
5-Thioglucotropaeolin and 5-thioglucobrassicin were prepared following standard steps [72] and it was further shown that myrosinase was unable to recognize such GL-mimics.
4.2.2 Synthesis of 5a-carbaglucotropaeolin
Puzzled by the lack of inhibitory activity displayed by C-glucosinolates (see § 3.3.3), Rollin et al. decided to undertake development of a synthetic route to 5a-carbaglucosinolates, inspired by established methodologies [73]. Through a six-step sequence (Scheme 29), the known 5a-carba-DL-glucal [74] was converted into the key 2,3,4,6-tetra-O-benzoyl-1-thio-5a-carba-β-DL-glucopyranose, which finally led to 5a-carbaglucotropaeolin in racemic form [75].
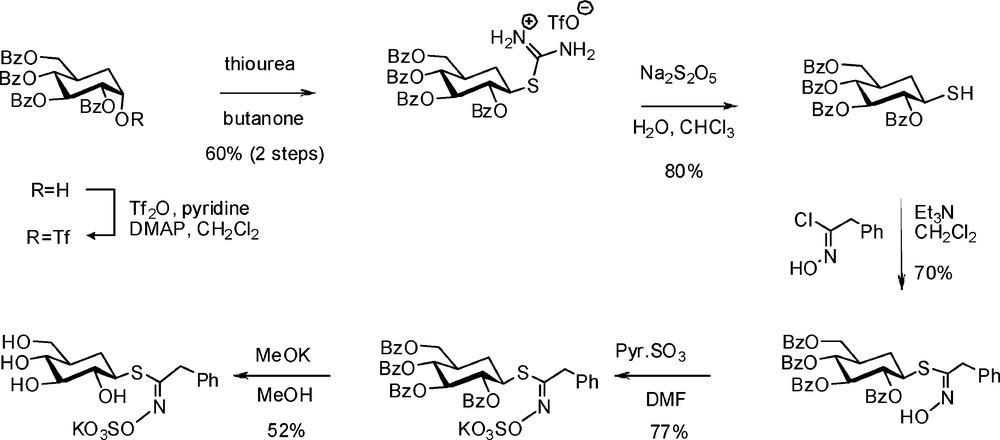
This non-hydrolysable GL showed a millimolar range inhibitory power towards myrosinase and allowed X-ray crystallographic direct observation of an enzyme-substrate analogue complex.
4.2.3 Miscellaneous artificial glucosinolates and surrogates
Replacing in a GL structure the aglycon chain by another saccharidic moiety requires construction of hydroximoyl chlorides on a sugar framework. Five representatives of a new family of pseudo-disaccharides were thus synthesized [76].
The negative charge of GLs being most important in the recognition process by myrosinase, modified anionic subunits were grafted onto the anomeric sulphur in place of the O-sulfated thiohydroximate. Several thioglucosides bearing a terminal sulfate or sulfonate were thus prepared and submitted to myrosinase inhibition tests [75].
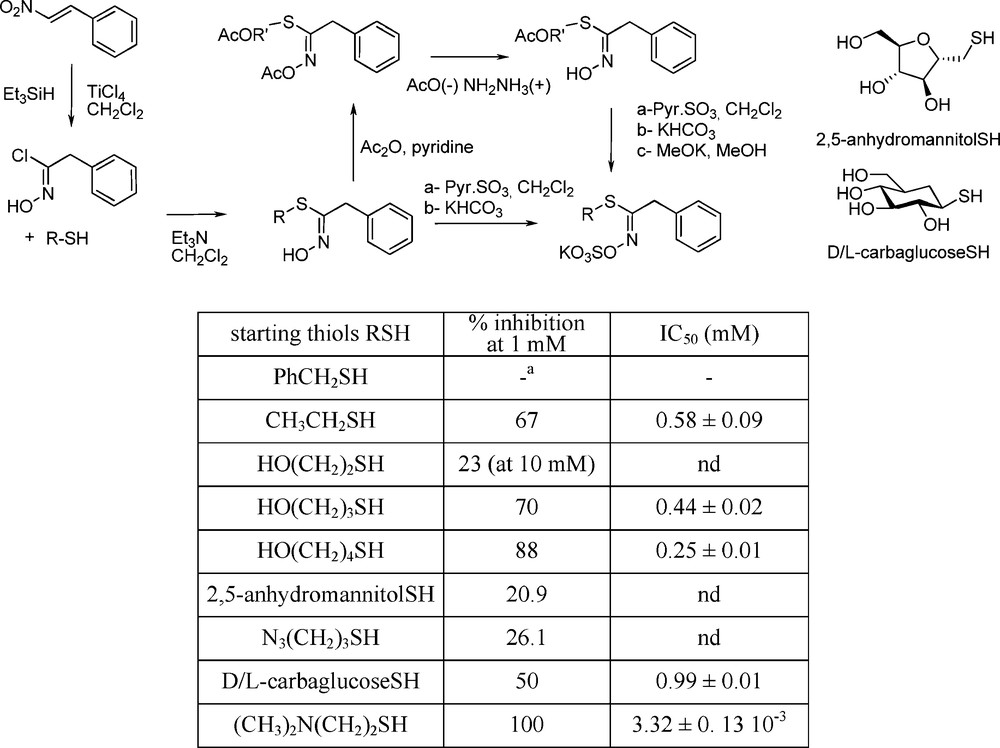
From the results obtained by replacing in a GL the glucopyrano part by its carba-analogue (see § 4.2.2), new insights into myrosinase-substrate interactions had come to light, in showing the unimportance of the D-gluco moiety during the initial interaction with the active site [75]. Many thiohydroximate-based inhibitors with a simplified structure were developed by Tatibouët et al. who synthesized a range of pseudo-glucotropaeolins in which the saccharidic frame is replaced by simple alkyl [75] or arylalkyl [77] chains. A zwitterionic system bearing a N,N-dimethylammonium segment was shown to be the first micromolar range myrosinase inhibitor (Scheme 30):
5 Concluding remarks
Although extraction of naturally-occurring GLs from appropriate vegetable sources remains the method of choice to obtain those molecules in amounts that allow diversified biological testing, this frequently inconvenient and delicate approach requires highly specialized expertise and know-how [4]. On the other hand, the chemical synthesis approach has appeared to be a more general way to access natural GLs, although differential difficulties can arise with some functionalized aglycons (indole-type, methylsulfanylalkyl-type…). In other respects, the synthetic approach evidently stands as the only way to access tailor-made artificial GLs which are mostly required by biochemistry and biology. Over the past 50 years, a limited number of chemical methods have been developed to produce GLs of many types, but improved procedures to circumvent labile intermediates would still be welcome. Due to limited knowledge and mastery of the enzymatic sequences involved in their biosynthesis, a biotechnological vision of GLs production still is remote [78]. However, combination of extractive and synthetic methodologies can be considered in close future: some scarce examples of the hemi-synthetic approach have already been reported, namely using oxidative processes [79].
Acknowledgements
The authors are grateful to all co-workers involved over the past 20 years in either natural or artificial glucosinolates synthesis.