

1 Introduction
For a long time, tautomerism has remained an intriguing phenomenon in physical organic chemistry. Hydrogen bonding appears to be one of the most important factors governing tautomeric equilibrium [1]. Resonance-assisted hydrogen bonding (RAHB) is a type of strong hydrogen bond commonly observed in systems where two oxygen atoms are connected by a conjugated spacer containing single and double bonds, such as carboxylic acid dimers or β-enolones, with O···O distances of 2.39–2.55 Å [2]. Over the last few decades, much interest has been focused on the nitrogen and sulfur analogues of such systems, particularly those with strong intramolecular RAHB related to the ···O = R3NH··· ↔···HOR3 = N··· tautomeric equilibrium, where R3 is a resonant spacer of three atoms [3]. In RAHB, the delocalization of electrons increases the strength of the hydrogen bond by increasing both the acidity of the hydrogen bond donor and the basicity of the hydrogen bond acceptor [4]. Similar to other aromatic azaheterocycles with an acylmethyl substituent [5], tautomerism in acylmethyl-1,3,5-triazines A (Fig. 1) can potentially result in the formation of two tautomers stabilized by intramolecular RAHB i.e. enol B and enaminone C. The six-membered hydrogen bond chelate may involve the triazine nitrogen atom acting as a proton acceptor and oxygen atom as proton donor (in form B) or vice versa (in form C). The tautomeric equilibrium depends on structural specificity and various external conditions. However, these factors have not been systematically studied for acylmethyl-1,3,5-triazines and some questionable arbitrarily chosen assignments have been appearing [6,7]. In some reported cases, the potential existence of enaminone form C was ignored [8,9] even though this form appeared to be predominant in other studies [10,11] and was the only one found in the solid state by X-ray crystallography [12].
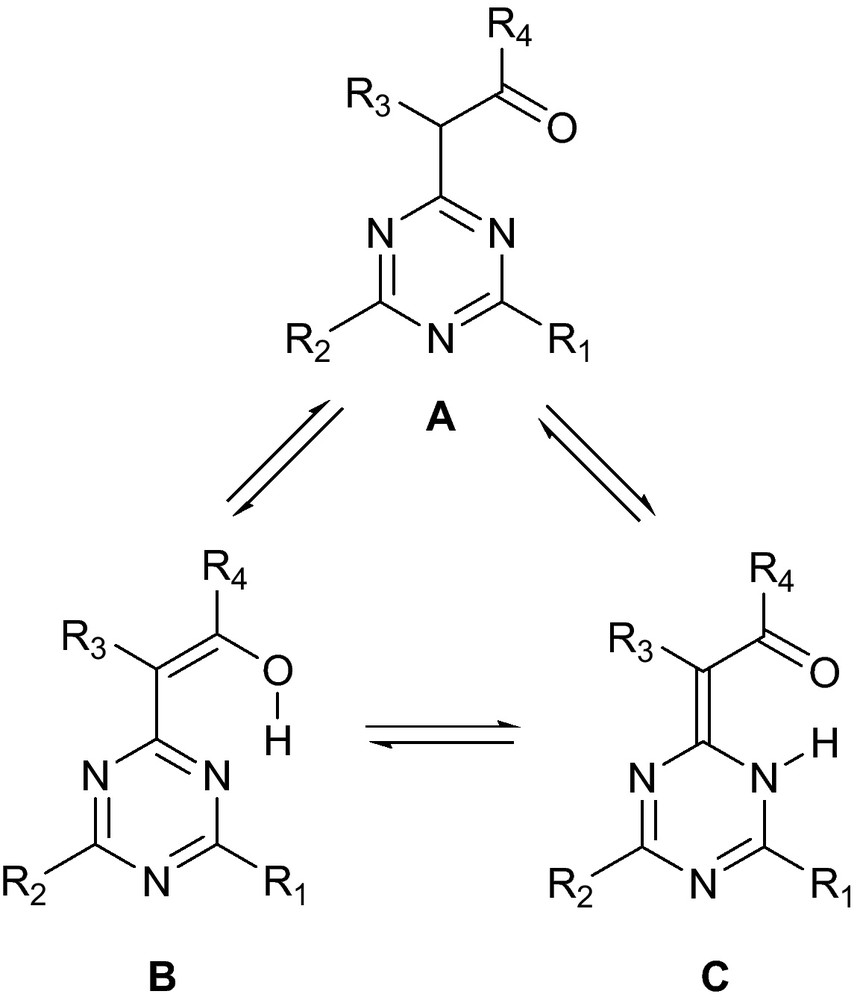
Tautomerism in acylmethyl-1,3,5-triazines.
Acylmethyl-1,3,5-triazines can be prepared using two general approaches: (1) derivatisation of 1,3,5-triazines using methylene active carbonyl compounds via nucleophilic substitution of chlorotriazines [6,13] or by addition to triazines followed by oxidative aromatisation [6], and (2) the 1,3,5-triazine ring formation via heterocyclization of biguanides or their analogues using β-keto esters [14].
However, using unsubstituted biguanide in the second approach may result in the formation of alternative products – guanidinopyrimidines [15] or their mixtures with acylmethyl-1,3,5-triazin-2,4-diamines [14,16].
In our continuous investigations [17] on azaheterylguanidines as valuable synthons for the preparation of bioactive compounds, we became interested in the synthesis of N-(6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl)guanidine (1). However, previously described [15c] reaction of biguanide with ethyl benzoylacetate resulted in the formation of additional product of interesting structure, namely 6-benzoylmethyl-1,3,5-triazin-2,4-diamine (2) together with the reported 1. Derivatives of the 2,4-diamino-1,3,5-triazine scaffold have been reported to exhibit antibacterial [18] and anticancer properties [19]. This prompted us to carry out a thorough structural characterization and biological investigation on 2. The present paper is devoted to the detailed structural investigations of 2, particularly tautomerism study using NMR spectroscopy, X-ray crystallography and theoretical calculations.
2 Result and discussion
2.1 Synthesis and structural assignments
In the reaction of biguanide with ethyl benzoylacetate in the presence of sodium methoxide, biguanide played the roles of triatomic and pentatomic synthons, affording N-(6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl)guanidine (1, 37%) and 6-benzoylmethyl-1,3,5-triazin-2,4-diamine (2, 26%), respectively (Scheme 1). The two compounds were effectively isolated from the reaction mixture using differential solubility in methanol. The structure of the products was established using NMR spectroscopy and X-ray crystallography.
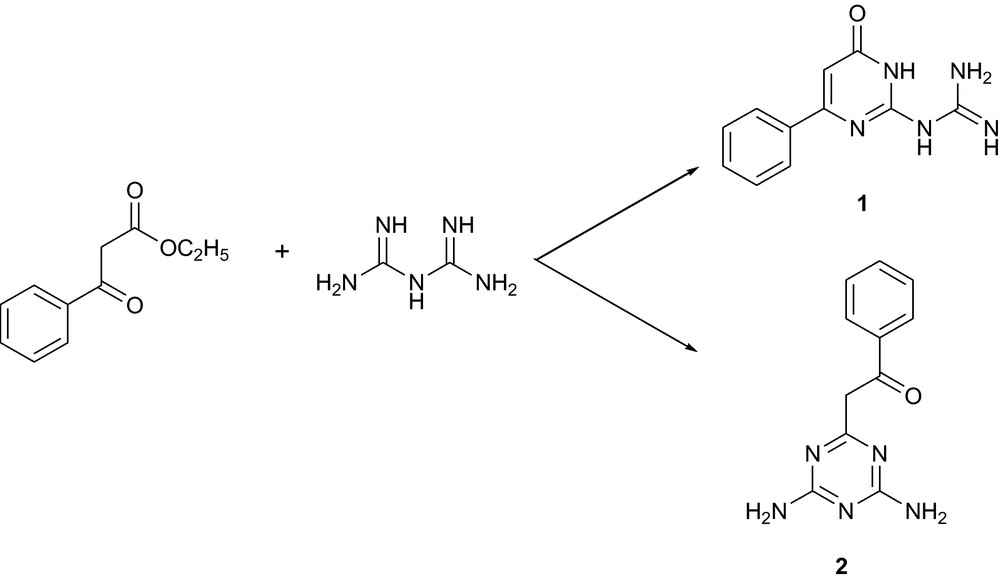
Reaction of ethyl benzoylacetate with biguanide.
Two distinct, unequally populated sets of signals were observed in the NMR spectrum of 2 at room temperature in DMSO-d6 solution indicating the existence of two tautomers in the proportion of 7:93. The singlet of methylene protons observed at 4.07 ppm in the 1H NMR spectrum along with the signals of methylene and carbonyl carbon atoms appearing at 48.8 ppm and 195.5 ppm in the 13C NMR spectrum corresponded to the structure of keto form A, which was found to be minor (7%) under these conditions (Fig. 2). The characteristic signals of the predominant tautomeric form (signals of methine proton and carbon at 5.67 and 89.2 ppm of 1H NMR and 13C NMR spectra, respectively as well as sharp proton peak at 15.22 ppm and carbon atom at 174.2 ppm) did not allow unambiguous differentiation between enol B and enaminone C. Moreover, the rapid NMR time scale in interconversion by intramolecular proton transfer between electronegative atoms via RAHB could not be excluded. Significant broadening of signal of quasiequivalent carbon atoms at 162.8 ppm in 13C NMR spectrum also suggested this situation.

Tautomerism in 6-benzoylmethyl-1,3,5-triazin-2,4-diamine and intramolecular RAHB.
2.2 Ab initio calculations
In order to clarify the structural assignments, the ab initio calculations of the relative energies of the tautomers A–C were preformed and the 13C chemical shifts were predicted using the gauge independent atomic orbital (GIAO) perturbation method [20].
The structures of the tautomeric forms were optimized at the B3LYP/6-31G(d,p) level of theory. Since there was no imaginary frequency in the vibrational spectra, all the tautomers were confirmed to exist at stationary points corresponding to the local minima on the potential energy surface. The relative energies at different level of theories were computed and compared for all the tautomeric forms (Table 1). Based on these results, tautomer A was found to be preferred in the gas phase and the order of stability of the tautomeric forms was calculated to be B > A ≈ C. However, the energy difference between the forms was found to be <10 kcal/mol.
Relative energies for the tautomeric forms of 2 according to ab initio calculations.
| Relative energies for tautomers, kcal/mol | |||
| Method | Keto form (A) | Enol form (B) | Enaminone form (C) |
| B3LYP/6-311G2d,2p | 4.50 | 0.00 | 3.65 |
| MP2/6-311G2d,2p | 3.52 | 0.00 | 9.13 |
The absolute shielding (σ) value of carbon in TMS was calculated at the B3LYP/6-311G2d,2p level of theory and was found to be 178.74 ppm. Using this value and absolute shielding values of ring carbon atoms for various tautomers obtained at same level of theory, the chemical shift values (δ) along with the deviation between experimental and predicted values (Δδ) have been calculated (Table 2). An excellent correlation (r2 = 0.9994) was found between the experimental and theoretical values for the keto tautomer A (Fig. 2) validating the calculation method. The experimentally obtained values of the 13C chemical shift δ for atoms connecting the triazine and phenyl ring of the major tautomeric form more closely resembled the calculated values for the enol form B (Table 2). It is in agreement with generally applicable distinction between enol and enaminone tautomers provided ∼20 ppm difference of the 13C chemical shift δ values for the carbonyl/enolic carbon atom [5b]. The experimental data were found to correlate better with theoretical values calculated for enolic form B rather than with those of enaminone tautomer C (cf. Fig. 3b and c). However, detailed comparison of the experimental and calculated values suggested that the experimental results appeared to be described more accurately as a weighted average of the two forms B and C. Therefore, the contribution of tautomer C cannot be excluded.
Experimental and calculated [B3LYP/6-311G(2d,2p)] chemical shifts (δ) in 13C NMR spectrum of 2 and deviation between experimental and predicted values (Δδ).
| Carbon atom | Experimental data, δ ppm | GIAO calculation data, δ(Δδ) ppm | |||
| Minor form | Major form | Keto form A | Enol form B | Enaninone form C | |
| C-2 | 166.9 | 167.8 (br) | 168.99 (−2.09) | 167.23 (0.57) | 164.92 (2.88) |
| C-4 | 166.9 | 162.8 (br) | 168.98 (−2.08) | 166.00 (−3.2) | 156.29 (6.51) |
| C-6 | 172.8 | 167.5 | 177.59 (−4.79) | 175.59 (−8.09) | 163.57 (3.93) |
| CH2/CH | 48.9 | 89.2 | 49.31 (−0.41) | 94.12 (−4.92) | 82.32 (6.88) |
| CO/COH | 195.52 | 174.2 | 196.83 (−1.31) | 172.88 (1.32) | 187.53 (−13.33) |
| C-1′ | 136.4 | 137.3 | 137.88 (−1.48) | 137.57 (−0.27) | 141.00 (−3.7) |
| C-2′ | 128.6 | 128.3 | 130.40 (−1.8) | 128.27 (0.03) | 128.30 (0.00) |
| C-3′ | n.i | 125.7 | 129.70 | 129.26 (−3.56) | 129.70 (−4.0) |
| C-4′ | 133.2 | 130 | 134.13 (−0.93) | 131.21 (−1.21) | 132.85 (−2.85) |
| C-5′ | n.i | 125.7 | 128.76 | 128.76 (−3.06) | 129.36 (−3.66) |
| C-6′ | 128.6 | 128.3 | 129.64 (−1.04) | 126.92 (1.38) | 127.97 (0.33) |

Tautomerization of 2 in the DMSO-d6 solution with time after addition of D2O (20%) at 27 °C. 1H NMR spectra (the phenyl groups signals) depict shift of the equilibrium towards keto form A (indicated with asterisk).
2.3 Effects of solvent and temperature on the tautomerism of 6-benzoylmethyl-1,3,5-triazin-2,4-diamine
It has been well recognized that the relative stability of the keto, enol and enaminone tautomers in the solution are dependent on the properties of the solvents [21]. We studied the effect of the solvent on the tautomerism of 2 using 1H NMR spectroscopy. DMSO-d6, acetone-d6 and methanol-d4 were used as solvents in the experiments. In all cases, keto form A was the minor tautomer (Table 3). The equilibrium was established instantly after dissolving the sample and remained unchanged after 48 h. The tautomeric equilibrium could be shifted by changing solvent composition. The addition of D2O to the solution of 2 in DMSO-d6 (1:4 - D2O:DMSO-d6) showed along with the instant suppression of signals of two amino groups and OH/NH singlet, the time dependent suppression of the methine proton peak at 5.67 ppm and methylene signal at 4.07 ppm in the 1H NMR spectrum. At the same time, a gradual shift of the tautomeric equilibrium towards keto form A was observed (Fig. 3). This time-dependent process was monitored at different temperatures by 1H NMR spectroscopy and the sample compositions were estimated by integration of the phenyl group signals of the tautomers with 5 min intervals until the equilibration was achieved (Fig. 3, Table 3). Heating was found to shift equilibrium towards keto form A (Fig. 4).
Kinetic and thermodynamic parameters of D2O induced tautomerization.
| Solvent | Temperature (°C) | KT = [A]/[B + C] | ΔG (Kcal/mol) | k1 + k−1 (10−2 min−1) | k1 (10−3 min−1) | ΔH (kcal) | ΔS (cal/deg) |
| DMSO d6: D2O 4:1 | 27 | 0.201 | 0.957 | 1.1 | 1.2 | - | - |
| DMSO d6: D2O 4:1 | 32 | 0.222 | 0.896 | 2.5 | 4.7 | 2.389 | 5.691 |
| DMSO d6: D2O 4:1 | 37 | 0.243 | 0.844 | 6.7 | 13 | - | - |
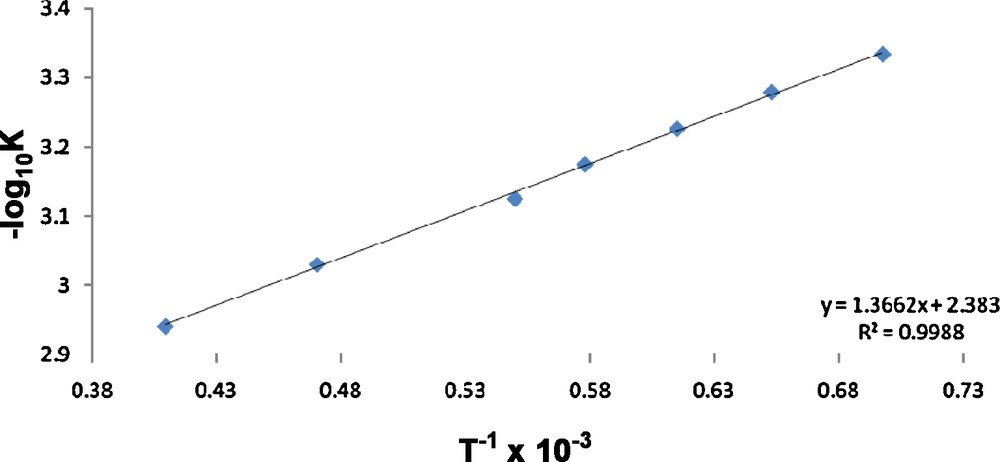
Plot of equilibrium constant vs temperature.
The kinetic and thermodynamic parameters of the D2O induced tautomerization of 2 were calculated [22] (Table 3). A reversible first-order reaction was postulated and the data were fitted [23] to the rate equation to obtain the fittable parameters: (1) enol-enaminone (B + C) concentration at equilibrium – Ae, (2) difference in initial enol-enaminone (B + C) concentration and the concentration at the equilibrium–(Ao–Ae), and (3) effective rate constant – (k1 + k−1). The activation energies for the forward and the backward reaction, Ea and Ea’ respectively, were estimated using the Arrhenius plot. The Ea and Ea’ were obtained as 38.2 and 35.8 kcal mol−1 respectively (Table 4).
Solvent effect on tautomerism of 2.
| Solvent | Temperature (°C) | KT = [A]/[B + C] | ΔG (kcal/mol) |
| DMSO-d6 | 25 | 0.147 | 1.144 |
| Acetone-d6 | 25 | 0.135 | 1.193 |
| Methanol-d4 | 25 | 0.058 | 1.695 |
2.4 Crystal structure (solid state)
The tautomeric preference of 2 in solid state was determined by an X-ray crystallographic study (Table 5). The single crystal suitable for the analysis was grown by slow evaporation of a methanolic solution of 2. The asymmetric unit was found to contain two molecules of the compound, two water molecules and one molecule of methanol. Both triazine molecules existed in the crystal as enaminone tautomeric form C. The molecular structures of enaminone tautomer were stabilized by the resonance between phenyl and triazine rings connected with the methylene carbonyl bridge as well as by strong intramolecular hydrogen bonding (NH…OC) (Table 6). In one molecule, the triazine ring was essentially planar (r.m.s. deviation was 0.0244 Å) and almost coplanar with the phenyl ring (dihedral angle was 3.45 (5)°). The triazine ring of the second molecule was also planar with r.m.s. deviation of 0.0145 Å. However, the dihedral angle between the triazine and phenyl rings in this molecule was larger (12.59 (9)°). The comparison of the bond lengths of the six-membered OCCHCNH chelate of the molecules with the values for the corresponding tautomer obtained by the DFT calculations (Table 5) revealed a potential for intramolecular hydrogen transfer within the crystal. This process is likely to be observed at higher temperatures, but decomposition of the crystal lattice did not allow the experimental confirmation of the tautomerization to enolic tautomer B.
Calculated and experimentally obtained bond lengths and angles for 2-(4,6-diamino-1,3,5-triazin-2(1H)-ylidene)-1-phenylethanone.
| Form B (Theoretical) | Molecule 1 (X-ray) | Molecule 2 (X-ray) | |||
| Bond lengths Å | Bond lengths Å | Bond lengths Å | |||
| OC(8) | 1.2668 | O(1)C(7) | 1.2815(17) | O(2)C(18) | 1.2762(19) |
| N(1)C(2) | 1.3515 | N(1)C(10) | 1.3617(18) | N(6)C(21) | 1.3577(19) |
| N(1)C(6) | 1.3947 | N(1)C(9) | 1.3671(18) | N(6)C(20) | 1.3680(19) |
| N(1)H(1N) | 1.0475 | N(1)H(1N) | 0.90(2) | N(6)H(6N) | 0.91(2) |
| N(5)C(6) | 1.3513 | N(2)C(9) | 1.3413(18) | N(7)C(20) | 1.3423(19) |
| N(5)C(4) | 1.3321 | N(2)C(11) | 1.3437(18) | N(7)C(22) | 1.346(2) |
| N(3)C(2) | 1.3216 | N(3)C(10) | 1.3293(18) | N(8)C(21) | 1.3283(19) |
| N(3)C(4) | 1.3658 | N(3)C(11) | 1.3556(19) | N(8)C(22) | 1.3554(19) |
| C(2)NH2 | 1.3523 | C(10)N(4) | 1.3241(19) | C(21)N(9) | 1.322(2) |
| C(4)NH2 | 1.3522 | C(11)N(5) | 1.3335(19) | C(22)N(10) | 1.332(2) |
| C(9)C(8) | 1.5034 | C(6)C(7) | 1.5038(19) | C(17)C(18) | 1.501(2) |
| C(7)C(8) | 1.4259 | C(8)C(7) | 1.396(2) | C(19)C(18) | 1.397(2) |
| C(7)C(6) | 1.3925 | C(8)C(9) | 1.4024(19) | C(19)C(20) | 1.401(2) |
| Bond angles,° | Bond angles,° | Bond angles,° | |||
| C(2)N(1)C(6) | 120.27 | C(10)N(1)C(9) | 120.39(12) | C(21)N(6)C(20) | 120.80(13) |
| C(6)N(5)C(4) | 116.29 | C(9)N(2)C(11) | 116.44(12) | C(20)N(7)C(22) | 116.54(13) |
| C(2)N(3)C(4) | 114.08 | C(10)N(3)C(11) | 115.20(12) | C(21)N(8)C(22) | 115.48(13) |
| C(11)C(9)C(8) | 117.86 | C(1)C(6)C(7) | 118.86(13) | C(12)C(17)C(18) | 117.97(15) |
| C(10)C(9)C(8) | 123.52 | C(5)C(6)C(7) | 122.95(13) | C(16)C(17)C(18) | 123.58(15) |
| OC(8)C(7) | 123.34 | O(1)C(7)C(8) | 123.06(13) | O(2)C(18)C(17) | 123.72(14) |
| OC(8)C(9) | 117.18 | O(1)C(7)C(6) | 117.73(12) | O(2)C(18)C(17) | 117.06(13) |
| C(7)C(8)C(9) | 119.48 | C(8)C(7)C(6) | 119.21(13) | C(19)C(18)C(17) | 119.22(14) |
| C(8)C(7)C(6) | 122.74 | C(7)C(8)C(9) | 123.82(13) | C(18)C(19)C(20) | 122.88(14) |
| N(5)C(6)N(1) | 118.76 | N(2)C(9)N(1) | 119.74(12) | N(7)C(20)N(6) | 119.46(13) |
| N(5)C(6)C(7) | 123.31 | N(2)C(9)C(8) | 120.88(13) | N(7)C(20)C(19) | 121.48(14) |
| N(1)C(6)C(7) | 117.92 | N(1)C(9)C(8) | 119.38(13) | N(6)C(20)C(19) | 119.05(13) |
| N(15)C(2)N(3) | 119.17 | N(4)C(10)N(3) | 120.79(13) | N(9)C(21)N(8) | 121.13(14) |
| N(15)C(2)N(1) | 118.07 | N(4)C(10)N(1) | 117.73(13) | N(9)C(21)N(6) | 117.55(13) |
| N(3)C(2)N(1) | 122.76 | N(3)C(10)N(1) | 121.46(13) | N(8)C(21)N(6) | 121.32(14) |
| N(16)C(4)N(5) | 117.18 | N(5)C(11)N(2) | 116.67(14) | N(10)C(22)N(7) | 117.26(14) |
| N(16)C(4)N(3) | 114.98 | N(5)C(11)N(3) | 116.95(13) | N(10)C(22)N(8) | 116.50(14) |
| N(5)C(4)N(3) | 127.83 | N(2)C(11)N(3) | 126.37(13) | N(7)C(22)N(8) | 126.23(13) |
Intramolecular hydrogen bonds for 2, Å and°.
| DH..A | d(DH) | d(H..A) | d(D..A) | <(DHA) |
| N(1)H(1N)...O(1)a | 0.90(2) | 1.89(2) | 2.6147(16) | 136.3(17) |
| N(6)H(6N)...O(2)a | 0.91(2) | 1.84(2) | 2.5934(16) | 139.6(18) |
3 Conclusion
A new product, namely 6-benzoylmethyl-1,3,5-triazin-2,4-diamine (2) was isolated from the reaction of biguanide with ethyl benzoylacetate together with the anticipated formation of N-(6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl)guanidine (1). The structure of 2 was studied in detail using NMR spectroscopy and X-ray crystallography. The effects of solvents and temperature on keto-enol-enaminone tautomeric equilibrium in 2 were explored. In DMSO, acetone and methanol, keto tautomeric form A was minor and its content was found to increase upon addition of D2O and on heating. The intramolecular RAHB played an important role in stabilization of enol B and enaminone C. The DFT calculations at the B3LYP/6-311G(2d,2p)//B3LYP/6-31G(d,p) level were also performed to get a clearer understanding of the phenomenon. In the crystal, the enaminone form C was identified exclusively.
4 Experimental
1H NMR spectra in solution were recorded on a Bruker 300 spectrometer. The samples were dissolved either in DMSO-d6, DMSO-d6:D2O 4:1, CD3OD and acetone d6. Kinetic parameters were calculated from the 1H NMR data obtained at 27 °C, 32 °C and 37 °C. Crystal data for 2 were collected on a Bruker APEX diffractometer attached to a CCD detector and graphite-monochromated MoKα radiation (λ, 0.71073 Å) using a sealed tube at 223(2) K. Absorption corrections were made with the program SADABS [24] and the crystallographic package SHELXTL [25] was used for all calculations.
4.1 General procedure
To biguanidinium sulfate (4.4 g, 20 mmol), sodium methoxide (prepared in situ from 1 g of sodium and 100 ml of methanol) was added, followed by slow addition of ethyl benzoylacetate (4.2 ml, 24 mmol). The reaction mixture was heated under reflux for 3.5 h. The precipitated solid 1 was filtered, washed with methanol and water and dried to obtain 1 as white flakes. The solvent was evaporated from the filtrate and solid thus obtained was recrystallized with methanol to obtain 2.
1-(6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl)guanidine/(3,4-Dihydro-4-oxo-6-phenyl-2-pyrimidinyl) guanidine (1) [15c]: Yield: 37%; mp 273 °C (decomposed); LC-MS (APCI) m/z = 229.1 (MH+); 1H NMR (300 MHz, DMSO-d6): δ 6.24 (1H, s, CH), 7.87 (2H, d, J = 7.9 Hz, H-2′ and H-6′), 7.38–7.49 (3H, m, H-3′, H-4′ and H-5′), 8.27 (4H, br. s, NHC(=NH)NH2), 11.53 (1H, s, NH); IR (KBr): ν NH 3335, CH 3052, 2940, 2665, C = O 1666, 1630, 1508, 1492, 1340, 1239, 1239.
2-(4,6-diamino-1,3,5-triazin-2-yl)-1-phenylethanone (2): Yield: 26%; mp 203 °C (EA); LC-MS (APCI) m/z = 229.1 (MH+); 1H NMR (300 MHz, DMSO-d6): δ 4.07*1 (2H, s, CH2), 5.67 (1H, s, CH), 6.73* (4H, br. s, two NH2), 7.09 (4H, br. s, two NH2), 7.39–7.47 (3H, m, H-3′, H-4′, and H-5′), 7.55* (2H, t, J = 7.6 Hz H-3′ and H-5′), 7.62* (1H, t, J = 7.5 Hz, H-4′), 7.78–7.88 (2H, m, H-2′ and H-6′), 7.98* (2H, d, J = 7.5 Hz, H-2′ and H-6′), 15.22 (1H, s, OH); 13C NMR (75 MHz, Me2SO-d6): 48.8* (CH2), 89.2 (CH), 125.7 (C-3′ and C-5′), 128.3 (C-2′ and C-6′), 128.6* (C-2′), 130.0 (C-4′), 133.2* (C-4′), 136.4* (C-1′), 137.3 (C-1′), 162.8 (C-4 and C-6), 166.9* (C-4 and C-6), 167.5 (C-2), 172.8* (C-2), 174.2 (COH), 195.5* (CO); IR (KBr): v NH2 3480, CH 3017, C = O 1693, 1580–1602 br., 1534, 1443, 1415, 1223, 1114. Anal. calcd. for C11H11N5O: C (57.63), H (4.84), N (30.55); found: C (57.58), H (4.70), N (30.34).
1H NMR (300 MHz, DMSO-d6:20% D2O): δ 5.68 (1H, s, CH), 7.37–7.50 (3H, m, H-3′, H-4′, H-5′), *7.51–7.69 (3H, m, H-3′, H-4′, H-5′), 7.78-7.90 (2H, m, H-2′ and H-6′), 7.98* (2H, d, J = 7.9 Hz, H-2′ and H-6′); 13C NMR (75 MHz, Me2SO-d6:20% D2O): 89.2 (CH), 125.8 (C-3′ and C-5′), 128.3* (C-6′), 128.5 (C-2′ and C-6′), 128.8* (C-2′), 130.5 (C-4′), 133.7* (C-4′), 136.1* (C-1′), 136.8 (C-1′), 162.5 (C-4 and C-6), 166.4* (C-4 and C-6), 167.2 (C-2), 172.9* (C-2), 174.6 (COH), 195.5* (CO).
1H NMR (300 MHz, CD3OD): δ 4.57* (2H, s, CH2), 5.76 (1H, s, CH), 7.35–7.46 (3H, m, H-3′, H-4′ and H-5′), 7.50* (2H, t, J = 7.7 Hz, H-3′ and H-5′), 7.62* (1H, t, J = 7.5 Hz, H-4′), 7.74–7.87 (2H, m, H-2′ and H-6′), 8.02* (2H, d, J = 7.5 Hz, H-2′ and H-6′); 13C NMR (75 MHz, CD3OD): 127.1 (C-3′ and C-5′), 129.4 (C-2′ and C-6′), 129.6, 129.8, 131.5 (C-4′), 134.7, 138.4 (C-1′), 165.0 (C-4 and C-6 br.), 168.3, 170.2 (C-2), 176.6 (COH).
1H NMR (300 MHz, (CD3)2CO): δ 4.06 (1H, s, CH2), 5.81 (1H, s, CH), 6.13* (4H, br. s, NH2), 6.41 (4H, br. s, NH2), 7.42–7.45 (3H, m, H-3′, H-4′ and H-5′), 7.46–7.68* (3H, m, H-3′, H-4′ and H-5′), 7.82–7.87 (2H, m, H-2′ and H-6′), 8.02 (2H, d, J = 7.1 Hz, H-2′ and H-6′), 15.12 (1H, s, OH); 13C NMR (75 MHz, (CD3)2CO): 94.1 (CH), 126.6 (C-3′ and C-5′), 129.2 (C-2′ and C-6′), 130.9 (C-4′), 137.0 (C-1′), 170.2 (C-2), 172.7 (COH).
Crystal Data for 2-(4,6-diamino-1,3,5-triazin-2-yl)-1-phenylethanone (2): C11.50 H15 N5 O2.50, Mr = 263.29, triclinic space group P-1, a = 7.1343(4) Å, b = 11.6996(6) Å, c = 15.4502(8) Å, α = 81.8910(10)°, β = 77.3160(10)°, γ = 87.0080(10)°, V = 1245.25(11) Å3, Z = 4, ρcalcd = 1.404 mg/m3, μ = 0.103 mm−1, T = 223(2)K, R1 = 0.0517, wR2 = 0.1357, GOF = 1.038 for 16303 reflections with I > 2σ(I).
4.1.1 Computational details
It is generally recognized that the H-bond geometry of RAHB molecules cannot be reproduced at the Hartree-Fock level [26] and that electron correlation can be satisfactorily accounted for both by ab initio MP2 (or higher) methods [27] and by DFT calculations [28] with hybrid functional, in particular B3LYP [29,30]. Single point calculations at different levels of theory namely, B3LYP/6-311G(2d,2p), MP2/6-311G(2d,2p) were performed using the B3LYP/6-31G(d,p) optimized geometries and the relative energies (kcal/mol) of tautomers were calculated. The chemical shifts of 13C NMR were also calculated at the B3LYP/6-311G(2d,2p) level using GIAO perturbation method [20]. For valid comparison, the chemical shift of the reference compound (TMS) was carried out at the same level of theory as that for the tautomers. All the calculations have been performed using Gaussian computational package [31].
4.2 Supporting Information
Crystallographic data for the structure reported in this article have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 771460) as supplementary publication. Copies of the data can be obtained free of charge on application to CCDC 12 Union Road, Cambridge CB21 EZ, UK (Fax: (+44)1223 336-033; email: data_request@ccdc.cam.ac.uk).
Acknowledgement
This work is supported by Academic Research Fund (WBS R-148-000-069-112) from the National University of Singapore. One of the authors (N.S.) thanks the National University of Singapore for a research scholarship. We also acknowledge Geok Kheng Tan and Lip Lin Koh (Chemistry, NUS) for crystallographic support. The authors also wish to acknowledge help of Janardan Krishnamurthy (DBS, NUS) and Krishnan Chandrashekharan (Chemistry, NUS).
1 *: peaks corresponding to minor keto form A.