

1 Introduction
Britholites, which are phosphosilicated apatites, are the subject of numerous studies both at the fundamental and the experimental levels. Since the discovery of the Oklo site [1,2], they have been considered to be particularly promising for use as matrices for the confinement of nuclear waste [3–9]. Indeed, this discovery showed that these materials can retain fission products over several million years without suffering damage [10]. Furthermore, several studies have shown that these compounds present a good resistance to aqueous alteration [11–14] and irradiation [15,16].
Several processes have been developed for the preparation of britholites. Usually, they are obtained via a solid state reaction at high temperature [3–5,17,18]. Therefore, it seems interesting to attempt the preparation of these britholites by a mechanochemical synthesis.
The mechanochemical synthesis was originally developed by Benjamin and co-workers in the early 1970s to obtain metallic compounds whose elaboration with the conventional methods is often ineffective [19]. Since the 1980s, the works of Yermakov et al. [20] and Koch et al. [21] have permitted to extend the use of this method for the preparation of a wide range of materials.
In the mechanochemical synthesis, a mixture of elementary powders is submitted in a mill to intense successive shocks with adjustable frequencies and energies. These shocks by creating punctual, linear or surface defects induce an increase in the free energy of the initial material. Therefore, to minimize the free energy, change and transformation phases occur in the system leading to the desired product [22,23]. Thus, this method, which involves only a solid-state reaction at room temperature, seems quite appealing. Indeed, besides its simplicity, speed and low power consumption, it is a valuable method for industrial production. Moreover, it allows one to obtain powders with a large specific surface area, permitting a lowering in the sintering temperatures [24–26].
In this work, we attempt to prepare fluorbritholites with the chemical formula of Sr10 − xLax(PO4)6 − x(SiO4)xF2 with 0 ≤ x ≤ 6, by a mechanochemical synthesis. In the following sections, the samples will be assigned as Sr10 − xLax where x designs the used La content.
The phase evolution with milling duration was followed by using the X-ray diffraction (XRD) and infrared spectroscopy (IR).
2 Experimental procedure
2.1 Powders preparation
The starting materials were of an analytical grade: strontium carbonate (SrCO3) (> 96% Riedel de Haen), lanthanum oxide (La2O3) (> 99.5% Prolabo), silica (SiO2) (> 99.5% Alfa), strontium fluoride (SrF2) (> 99.5% Prolabo), and strontium diphosphate (Sr2P2O7).
2.1.1 Preparation of Sr2P2O7
The strontium diphosphate was prepared by using the solid state reaction at 900 °C for 10 h, according to the following equation:
| (1) |
2.1.2 Preparation of britholites
Britholites with the chemical formula of Sr10 − xLax(PO4)6 − x(SiO4)xF2 with 0 ≤ x ≤ 6 were prepared according to the following equation:
| (2) |
Before use, the lanthanum oxide was calcined at 1000 °C for 2 h. Indeed, when light rare earth oxides are exposed to air, there is formation of the corresponding hydroxides [27].
The mechanochemical synthesis was performed in a Fritsch Pulverisette 7 planetary micro mill using stainless steel balls with a diameter of 12 mm and a stainless steel cell of 45 cm3. The cell was loaded with several balls and raw materials at appropriate amounts to obtain stoichiometric apatites as given in the Eq. (2). The mass of the powder was 1 g and the balls-to-powder mass ratio was 34:1. The rotating disc speed and cell speed were 500 and 1000 rpm, respectively. The experimental conditions were a shock kinetic energy of 0.151 J/shock, a shock frequency of 100 Hz and an injected power shock of 15.1 Watt/g. The milling was carried out without any additives for different durations, and the powders were heat-treated under an argon flow at 900 and 1100 °C for 12 h with a heating rate of 10 °C/min.
2.2 Powder characterization
XRD patterns of both as-milled and heat-treated powders were carried out on an X’Pert Pro, PANalytical diffractometer using Cu Kα radiation. Scans were run from 20 to 65° (2θ) with a step size of 0.03°, and a counting time of 1 s per step. The experimental patterns were compared to standards compiled by the Joint Committee on Powder Diffraction and Standards (JCPDS) using the X’Pert HighScore Plus software. The lattice parameters of both as-prepared and heat-treated samples were determined by Rietveld refinement of the XRD data using the Fullprof program [28]. The refinements were based on the structural data of the JCPDS card No. 00-050-1744 using the space group P63/m.
Infrared spectra were recorded in the range of 4000–400 cm−1 with an EQUINOX 55 TF-IR spectrometer equipped with OPUS/IR software, and a PerkinElmer Spectrum 100 spectrometer, using the KBr pellets technique.
3 Results
3.1 X-ray diffraction
As can be observed from Fig. 1, after 5 h of milling, the XRD patterns show reflections of a dominant apatite phase indexed in the hexagonal system (space group P63/m) based on the strontium fluorapatite pattern (JCPDS #00-050-1744). The other reflections correspond to the starting materials, identified by their reflections: SiO2, at 20.85° (JCPDS #01-085-0798), La2O3, at 52.49° (JCPDS #01-074-2430) and SrCO3, at 44.38° (JCPDS #01-074-1491). The two latter reagents were only observed for x ≥ 5, while silica was detected from x = 2. Nevertheless, the presence of other reactants and/or intermediate phases could not be excluded. They were not detected probably due to their low amounts or because they were in an amorphous phase. If we assume – in agreement with the XRD patterns – that all the reagents had reacted, it seems possible to obtain in a pure state the monosubstituted britholite and obviously the pure fluorapatite by a simple milling for 5 h. However, for the compositions richer in Si, a prolonged milling over 5 h is required.

XRD patterns of all the samples milled for 5 h.
Previous studies have demonstrated that the formation enthalpy of the series of Ca-La-fluorbritholites (Ca10 − xLax(PO4)6 − x(SiO4)xF2, with 0 ≤ x ≤ 6) increased when the incorporated amounts of lanthanum and silicate rose [29]. Furthermore, it was shown that the monosilicate britholite is not affected by the metamictisation due to the alpha particles and recoil nuclei, despite the intense internal and external irradiation doses [15,16]. Thus, a selected composition, Sr8La2(PO4)4(SiO4)2F2 was chosen to conduct a more detailed study. Indeed, if this compound is obtained in a pure state, the preparation of the monosubstituted sample is therefore possible.
Fig. 2 shows the XRD patterns of the Sr8La2 sample milled between 0.5 and 20 h. From these patterns, we notice that for 0.5 h of milling, practically all the peaks belong to the reagents. Nonetheless, one can see the emergence of peaks as shoulders located at 2θ angles of 21.97°, 24.42° and 31.87°, indicating a beginning of the formation of an apatite phase. As the milling process progressed, the intensities of the peaks of the starting materials gradually decreased and those of the apatite peaks increased. All the reactants disappeared for a milling duration higher than 1 h, except La2O3 and SiO2. If La2O3 disappeared for a milling duration higher than 3 h, SiO2 remained present for up to 10 h. After milling for 15 and 20 h, XRD pattern show the peaks corresponding only to britholites, and no other phases such as La2O3 and SiO2 were detected.
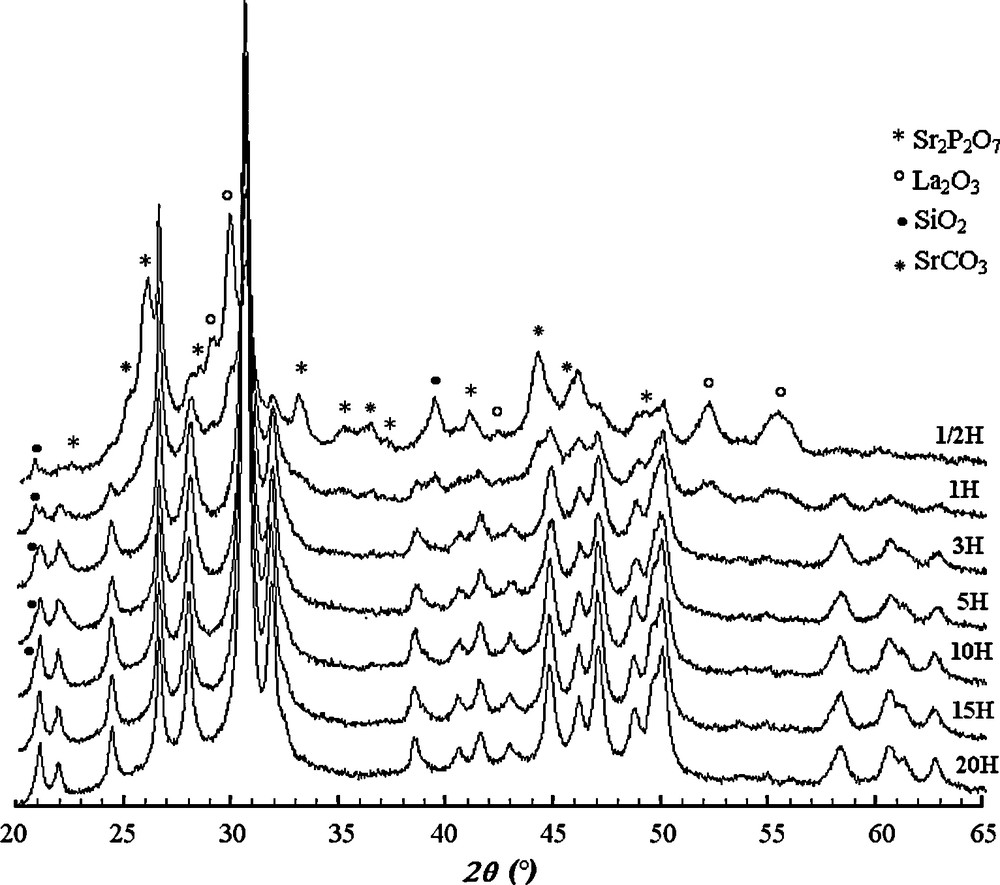
XRD patterns of the Sr8La2 sample milled between 0.5 and 20 h.
Figs. 3 and 4 give the XRD patterns of samples with 0 ≤ x ≤ 6, milled for 20 h. For x ≤ 3, The XRD patterns showed only the peaks belonging to the apatite (Fig. 3), while for x > 3, the XRD patterns contained in addition to the peaks of the britholite those of SiO2, the intensities of these latter increased as x increased (Fig. 4). As not all SiO2 was incorporated into the apatite structure, according to Eq. (2), a certain amount of La203 would also remain in the powder after reaction.
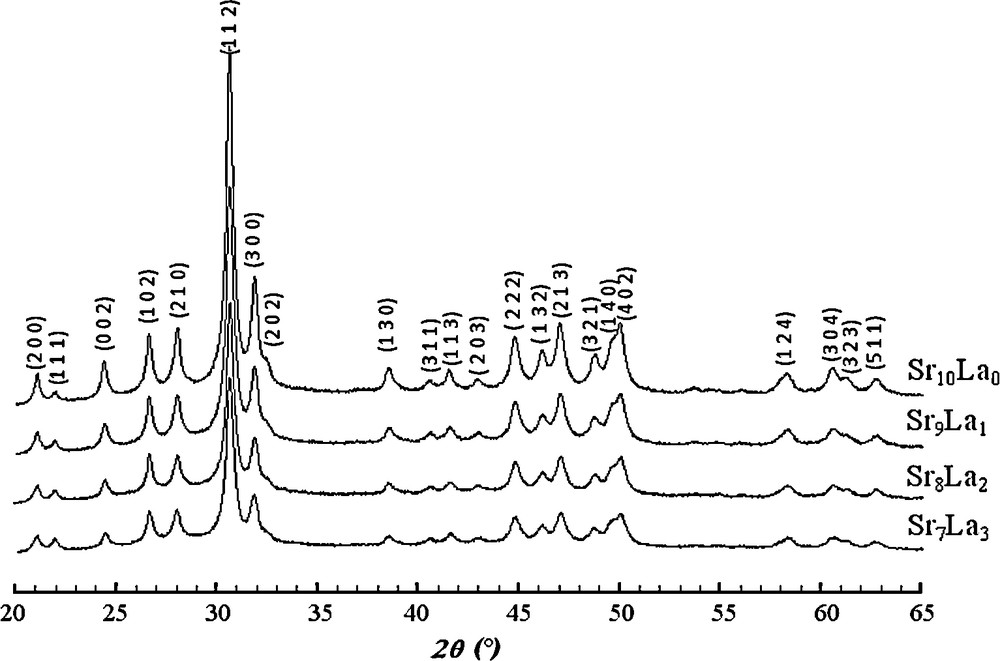
XRD patterns of samples with x ≤ 3 milled for 20 h.
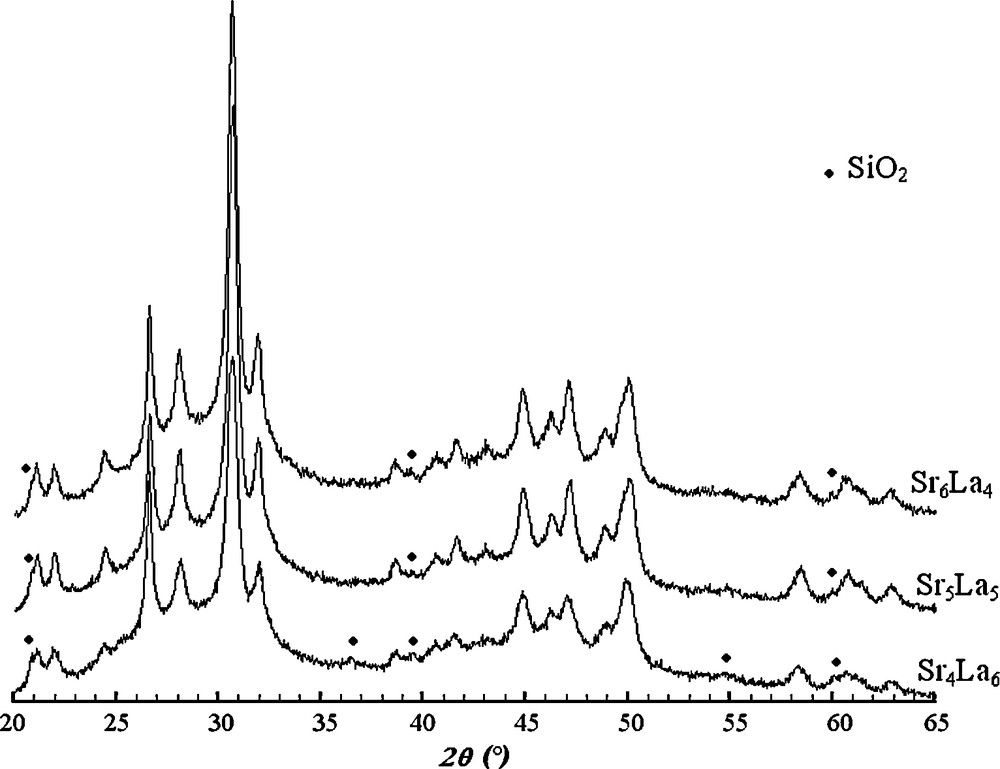
XRD patterns of samples with 4 ≤ x ≤ 6 milled for 20 h.
Fig. 5 shows the XRD patterns of the Sr8La2 sample heat treated at 900 and 1100 °C for 12 h after milling for 20 h. As can be noticed, the patterns presented only the peaks of the apatite and no peaks corresponding to other compounds could be detected.
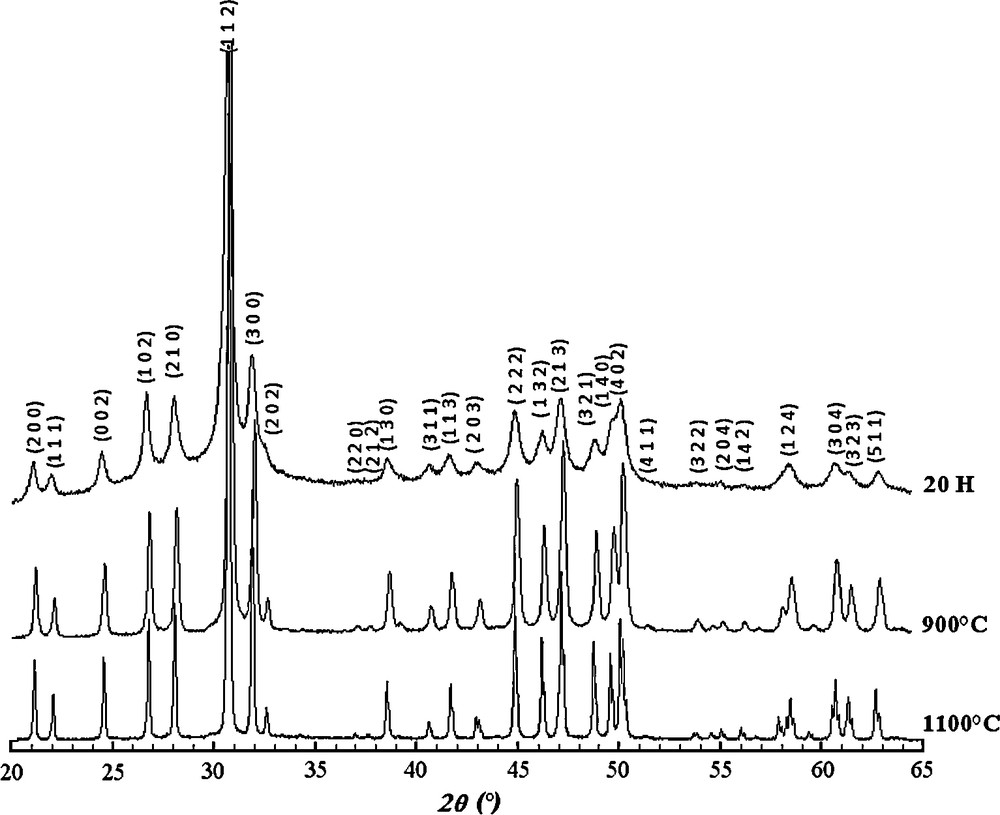
XRD patterns of the Sr8La2 sample heat-treated at 900 and 1100 °C after milling for 20 h.
3.2 Infrared spectroscopy
The IR absorption spectra of the samples obtained after 5 h of milling are shown in Fig. 6. These spectra continuously changed with the increase of the amount of silicate used. The attribution of the bands associated PO4 and SiO4 groups were carried out by comparison with the spectra of apatite phases with similar compositions, reported in the literature [4–7,9,30–32].
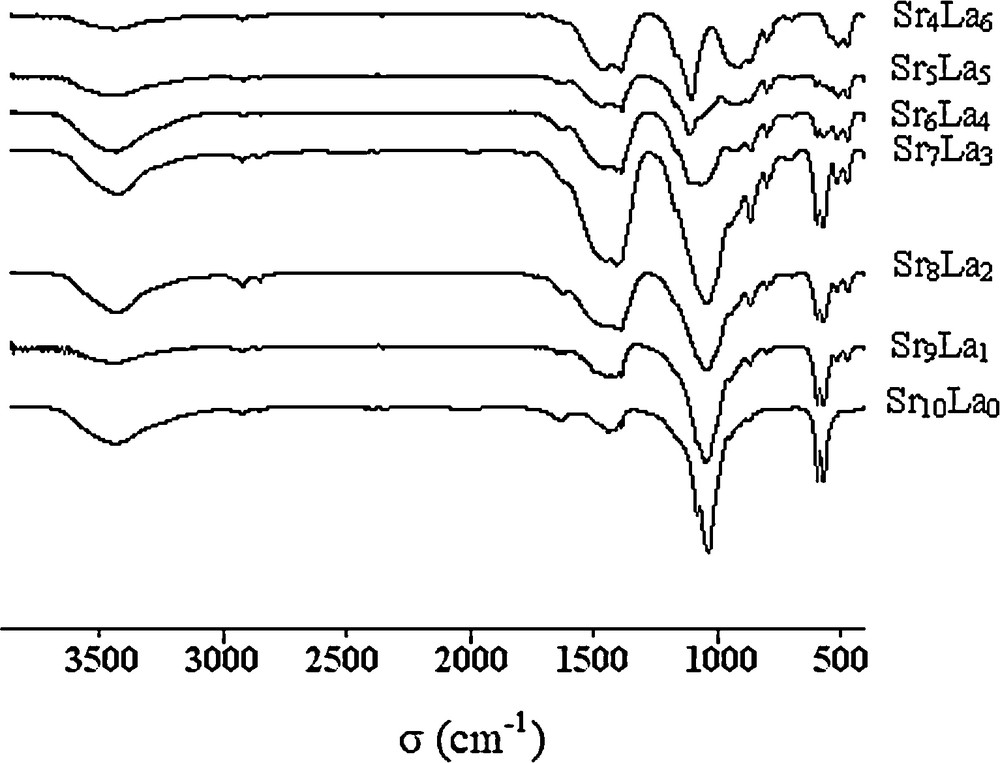
FTIR spectra of all the samples milled at 5 h.
For the nonsubstituted sample, the spectrum exhibited the characteristic bands of PO43− ions at 1080–1034 cm−1 attributed to the asymmetric stretching modes P-O (ν3), 948 cm−1 associated to the symmetric stretching mode P-O-P (ν1), 567 and 592 cm−1 assigned to asymmetric bending modes O-P-O (ν4). Besides, there are some additional bands. The strong doublet appearing at 1438 and 1417 cm−1 and the band at about 866 cm−1 were attributed to the stretching vibrations and deformation vibrations, respectively, of the C-O bonds in the CO32− anions, which were located in the PO43− sites (B-type substitution) [33,34]. The high intensities of these latter bands indicate that a significant proportion of carbonates resulting from the raw materials were incorporated into the apatite phase. The bands appearing at 3441 and 1634 cm−1 correspond to the adsorbed water on the particle's surface.
As far as the substituted samples are concerned, the FTIR spectra showed significant changes compared with the pure apatite. Besides those corresponding to PO43−, and obviously to CO32− groups, additional bands were detected. For x = 1, the bands at 469 and 513 cm−1 could be assigned to the SiO44− groups [35], while the bands observed at 799, 778 and 690 cm−1 were ascribed to SiO2 [36,37]. This latter compound was not detected by the XRD analysis because of its low amount or its presence in the amorphous or nanocrystallized forms. With the increase in the substituent amounts, we notice a decrease in the intensities of PO4 groups’ bands and an increase of those of SiO4. The bands’ frequencies associated with these two latter groups for the different compositions are presented in Table 1.
Assignment of the IR absorption bands for the Sr10 − xLax(PO4)6 − x(SiO4)xF2 samples milled for 5 h.
| Composition | PO43− | SiO44− | ||||
| ν1(cm−1) | ν3(cm−1) | ν4(cm−1) | ν1(cm−1) | ν3(cm−1) | ν4(cm−1) | |
| Sr10(PO4)6F2 | 948 | 1034 | 592 | - | - | - |
| 1080 | 567 | |||||
| Sr9La(PO4)5(SiO4)F2 | 946 | 1044 | 593 | 866 | 915 | 513 |
| 1080 | 567 | 469 | ||||
| Sr8La2(PO4)4(SiO4)2F2 | 943 | 1041 | 593 | 863 | 915 | 512 |
| 1083 | 566 | 467 | ||||
| Sr7La3(PO4)3(SiO4)3F2 | 945 | 1042 | 593 | 862 | 916 | 512 |
| 1081 | 567 | 469 | ||||
| Sr6La4(PO4)2(SiO4)4F2 | 942 | 1058 | 595 | 863 | 917 | 546 |
| 569 | 1103 | 511 | ||||
| 468 | ||||||
| Sr5La5(PO4)(SiO4)5F2 | 939 | 1061 | 594 | 870 | 915 | 544 |
| 569 | 1110 | 511 | ||||
| 469 | ||||||
| Sr4La6(SiO4)6F2 | - | - | - | 869 | 917 | 543 |
| 1103 | 508 | |||||
| 471 |
The IR absorption spectra for the Sr8La2 sample milled between 0.5 and 20 h are given in Fig. 7. These spectra clearly show the progression of the formation of the apatite phase. However, the complexity of the system due to the many used reagents made the assignment of the observed bands difficult. After 3 h of milling (Fig. 7(a)), the spectrum exhibited the vibration bands of an apatite phase and the disappearance of the vibration bands corresponding to the raw materials. In comparison with the spectrum of the sample milled for 0.5 h, we noticed the disappearance of bands positioned at 469, 486, 501, 546, 704, 747, 778, 799, 860, 980, 1013, 1106, 1142, 1385 and 1461 cm−1 which correspond to reactants. The bands appearing at 1047, 947, 592 and 563 cm−1 were assigned to PO4 groups, while the band centered at 512 cm−1 was assigned to O-Si-O asymmetric bending ν2 in the SiO4 groups [35]. This latter band serves as an evidence for the incorporation of SiO4 groups into the apatite structure. The bands appearing at 1469 and 1408 cm−1 were unambiguously attributed to carbonate groups. When the powder was subjected to a longer milling duration, the spectra still remained practically identical. We notice that the characteristic bands of the carbonate rmained present even after 20 h of milling (Fig. 7(b)).
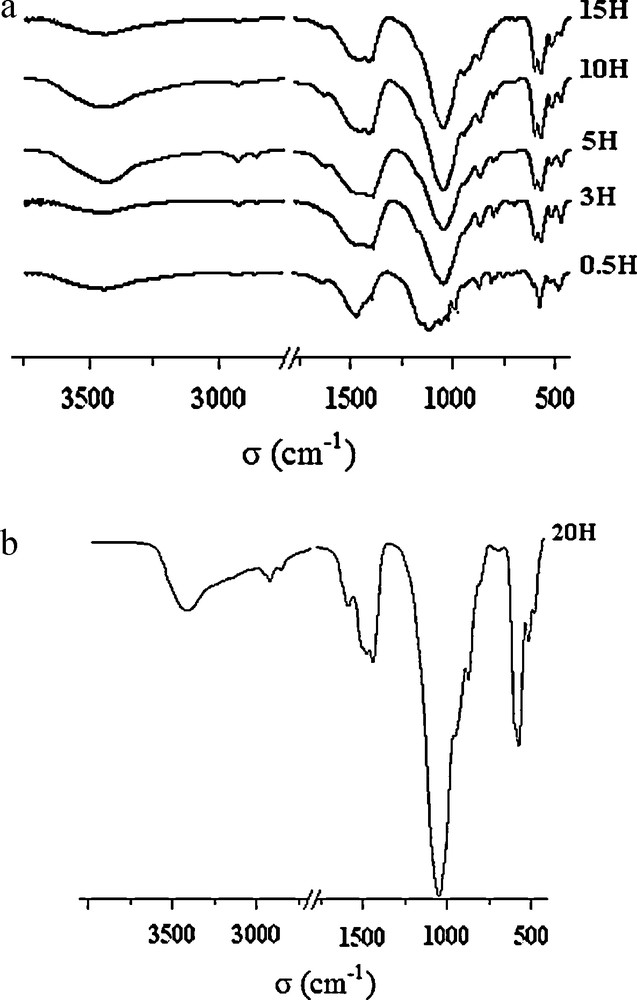
FTIR spectra of the Sr8La2 sample milled between 0.5 and 15 h; (b) for 20 h.
The IR spectra of the Sr8La2 sample heat-treated at 900 and 1100 °C after milling for 20 h are shown in Fig. 8. As observed, the heat treatment caused a significant change. The absorption bands became sharp and intense. Furthermore, at 900 °C, a decrease in intensities of peaks associated to the carbonate groups and SiO2, and concomitantly an increase in the intensities of the bands due to SiO4 groups were observed. At 1100 °C, the doublet at 1407 and 1443 cm−1 was absent, follows the removal of CO2 gas. Also, we noticed the disappearance of the characteristic bands associated to SiO2, observed previously for the milled powder such as the bands at 799, 778 and 690 cm−1. This indicates the formation of a pure apatitic powder. Thus, typical absorption bands of PO43− vibrations ν1, ν3 and ν4 were observed at 946, 1080–1040 and 593–568 cm−1, respectively. However, the fundamental vibrational modes of SiO44− groups in an apatitic environment are witnessed in the region around 918 cm−1 (ν3), 866–840 cm−1 (ν1) and at 544–507 cm−1 (ν4). In addition, the band positioned at about 866 cm−1, which was attributed to the CO32− groups for the milled powder, has dramatically decreased in intensity after heat-treatment and departure of CO2, but it was still present after heat treatement. Therefore, it was also assigned to SiO44− groups [35].
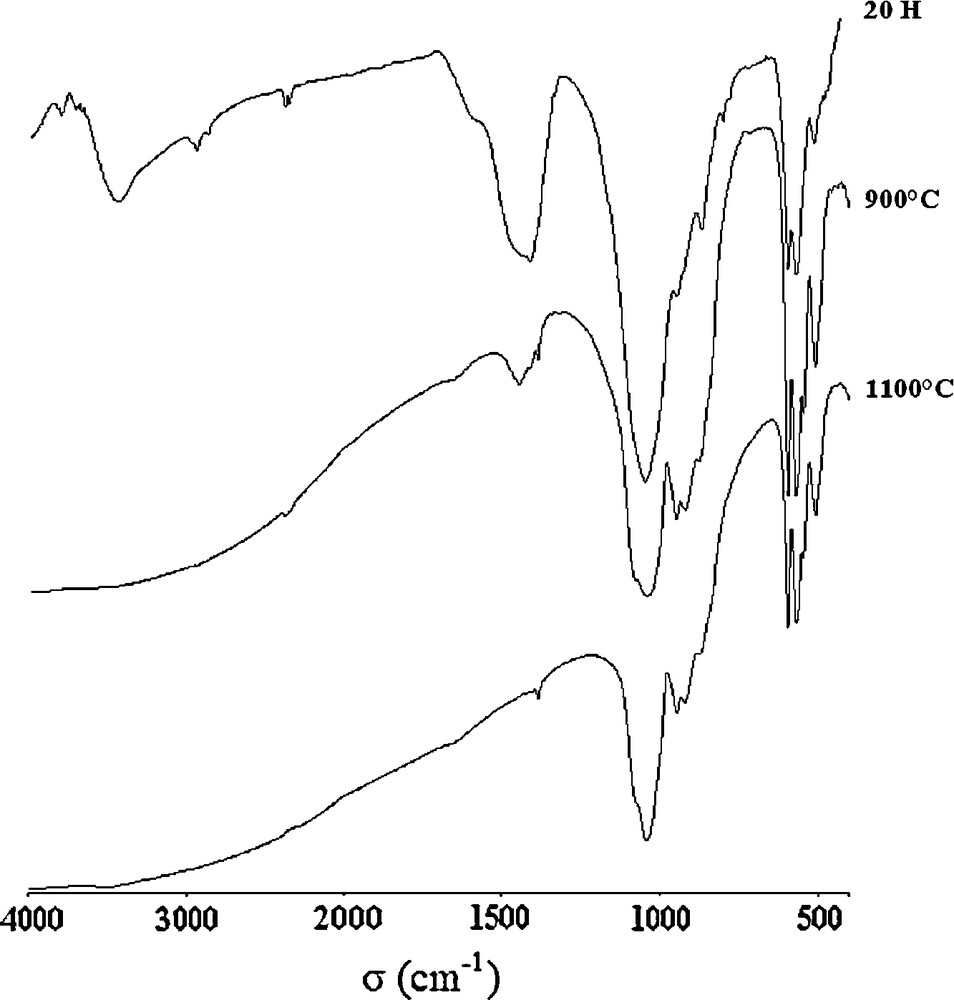
FTIR spectra of the Sr8La2 sample milled at 20 h and calcined at 900 and 1100 °C.
3.3 Lattice parameters
The lattice parameters for both as-prepared and heat-treated Sr8La2 sample determined by Rietveld refinement of the XRD data using the Fulprof program are presented in Table 2. For the as-prepared sample, the parameters are quite different from those of the same composition obtained by the solid state reaction at high temperature, a = 9.727(1) Å and c = 7.277(2) Å [18]. This result can be explained by the substitution of PO43− ions (the average lengths of P-O is 1.51 Å) by both more and less voluminous ions, SiO44− (the average lengths of Si-O is 1.62 Å) [38] and CO32− (the average lengths of C-O is 1.25 Å) [38], respectively, on the one hand, and the creation of vacancies in the Sr-sites following the substitution of PO43− ions by CO32− ones, on the other hand. It is worth noting that the charge balance following the substitution of PO43− ions by SiO44− ones is performed by the substitution of La3+ ions for Sr2+ ones. The sum of these effects results in a decrease of the lattice parameters, with respect to the sample obtained at a high temperature.
lattice parameters of the Sr8La2 sample heat-treated at 900 and 1100 °C after milling for 20 h.
| Sr8La2 | |||
| a(Å) | c(Å) | V(Å3) | |
| As-milled | 9,708(3) | 7,265(2) | 593,04(4) |
| Heat-treated at 900 °C | 9,713(2) | 7,267(4) | 593,77(3) |
| Heat-treated at 1100 °C | 9,740(4) | 7,272(1) | 597,47(2) |
When the powder was heat-treated at 1100 °C, the obtained results have indicated that the powder was formed by a pure apatite phase, and no secondary phases were detected. The Rietveld analysis has shown an increase in the lattice parameters, with respect to the as-prepared sample (Table 2). These values agree with those obtained in ref. [18]. These changes in the lattice parameters resulted from the elimination of the carbonates, as it will be shown below, and the incorporation of SiO2 and La2O3, which were not inserted into the apatite structure during the milling.
4 Discussion
4.1 As-prepared powder
For the synthesis, the amount of reactants was calculated by assuming that the silicate ions substituted the phosphate ions, according to the Eq. (2). During milling, the decrease of the diffraction peaks of SiO2 and La2O3 served as evidence that SiO44− and La3+ entered into the apatite lattice, especially since any other compound containing the latter species was detected even after heat treatment, as it will be show below. However, the IR spectroscopic analysis showed that all the used SiO2 amount was not completely consumed by the synthesis reaction during the milling. Furthermore, this technique revealed that some carbonate ions were inserted into the apatite's lattice in the B-site. Thus, both anionic ions, SiO44− and CO32−, were substituted for PO43− into the apatite's framework. This substitution can be written as follows [39]:
| (3) |
One has to bear in mind that the B-sites must be fully occupied, i.e., the total number of the anionic groups must be equal to six and at most u is equal to 3, according to Eq. (2). In these conditions, an amount of SiO2, equivalent to that of the incorporated carbonate into the apatite framework, must remain in the milled powder in its original form or in a secondary phase. According to the XRD analysis, SiO2 was remained in the powder after milling.
Depending on the incorporated carbonate amount, two cases can be observed:
(i) The incorporated amounts of SiO44− and CO32− are equal (y = u and x = 2y). So, there is a mutual charge balance. Using Kröger-Vink notation, this can be written as follows:
| (4) |
Nonetheless, the substitution of La3+ for Sr2+ implies the creation of cationic vacancies to compensate for the electric neutrality. If we assume that all the introduced strontium has participated in the reaction, the cationic vacancies were created according to the following mechanism:
| (5) |
Considering the electric neutrality requirement, the global substitution mechanism is given by:
| (6) |
The corresponding generic formula is:
| (7) |
According to the above mechanism, y mol of SiO2 and y/3 mol of La2O3 remained in the mixture in their original form or in a secondary phase. As mentioned previously, the characterization results showed that no secondary phase was formed, but a certain amount of SiO2 remained in the powder after milling. In these conditions, La2O3 must also be found in the powder. It was not detected by XRD probably because of its very low amount.
(ii) The incorporated amounts of SiO44− and CO32− were different. Two cases can be distinguished:
−y > u, i.e., an excess of silicate (w = y − u and x = 2u + w), with respect to the carbonate, was incorporated into the apatite. The resulting negative charge excess (w) is compensated by an equivalent number of La3+ ions, according to the following equation:
| (8) |
Taking into account the Eqs. (6), (7) and (8), the global substitution mechanism is given by:
| (9) |
The corresponding generic formula may be written as follows:
| (10) |
In this case, the quantities of SiO2 and La2O3 not incorporated into the apatite correspond to u mol and u/3 mol, respectively.
y < u, i.e., the number of the incorporated carbonate ions (u) was higher than that of the silicate ions (y). The excess of the carbonate ions is given by: z = u − y and x = 2y + z.
In these conditions, following the substitution of SiO44− and CO32− for PO43−, the charge compensation occurred by the creation of vacancies in the Sr-sublattice, according to the following equation:
| (11) |
Disregarding this latter equation, and considering the electrical neutrality, the substitution of La3+ for Sr2+ can be formulated as:
| (12) |
Finally, the global mechanism can be described as follows:
| (13) |
| (14) |
The quantities of SiO2 and La2O3 not incorporated into the apatite correspond to (y + z) (i.e. u) and (4y + 5z)/12 (i.e. (5u − y)/12) mol, respectively.
4.2 Powders’ calcination
The calcination did not appear to significantly affect the XRD patterns except for a narrowing of the peaks, indicating a higher crystallinity of the powder. Furthermore, no secondary phases could be detected other than the apatite. However, the IR spectra significantly changed after calcination at 1100 °C. No bands corresponding to the carbonate groups were detected and there was an increase in the intensities of the bands associated with the SiO44− groups. As no secondary phases were formed after calcination, the pure apatite could result from the reaction between the carbonated apatite and the SiO2 and La2O3 amounts remaining in the powders, following three equations, depending on the incorporated silicate and carbonate amounts:
In the case where the incorporated SiO2 and La2O3 had an identical molar number, the reaction leading to the pure britholite might be written as follows:
| (15) |
| (16) |
However, when the molar numbers of SiO44− and CO32− are different (y ≠ u), the two following cases must be considered:
For y > u, the following reaction would occur:
| (17) |
| (18) |
For y < u, the reaction leading to the pure britholite is:
| (19) |
The corresponding mechanism can be formulated as:
| (20) |
The IR spectroscopy, has revealed the presence of strong absorption bands associated to the carbonates. Nonetheless, for the powders milling for 5 h, although the IR spectra have shown that the intensities of the bands attributed to carbonates decreased when the milling progressed, it seems that the incorporated amount of this species was higher than that of silicates. In these conditions, the mechanism leading to the formation of carbonated britholites and pure britholites during calcination would correspond to Eq. (13) and Eq. (20), respectively.
5 Conclusion
In this study, we attempted to prepare strontium fluorbritholites with the chemical formula Sr10 − xLax(PO4)6 − x(SiO4)xF2 (where 0 ≤ x ≤ 6) from SrF2, SrCO3, Sr2P2O7, La2O3 and SiO2 by the mechanochemical synthesis. The X-ray diffraction and FTIR spectroscopy identified the resultant powders as carbonated britholites. An amount of silica equivalent to that of the incorporated carbonates and some amount of lanthanum oxide remained in the powders after milling. The obtaining of pure britholites needed a heat-treatment at 1100 °C. The mechanism leading to the formation of the pure britholites was also discussed. The obtained results suggest that the use of raw materials containing no carbonate should lead, by a suitable grinding at room temperature, to pure britholites.
Acknowledgements
The authors would like to thank F.H. Ayedi for his useful discussions, and A. Hadroug for his help with English.