

1 Introduction
The Heck reaction, based on the palladium catalysed olefination of aryl halides, is an important C-C coupling in the synthesis of pharmaceuticals and conjugated molecular systems for materials science applications [1]. According to this and in view of industrial applications, many efforts have been devoted to the development of new Pd-based catalysts, either homogeneous or heterogeneous, which could be operated under mild conditions and a simple operating process. The C-C coupling reactions based on the use of homogenous catalysts is well understood and widely investigated [2]. However, phosphine and phosphane ligands are used to stabilize and activate in situ the catalytically active Pd(0) and to prevent formation of particles aggregates and catalytically inactive “palladium black” [3].
Recently, some reports have focused on the development of new “ligand-free” catalysts using stabilized palladium nanoparticles such as metallic palladium and palladium nanoclusters immobilized on solid supports. Few examples of stable palladium nanoparticles which are active in Heck cross-coupling reactions have been reported [4]. Ying and co-workers have shown that palladium-grafted molecular sieves prepared by vapour grafting method exhibit a high activity for the studied reaction [5]. Highly active palladium species were also generated in situ by dissolution of the Pd active phase from the MOx support (TiO2, Al2O3, NaY) during the course of the reaction [6]. These last leached palladium species, however, were considered as homogeneous-like catalysts which re-precipitates onto the surface of the support during the reaction. According to the other studies in the area, the leached palladium species are considered as the real active phase in the Heck reaction as well [7]. In order to obtain a stable heterogeneous catalyst the choice of a support which interacts strongly with the active phase is necessary.
Due to their specific chemical and physical properties, carbon nanotubes and nanofibers (CNTs, CNFs) are amongst the most explored new generation nanomaterials [8]. Their large surface areas (> 100 m2.g−1), well-defined structures and the absence of micropores which could induce diffusion phenomena are interesting properties to be studied in catalysis. CNTs and CNFs have been studied as catalyst supports for noble metals in several and mostly in the hydrogenation and oxidation reactions [9].
These graphitic carbon nanomaterials have shown improved catalytic efficiency compared to activated charcoal or alumina-supported catalytic systems [10]. Recently, Yuan and co-workers have investigated a new stable carbon nanofiber-supported palladium and pointed that the high stability of this catalyst was related to the presence of the small pores in this mesoporous system behaving as containers for the metal particles [11]. Previous works from us and Corma's group have also reported highly active and stable palladium supported-CNTs catalysts for C-C coupling reactions [12].
Microwave (μW) heating has received an ever-increasing interest during the last decade as it allows a significant improvement of reaction conversion and selectivity along with a drastic reduction of its duration. Up to now, the field in which microwaves were mostly employed deals with organic synthesis where short reaction time and usually high product yield are gained [13]. Microwave-assisted organic synthesis (MAOS), applied to C-C coupling reactions, allowed a significant reduction of the reaction time from several hours to a few minutes [14]. If the reagents or the solvent do not absorb the μW, sensitizers such as graphite [15] or SiC materials are used [16]. Carbon materials, possessing high electrical and thermal conductivity, are rapidly heated under microwave irradiation. It is expected that their combination with the metal active phase in μW conditions should give rise to highly efficient catalytic processes [12b,17].
In the present contribution, we were interested in the use of the CNTs macroscopic pattern, i.e. vertically-aligned carbon nanotubes (VA-CNTs) to support palladium particles and apply them as catalyst in the Heck reactions using the microwave (μW) heating mode. The macroscopic form of the catalyst avoids a cost incentive post-reaction filtration which is a step forward with respect to the separation and recycling problems of the catalyst, especially when the active phase is constituted of toxic or precious metal [18]. The catalytic activity of Pd/VA-CNTs was compared with a commercial Pd supported on activated charcoal (AC).
2 Experimental
2.1 Catalyst preparation
Pd/AC with a Pd loading of 10 wt.% is commercially available (Alfa Aesar GmbH & Co KG) and was used as received.
The aligned carbon nanotubes were obtained by catalytic chemical vapour deposition (CCVD), using ferrocene as an iron-catalyst precursor and toluene as a carbon source [8b-e,19]. Ferrocene concentration in toluene solution (Fe(C5H5)2/C7H8) was fixed at 20 g.L−1. The liquid mixture was injected into the hot zone of the furnace, equipped with a quartz tube reactor (diameter × length, 4 × 80 cm) in flowing argon (1.5 L.min−1) at 850 °C. After the reaction the reactor with VA-CNTs product was cooled down under an argon flow to room temperature. The product was stripped from the inner walls of the quartz tube in pieces of average size of 3 cm2. The CNTs were further treated with nitric acid at 120 °C for 2 h in order to remove the remaining Fe catalyst, washed several times with distilled water and dry for 48 h at room temperature and for 2 h at 100 °C.
The palladium catalyst was prepared by an incipient wetness impregnation method. Twenty-five milligrams of aligned nanotubes (0.5 cm2 of VA-CNTs plate) were impregnated with a EtOH:H2O solution (1:1) containing Pd(NO3)2 and dried for 20 h at RT and for 2 h at 100 °C. The sample was further calcinated in air at 300 °C, in order to transform the Pd salt into Pd oxide and next, reduced in flowing H2 at 400 °C for 2 h to obtain Pd (0).
2.2 Catalyst characterization
The specific surface area of the aligned CNTs was measured by the BET method using N2 as adsorbant at liquid nitrogen temperature (TriStar sorptometer). Before measurement the sample was outgassed at 300 °C overnight in order to desorb some moisture and impurities from its surface.
The XPS measurements of the CNTs support were performed on a MULTILAB 2000 (THERMO) spectrometer equipped with an Al kα anode (hν = 1486,6 eV) with 10 min of acquisition. Peak decomposition was made with the “Avantage” program from Thermoelectron Company.
The morphology of the solid was examined by scanning electron microscopy (SEM) on a JEOL 6700-FEG microscope. The solid was fixed on the sample holder by a graphite paste for examination.
The microstructure of the support and the average particle size of the active phase were examined by transmission electron microscopy (TEM) on a Topcon 002B-UHR microscope working with an accelerated voltage of 200 kV and a point-to-point resolution of 0.17 nm. The sample was sonically dispersed in an ethanol solution during 5 min and a drop of the solution was deposited onto a perforated carbon membrane on a copper grid for observation.
Zeta potential measurements and pH titration were carried out on the Nano-ZS (multipurpose titrator, Malvern Instruments). Samples were sonicated in aqua NaClO4 electrolyte solution for 30 min before measurements. Titrations of the CNTs were performed using 10−1 M NaOH and HCl aqua solutions. The Zeta potential values were determinate from the particle velocities according to the Helmholtz-Smoluchowski equation: ζ = 4πμη/D where μ is the electrophoretic mobility, η is the viscosity, and D the dielectric constant of the liquid in the boundary layer.
2.3 Microwaves irradiation experiment
Microwaves irradiation experiments were performed using a multi-mode Mars System from CEM Corporation using standard Pyrex vessel. The temperature profiles for microwaves experiments were recorded using a fiber-optic probe protected by a sapphire immersion well inserted directly into the reaction mixture. Pressure profiles were recorded with a pressure sensor directly connected to the reaction vessel.
2.4 General procedure for the coupling between aryl iodide and terminal olefins
Terminal olefin (2.007 mmol), triethylamine (2.409 mmol, 333 μL) and Pd(10%)/VA-CNTs (25 mg, 0.5 cm2 plate) or Pd/AC (25 mg) were added to a solution of aryl iodide (0.803 mmol) dissolved in 10 mL of EtOH. The mixture was subsequently heated at 160 °C with stirring during the appropriate time (300 W, 12–13 bars) in a 50 mL reactor vessel. After this, the mixture was cooled down to 60 °C, Pd(10%)/VA-CNTs plate was extracted with tweezers or separated by filtration in the case of Pd/AC and the remaining solution was evaporated to obtain a dark red oil. The residue was purified by flash chromatography on silica gel eluting with a mixture of dichloromethane and petroleum ether to provide the desired product.
2.5 Ethyl (E)-3-(4-nitrophenyl)-2-propenate
The general procedure was followed using 4-iodo-nitrobenzene (0.803 mmol, 200 mg) and ethyl acrylate (2.007 mmol, 218 μL). The reaction was heated at 160 °C during 5 min. Purification by flash chromatography on silica gel (40:60 dichloromethane:petroleum ether) afforded the compound as a white solid (141 mg, 80%): 1H NMR (300 MHz, CDCl3): δ 8.25 (d, 2H, J = 8.7 Hz), 7.71 (d, 1H, J = 16.2 Hz), 7.69 (d, 2H, J = 8.7 Hz), 6.57 (d, 1H, J = 16.2 Hz), 4.30 (q, 2H, J = 7.2 Hz), 1.35 (t, 3H, J = 7.2 Hz). 13C NMR (75 MHz, CDCl3): δ 165.9, 148.3, 141.5, 140.5, 128.5, 124.0, 122.5, 60.9, 14.1.
2.6 (E)-1-nitro-4-styrylbenzene
The general procedure was followed using 4-iodo-nitrobenzene (0.803 mmol, 200 mg) and styrene (2.007 mmol, 230 μL). The reaction was heated at 160 °C during 10 min. Purification by flash chromatography on silica gel (30:70 dichloromethane: petroleum ether) afforded the (E)-1-nitro-4-styrylbenzene as a yellow solid (108 mg, 60%), and (Z)-1-nitro-4-styrylbenzene as a yellow solid (16 mg, 9%).
(E)-1-nitro-4-styrylbenzene: 1H NMR (CDCl3, 300 MHz): δ = 8.23-8.20 (m, 2H), 7.65-7.61 (m, 2H), 7.56-7.54 (m, 2H), 7.43-7.31 (m, 3H), 7.27 (d, J = 16.3 Hz, 1H), 7.14 (d, J = 16.3 Hz); 13C NMR (CDCl3, 75 MHz, ppm): δ = 146.9, 144.0, 136.3, 133.4, 129.0, 127.1, 127.0, 126.4, 124.2.
(E,E’)-1-nitro-4-styrylbenzene: 1H NMR (CDCl3, 300 MHz): δ = 8.21 (d, 2H, J = 7.9 Hz), 7.50 (d, 2H, J = 8.2 Hz), 7.37-7.33 (m, 3H), 7.32-7.26 (m, 2H), 5.62 (s, 1H), 5.55 (s, 1H); 13C NMR (CDCl3, 75 MHz, ppm): δ = 146.9, 144.2, 135.9, 133.2, 129.4, 129.5, 126.9, 127.0, 126.3, 122.1.
3 Results and discussion
3.1 VA-CNTs characteristics
The self-supported vertically aligned nanotubes were obtained in high yield in the form of macroscopic plates (c.a. 3 cm2) with high mechanical strength (Fig. 1A). SEM micrographs demonstrated aligned carbon nanotubes with an average diameter of 80 nm and no carbon nanoparticles, indicating a high selectivity of the synthesis method (Fig. 1B). A few experiments showed that the thickness of VA-CNTs carpet can be controlled by varying a duration of the CCVD process without modification of neither the diameter nor the alignment of the tubes. The SEM micrograph on the Fig. 1C corresponds to the 0.6 mm thick carpet of VA-CNTs, used subsequently in the Heck coupling reaction. The as-synthesized VA-CNTs were treated in an acid medium (HNO3, 1 M, 120 °C) in order to remove the residual iron-based catalyst. As one can see in Fig. 1A, patterned CNTs were mechanically resistant which allowed an easy manipulation with a pair of tweezers. The macroscopic form of nanotubes avoided as well the formation of nanotube dust in the atmosphere during the manipulation which was good for health safe. The pattern of VA-CNTs remained unaltered after the acid treatment according to the optical photos (Fig. 1A). Similar mechanical resistance of VA-CNTs have also been reported by Ajayan and co-workers [8d] during their study of CNTs based filters. This very important mechanical resistance was attributed to high degree of tube entanglement and the van der Waals force between the tubes.

The aligned carbon nanotubes obtained by CVD method using ferrocene/toluene mixture. A. Macroscopic plate of aligned carbon nanotubes with high mechanical strength obtained by stripping them from the inner walls of the reactor. B. High-resolution SEM images showing carbon nanotubes with a homogeneous diameter centered at around 80 nm and no carbon particles. C, D. The CNTs carpet with a height of 0,6 mm before (C) and after (D) μW-irradiation. No damage of the aligned structure was observed.
The morphology of the nanotubes from VA-CNTs carpet was investigated by TEM. The TEM micrograph (Fig. 2A) confirmed the presence of nanotubes with an average diameter of 80 nm. The tubes consisted of relatively thick walls with a narrow channel in the middle of c.a. 10 nm in diameter. High-resolution TEM analysis (Fig. 2B) indicated a high graphitisation degree of tubes walls and the presence of small amount of defects on the CNTs surface, which was in agreement with the low specific surface area c.a. 30 m2.g−1. The high crystallinity of the material with low topological defects along the tube axis was related to the relatively high synthesis temperature. High-resolution TEM micrograph (Fig. 2B) also highlighted the almost absence of amorphous carbon on the tube surface.

A. TEM micrographs of carbon nanotubes obtained by pyrolysis of a mixture of ferrocene and toluene at 850 °C. B. High graphitization degree and small amount of defects of the nanotube wall were observed.
The following acid treatment of the synthesized CNTs introduced oxygenated groups on the nanotubes surface. The presence of the oxygenated groups rendered the CNTs surface more hydrophilic, and thus increased the surface wetability towards the polar solvents - used during the subsequent impregnation step. The oxygenated groups also provided anchorage sites for the active phase precursor which resulted in a better dispersion and stability of the Pd particles in the catalytic system [10a,20]. The percentage of oxygen atoms decorating the CNTs surface, estimated by XPS spectroscopy, steadily increased after acid treatment from 2 to 16 at. %. According to the C1s and O1s XPS spectra, a different type of the oxygen species on the nanotubes surface was observed (Fig. 3A and B).
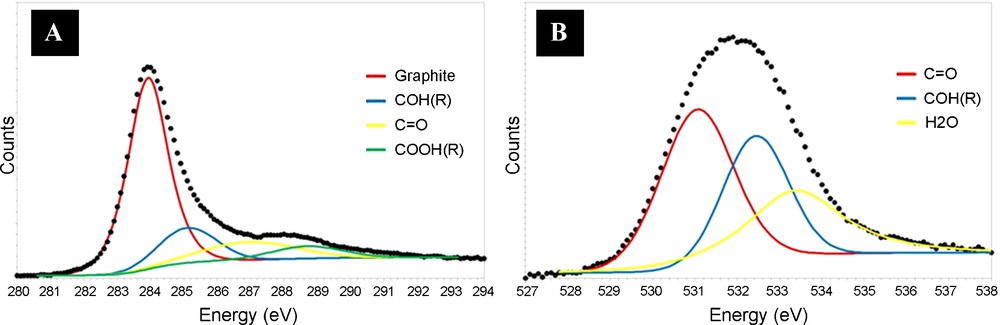
C1s (A) and O1s (B) XPS spectrum of the carbon nanotubes after acid treatment at 120 °C for 2 h showing the presence of several oxygenated functional groups on the solid surface.
The presence of oxygen groups was also reflected by the nanotubes surface charge modification observed by Zeta potential measurements. The results for untreated and acid treated CNTs as a function of pH of the nanotube aqua suspension are presented in Fig. 4. The Zeta potentials of pristine CNTs were negative in the major range of pH and decreased gradually with increasing pH, with an isoelectric point (IEP) at around 3.7 pH unities. The acid treated CNTs displayed negative Zeta potential values over the whole pH range demonstrating the presence of a high density of acidic sites [21]. A negligible change of Zeta potential values along the pH axis means a better solvatation by polar solvents and higher stability of such a suspension. The negatively charged acid treated CNTs surface at the pH of subsequent infiltration process (pH of nitrate of palladium solution), ca. pH = 1.0 favored the anchorage of palladium cations, when one should expect repulsive interactions if the positively charged (untreated by acid) CNTs are used.
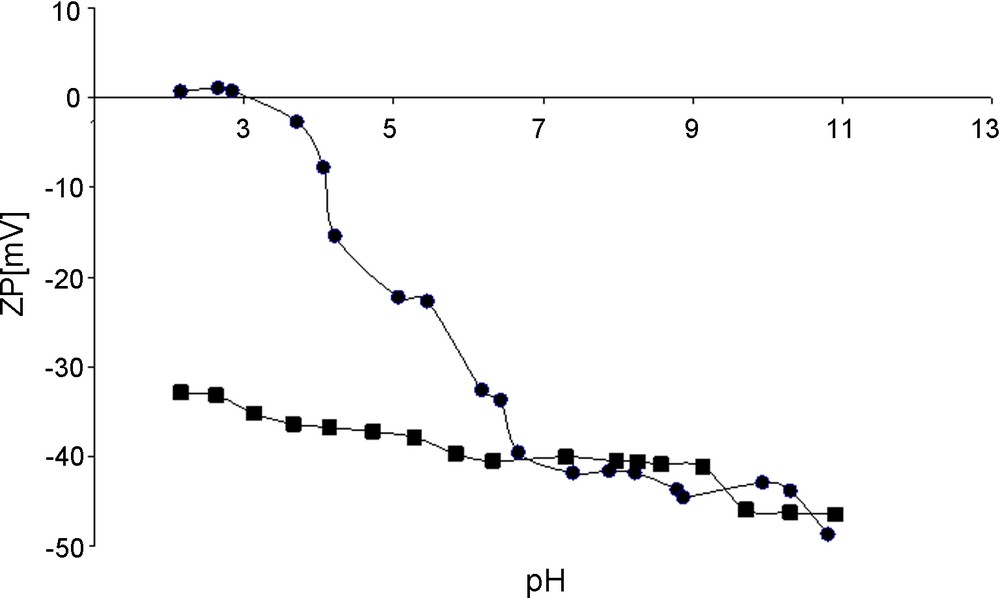
Zeta potentials for pristine CNTs (-≏-) and acid treated CNTs (-■-) as a function of pH of nanotubes aqua suspension.
3.2 Pd/VA-CNTs catalyst characteristics
A deposition of Pd nanoparticles on the CNTs surface was carried out by the incipient wetness impregnation method with an aqueous/ethanol solution containing palladium nitrate, followed by appropriate calcinations and reduction processes (see Experimental section). The TEM micrographs indicated the formation of relatively small palladium particles on the surface of the carbon nanotubes. The average palladium particle size deduced from TEM analysis ranged between 5 and 10 nm and no aggregates were observed on the sample (Figs. 5 and 6A).

TEM micrographs of the CNTs supported Pd after calcinations and reduction. (Middle-top) HR-TEM of palladium particle on the CNT surface. The average size of palladium particles are 5-10 nm.

A. COMPO – SEM micrograph of aligned CNTs supported palladium. The high dispersion of metal particles was attributed to the presence of oxygenated functional groups on tubes surface and homogeneous morphology of the tubes. B. Low magnification TEM micrograph showing the weak dispersion of Pd particles on activated charcoal (AC).
The HR-TEM micrograph in the middle-top on Fig. 5 shows a palladium particle strongly attached to the surface of the CNT. Previous results on palladium supported on commercial carbon nanotubes reported by our group have evidenced the strong interaction between the palladium particles and the tube surface through oxygenated functional groups, leading to relatively high palladium dispersion [22]. The homogeneous graphitic morphology of CNTs is also an important factor which influences the dispersion of metal on the tube surface.
On another hand, representative TEM micrograph (Fig. 6B) of the commercial Pd/AC (Pd/activated charcoal) catalyst reveals a heterogeneous dispersion of palladium particles. Two different populations of Pd particles are observed: the small ones and aggregates of large size, despite much higher specific surface area of AC compared to that of CNTs (Fig. 6A). Similar results were observed in previous works [22] and the heterogeneous dispersion was attributed to the heterogeneous surface character of the support. One can easily find a portion of the support, free of Pd (left part in Fig. 6B) and others with large Pd agglomerates (right part in Fig. 6B).
3.3 Catalytic evaluation
To test Pd/VA-CNTsas catalyst in the Heck reaction, p-iodonitrobenzene as an aryl halide was chosen to react with styrene (Scheme 1) and ethyl acrylate (Scheme 2) as an olefin. For comparison, commercial Pd supported on an activated charcoal (Pd/AC) catalyst was also tested in the same reaction conditions. All experiments were performed under microwave irradiation which provides a rapid heat supply to the catalyst regardless of the surrounding liquid medium. A preliminary test, carried out in water, resulted in the decomposition of ethyl acrylate. Thus water was replaced by ethanol and the mineral base -K2CO3 by triethylamine. The reaction pathways are sketched in Schemes 1 and 2.
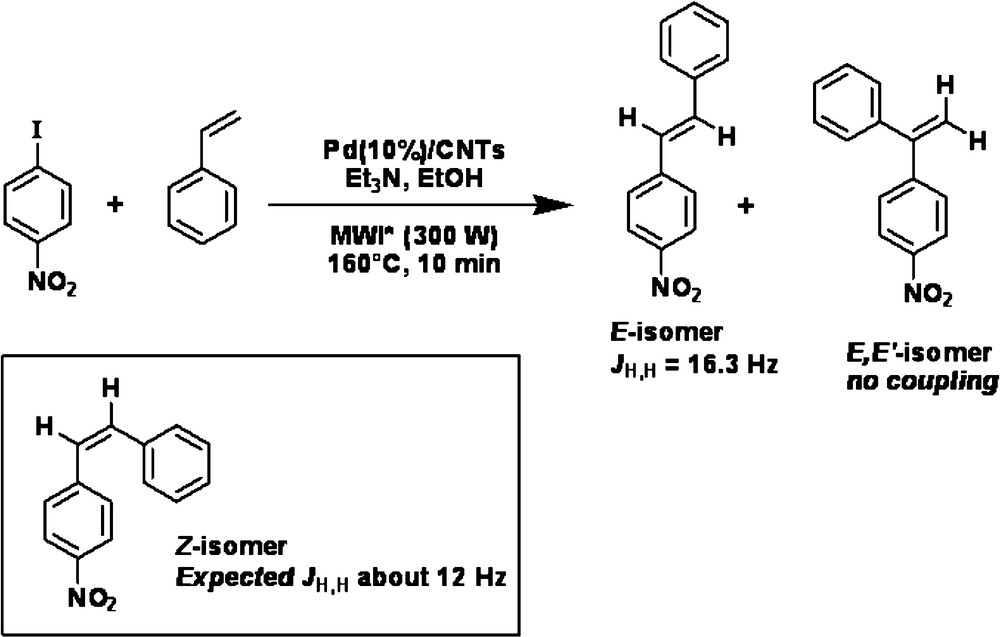
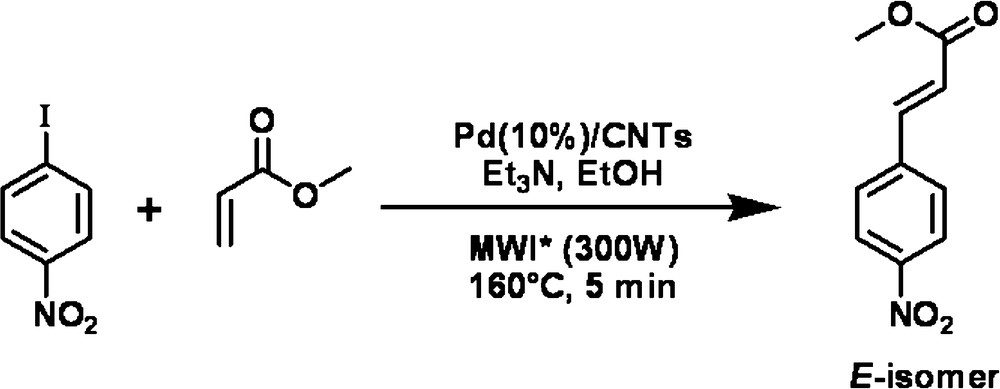
The complete conversion of p-iodonitrobenzene was obtained after 10 min of reaction at 160 °C with styrene and after 5 min at 160 °C with ethyl acrylate when the Pd/VA-CNTs catalyst was used. The difference in reaction time was likely due to a different reactivity of the olefins.
The yields of the Heck coupling products performed with both catalysts are presented in Table 1. The yield of product from reaction with ethyl acrylate approached 80% on Pd/VA-CNTs whereas the same product has been isolated with the yield of 70% for the Pd/AC catalyst. The reaction ran stereoselectively, since only the most thermodynamically stable “E” isomer was isolated.
The yields of the Heck reactions products obtained with Pd/VA-CNTs and Pd/AC catalysts.
| Catalyst | Yield (%) | Yield (%) | Yield (%) |
| (E)-3-(4-nitrophenyl)-2-propenate | (E)-1-nitro-4-styrylbenzene | (E,E’)-nitro-4-styrylbenzene | |
| Pd-10%/VA-CNT | 80 | 60 | 9 |
| Pd-10%/AC | 70 | 27 | 5 |
In the case of the reaction between p-iodonitrobenzene and styrene, two isomers were present in the reaction mixture with a larger amount of the “E” isomer. The first isomer, (E)-1-nitro-4-styrylbenzene and the second product, (E,E’)-nitro-4-styrylbenzene were isolated with the yield of: 60%-9% on Pd/VA-CNTs and 27%–5% on Pd/AC, respectively. The nature of the E,E’ isomer versus the potential Z isomer was ascertained by proton NMR. Two singlets were characteristic of the gem proton whereas two doublet of an AB system was expected for the Z isomer [23]. Furthermore the absence of any H,H coupling constant confirmed the E,E’ nature of the isomer.
Although the Pd/VA-CNTs catalyst exhibited similar selectivity compared to Pd/AC, its activity was much higher under similar reaction conditions: more than twice, when styrene was used as an olefin. It was worth noting that the reaction was very sloppy in the case of Pd/AC and several purification steps were required to isolate pure derivatives. In all cases the thermodynamically less stable ‘Z’ isomer was not formed. The palladium size, dispersion and stabilization against agglomeration were important factors for the catalytic efficiency, since it is known that the C-C couplings are sensitive to the nature of the active phase [24].
The low activity, in the C-C coupling reactions, of Pd/activated charcoal compared to Pd supported on SWNTs in the traditional heating mode was already reported [12b].
The Sheldon tests [25], performed after the catalytic reactions, indicated a small leaching of palladium from the tested catalysts, i.e. 1.0% from VA-CNTs and 3.8% from activated charcoal. After the catalytic experiment, the residue, obtained after separation of the catalyst and evaporation of the solvent was mineralised with concentrated acid. After evaporation of the acid, the resulting solid was re-dissolved in aqueous solution and analysed by ICP-mass spectroscopy. Different values of Pd particles leaching from CNTs and AC resulted from different metal-support interactions. CNT walls present a well-defined graphene structure and the important presence of oxygenated groups provides a higher anchorage site density for the deposited metal active phase than on AC. Activated charcoal is formed from different aromatic hydrocarbon platelets, connected through different heteroatom (O, N, S) bridges, and then weak interactions with Pd ions are partially responsible for the major Pd leaching during the course of the reaction and less homogeneous dispersion of Pd particles on the solid surface. At the same time high leaching and subsequent re-deposition phenomenon during the cooling of the system, called the “boomerang effect”, is often observed for the metal/C systems [26]. We inferred that superior stability of Pd/VA-CNTs catalyst over Pd/AC resulted from the stronger interaction between the active phase and the support, and the mesoporous character of CNTs [11]. It should be taken into account as well that a difference in the porosity of both “carbons” (CNTs versus AC) influences diffusion rate of reactants towards an active phase, which consequently modify the activity and selectivity of the catalyst. Additionally, the good conductivity of the carbon nanotubes favour the transport and vectorization of microwaves leading to a rapid and homogeneous heat transfer through the catalyst body.
After the catalytic tests, the platelet of Pd/VA-CNTs was easily recovered with tweezers from the reactor and re-tested four times without any additional loss of activity. The SEM analysis of re-used Pd/VA-CNTs showed no significant damage of the aligned CNTs structure (Fig. 1D) and no mass change. The Pd nanoparticle crystallinity or size distribution was hardly modified according to the XRD analysis before and after catalytic test (Fig. 7).
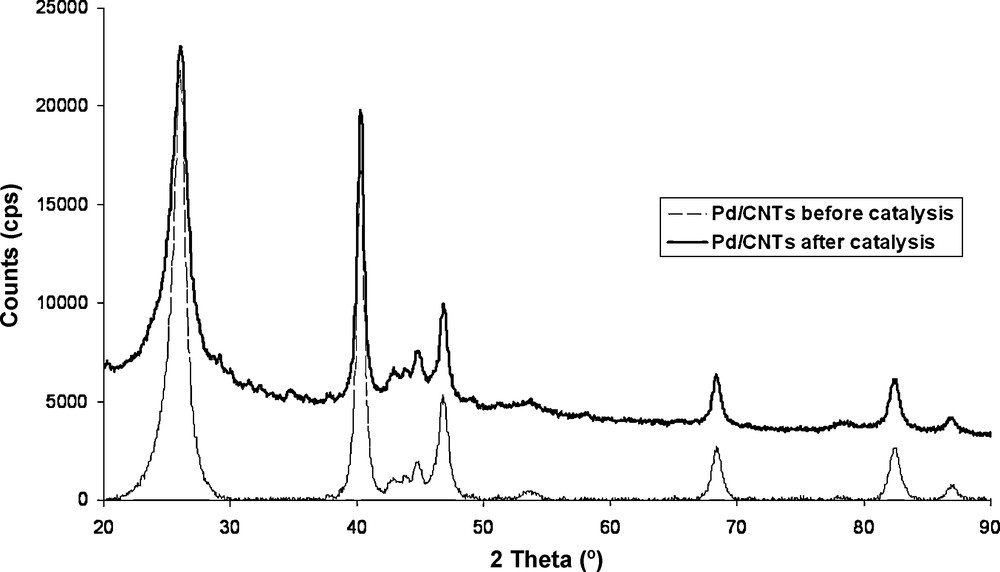
XRD spectra of Pd/VA-CNTs catalyst before and after Heck coupling reaction.
TEM observation of the catalyst after Heck reaction (not reported) shows no evidence of sintering of Pd particles; also no difference in Pd particles dispersion was observed.
4 Conclusion
The macroscopic platelet of vertically aligned carbon nanotubes (VA-CNTs) loaded with Pd (10 wt.%) catalyst, was prepared and tested in the liquid-phase Heck reaction under microwaves irradiation. The catalyst had demonstrated superior activity compared to commercial Pd/AC catalyst, in particular when styrene was used as an olefin source. The use of microwave irradiation resulted in a significant acceleration of the C-C coupling reaction rate, shortened the reaction time from hours to minutes. The macroscopic shape of the catalyst remained unaltered after the catalytic tests cycling. In addition, the palladium leaching remained extremely low on VA-CNTs catalyst compared to what occurred on activated charcoal. The cycling tests indicates the almost absence of deactivation of Pd/VA-CNT catalyst. The macroscopic patterned structure of the catalyst strongly facilitated the separation and the recycling of the catalyst after reaction, without any need for filtration, which represents a major advantage for liquid-phase reactions from an economic and environmental point of view.
Acknowledgements
Dr Thierry Dintzer (LMSPC) is gratefully acknowledged for SEM experiments. Dr Ovidiu Ersen (IPCMS) for the TEM analysis and Dr Dominique Begin for the BET measurements. The SEM and TEM characterizations were carried out at the Institut de physique et chimie des matériaux de Strasbourg (IPCMS, UMR 7504 du CNRS).