

1 Introduction
Solar energy conversion into electricity or fuels and chemicals is an important and active research area, because of the urgent need to shift from our polluting and exhaustible fossil fuels to environmentally friendly energy resources [1]. The conversion of light into electricity (photovoltaic) or its transduction into high energy compounds (artificial photosynthesis) can be achieved with the use of transition metal complexes as light collectors. For instance, the orange ruthenium tris-bipyridine complex (Rubpy3) and its derivatives have been most extensively studied and used as sensitizers for these purposes [2]. The reasons of this success lie in: i) its long-lived MLCT excited state, ii) light absorption in the visible region, iii) intense emission, iv) high photochemical and electrochemical stabilities and v) synthetic versatility. However, the ruthenium element presents significant drawbacks such as high cost, poor resource (the abundance of Ru in the earth's crust is 0.001 ppm) and toxicity. An attractive alternative is to use bis-diimine copper(I) complexes, because they also exhibit MLCT excited-state properties and copper is much cheaper, more abundant and less toxic than ruthenium. Another important distinction between ruthenium(II) and copper(I) complexes lies in their different preferential coordination geometries. Ru(II) complexes are generally six-coordinated in an octahedral geometry, while Cu(I) complexes are typically four-coordinated and tetrahedral (see below). This feature is particularly useful for the design of rod-like molecular arrays. During the past decade, there has been a renewed interest in bis-diimine copper(I) complexes as shown by the deeper understanding of the detailed deactivation pathway of the MLCT excited state of these complexes [3], their uses in light emitting diodes [4] and as sensitizers in dye-sensitized solar cells [5] and in photoinduced electron transfer schemes [6], and for photocatalysis [7]. The use of metal complexes for solar energy conversion is certainly pertinent if they are compared to organic dyes. They exhibit broader absorption bands, longer emission lifetimes and higher stabilities of the radicals, which is particularly appealing as transient radical ions of the dye are necessarily involved in redox reactions occurring in both artificial photosynthesis and photovoltaics. MLCT absorption bands of metal complexes are much broader than the π–π* transitions of organic dyes (π–π* transitions exhibit higher extinction coefficients but are much thinner) and their emissive state, typically triplet MLCT, much longer-lived than the fluorescent π–π* states of organic dyes (rarely longer than a few nanoseconds). These features are determinant to harvest the largest portion of the incident sunlight and to allow the desired charge transfer reaction to occur before the excited-state relaxes back to its ground state. The aim of this contribution is to illustrate the potential of bis-diimine copper(I) complexes with an emphasis on heteroleptic complexes via their recent applications as sensitizers for photoinduced charge separation and for dye-sensitized solar cells. The article is organized as follows: first, a short presentation of the HETPHEN strategy to prepare heteroleptic bis-dimime copper(I) complexes and secondly a summary of the current knowledge of the photophysical properties of bis-diimine copper(I) complexes. The two subsequent sections are devoted to the presentation of the few multicomponent systems designed for photoinduced electron transfer and then to recent and important developments of bis-diimine copper(I) complexes in the field of dye-sensitized solar cells (DSSC). Importantly, breakthroughs in the field of copper(I)-diimine sensitizers have recently been published [6(c),8], showing the amazing potential of these complexes for efficient photochemistry; these articles nevertheless concern homoleptic complexes and stand therefore outside the frame of the present review.
2 The HETPHEN approach for preparation of stable heteroleptic bis-diimine copper(I) complexes
Ruthenium polypyridine complexes are both thermodynamically very stable (i.e. exhibit high association constants usually over 1015 M−1) and kinetically inert (i.e. no ligand scrambling occurs in solution). Conversely, even if [Cu(diimine)2]+ complexes are thermodynamically very stable too, the diimine ligand sphere around the copper(I) usually exchanges rapidly, which generally makes it impossible to isolate pure heteroleptic complexes with two different diimine (bipyridine or phenanthroline) ligands. The wide use of copper complexes for applications in solar energy conversion devices has been certainly hampered by impossible preparation of heteroleptic complexes. However, this limitation has been overcome by Schmittel and coworkers who reported in 1997 the elegant HETPHEN concept (HETeroleptic PHENanthroline) [9]. This strategy relies on the utilization of one very encumbered 2,9-diarylphenanthroline such as 1, which prevents the formation of the symmetrical complex owing to steric reasons. When 1, or a similar very hindered ligand, is mixed with a non-encumbered phenanthroline and one equivalent of copper(I), the exclusive formation of the heteroleptic complex 2, in an almost quantitative yield, is observed driven by the maximum site occupancy principle and the interplay of π–π electronic interactions between the two ligands (Scheme 1).
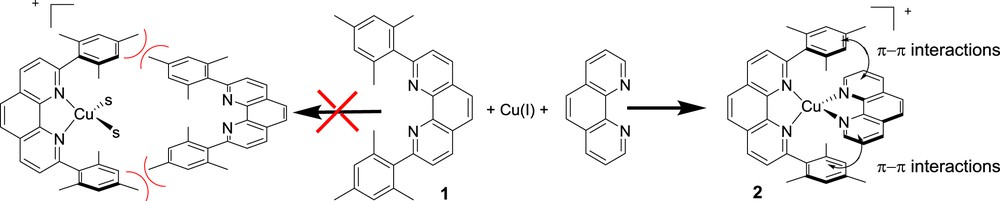
Illustration of the HETPHEN concept to prepare heteroleptic complexes. S = solvent molecule.
This approach has been used to prepare a whole range of metallo-supramolecular architectures [10], such as heteroleptic nanoladders, nanoboxes, nanogrids, and nanoracks; but, surprisingly, it has almost never been used to develop useful [Cu(diimineA)(diimineB)]+ complexes for applications for solar energy conversion. However, this concept could be extremely fruitful for the preparation of copper(I) complexes with very interesting and valuable photophysical and photoredox properties. The two main reasons for this assumption rely on the following observations. First of all, the possibility to assemble different ligands in the coordination sphere of the metal makes it possible to tune more accurately the spectroscopic and redox properties of the complex. In other words, the synthetic accessibility of heteroleptic complexes increases the number of variables that can be introduced around the metal via the two ligands and therefore the extent to which the photophysical and redox potentials can be modified. This is well demonstrated with the abundant use of heteroleptic ruthenium complexes for dye-sensitized solar cells (DSSCs), in which an anchoring ligand (such as dicarboxylic bipyridine) is associated with electron releasing ligands (such as thiocyananto or a bpy substituted with thienyl or oligophenylenethynylene chains) in order to reach the suitable electronic properties to inject electrons in TiO2 and to harvest the maximum number of photons in the solar spectrum [11]. Secondly, the preparation of heteroleptic bis-diimine copper(I) complexes opens the route to the development of rodlike multicomponent assemblies, which was not conveniently accessible with the octahedral ruthenium trisbipyridine complex [12]. This is straightforward from looking at Fig. 1, where the structures of Ru(bpy)3 and Cu(Phen)2 complexes are illustrated. While Ru(bpy)3 gives rise to stereoisomers and contains three bipyridine ligands that point to a D3d or Oh symmetry, Cu(I) complexes with imidazophenanthroline or dipyridophenazine ligands are essentially linear molecules. The replacement of bipyridine by terpyridine (tpy) ligands restores the linear arrangement, but the resulting Ru(tpy)2 complex displays extremely poor photophysical properties compared to Ru(bpy)3 (emission lifetime around 1 ns at 298 K) [12].
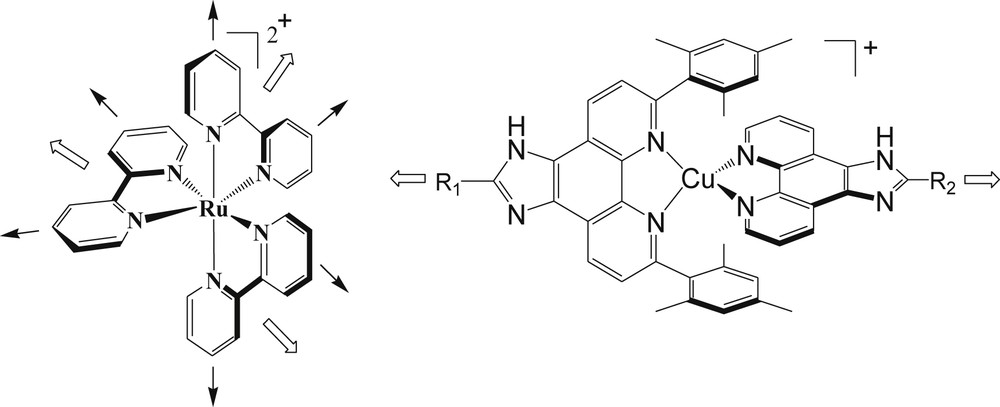
Potential geometrical organizations which are accessible with Ru(bpy)3 or Cu(Phen)2 complexes.
3 General photophysical properties of bis-diimine copper(I) complexes
Since the discovery of the room temperature luminescence of bis-diimine copper(I) complexes ([Cu(dmp)2]+, dmp = 2,9-dimethyl-1,10-phenanthroline) by Sauvage and McMillin [13], there has been a growing interest in the photophysical and structural properties of such compounds. The existence of emissive complexes opens the way to rich potential applications, which were not possible with the very short-lived MLCT excited states such as that of [Cu(phen)2]+ (phen = 1,10-phenanthroline). In the case of sterically challenged diimines, the lifetime of the triplet state for bis-diimine copper(I) complexes is long enough to envision photo-induced charge transfers with electron acceptors and/or donors.
In bis-diimine copper(I) complexes, the metal is characterized by a d10 electronic configuration. This implies the absence of metal centered states (MC or d–d states) which are natural born quenchers of the photogenerated singlet or triplet excited states owing to their close energy proximity in most complexes with the 1st row of transition metals. The completely filled external electronic shell results as well in a symmetrical distribution of the electron density, which favors tetrahedral arrangement of the ligands around the metallic ion.
All bis-diimine copper(I) complexes display a moderately intense absorption band in the visible (ca. 5 × 103 M−1 cm−1), ranging from 450 nm until 700 nm. This transition is a metal to ligand charge transfer (MLCT), similar to that of Ru(bpy)32+, and corresponds to the promotion of an electron from a 3d copper orbital to a low lying π* orbital on the diimine ligand. The shape of this band strongly depends on the geometric arrangement of the ligand coordination sphere. This band is an envelope of three distinct MLCT transitions (often classified as band I, band II and band III) of distinct relative probability [14]. The band I (λ > 500 nm) is proportional to the degree of flattening of the complex at the ground state. It is generally observed with diimine ligands substituted in α to the nitrogen positions with aromatic moieties, which force flattening towards D2 symmetry via π–π interactions with the ligand. The band II (430 > λ > 500 nm) is particularly intense with complexes having a symmetry very close to the pure tetrahedral geometry (ligand 2,9-ditertbutyl phenanthroline for ex. [15]) and it corresponds to a S2 ← S0 excitation. The band III (390 > λ > 420 nm) is usually weak and contains a vibronic structure. Concerning emission, the MLCT excited state lifetime in many bis-diimine copper(I) complexes is relatively short (lower than 100 ns). The accepted explanation for this behavior is the so-called exciplex quenching, proposed by McMillin and colleagues [4,16]. This phenomenon is due to the remarkably different geometry and coordination number of CuI and CuII complexes. In fact, as the lowest energy absorption has an MLCT character, in the excited state the copper(I) ion is formally oxidized to copper(II) and the complex undergoes a severe flattening from D2d to D2 symmetry, that opens the way to the attack by a coordinating molecule, such as the solvent or the counter-anion (Fig. 2). Moreover, according to Jörtner's energy gap law, the nonradiative rate constant increases exponentially with the decrease of the energy gap between the ground and the excited state. Bulky groups in alpha positions of the chelating nitrogen atoms of a diimine ligand efficiently increase the excited-state lifetime thanks to a double steric effect [6c,8]. On the one hand, they prevent the complex from flattening, and thus from reducing its energy content, and on the other hand they screen the copper ion from the attack of solvent molecules leading to exciplex formation.
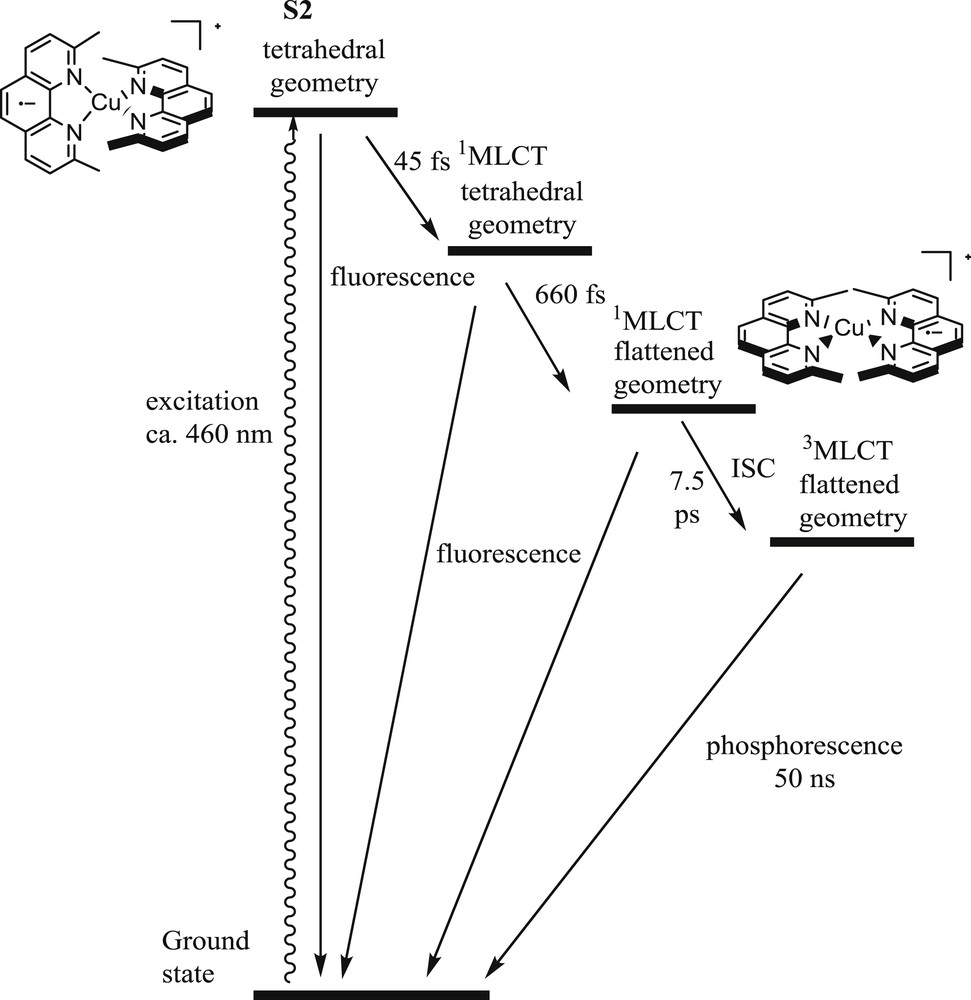
Diagram showing the photo-induced processes occurring upon excitation of the bis-2,9-dimethylphenanthroline copper(I) complex into the MLCT absorption band.
Ultrafast transient absorption spectroscopy and time-resolved fluorescence measurements helped to shed more light on the excited-state dynamic processes and the influence of bulky groups that rigidify the coordination sphere [3a,15,17]. Extensive studies conducted on the simple model [Cu(dmp)2]+ afforded a precise picture of the different processes occurring in the first moments after excitation (Fig. 2). The excitation of the complex in its MLCT absorption band leads to the S2 singlet excited state because the S1 ← S0 transition is generally not allowed. In its initial “tetrahedral” geometry of the Franck–Condon state, the S2 state relaxes to the S1 state with a circa 45 fs rate constant. Then, a flattening distortion (D2h → D2 structural flattening) occurs from this 1MLCT state with a short time constant of around 660 fs. When substituents in 2,9 positions are very bulky (such as tert-butyl), the flattening is inhibited and the 1MLCT decays directly via intersystem crossing (ISC) to 3MLCT [15]. The flattening is generally followed by ISC to the lowest triplet state with a timescale of about 10 ps. Finally, the triplet MLCT decays to the ground state with a phosphorescence of circa 50 ns for the bis-2,9-dimethylphenanthroline copper(I) complex. Interestingly, intersystem crossing occurs on a relatively much slower timescale than for tris-bipyridine RuII complexes (40 fs), due to the lower spin-orbit coupling for CuI complexes and to the structural distortion occurring in the 1MLCT state. Moreover, the rate constant of ICS is also controlled by the degree of flattening of the complex because the spin orbit coupling is governed by the overlapping of the copper orbitals with those of the ligand, which naturally depends on the geometry of the complex. The overall behavior of all bis-diimine copper(I) complexes, both homo-and heteroleptic, roughly follows the same scheme. A fine description of the luminescence properties of these complexes is outside the scope of the present review; we will focus instead on the redox properties of the 3MLCT excited state.
The ground state electrochemical properties of copper(I) complexes are fundamental to understand their behavior in photoinduced processes and they are strongly related to the different geometrical structures of CuI and CuII. The reduction of usual bis-diimine copper(I) complexes occurs at quite low cathodic potentials, usually between −1.5 and −1.7 V vs. SCE in acetonitrile. The extra electron is located in a ligand-centered π* orbital (as confirmed also by quantum mechanical calculations). The oxidation process consists of the removal of an electron from the copper-centered HOMO, which is lying in a higher energy than that of RuII complexes, as CuI is more electron rich into its oxidation state +1. Consequently, the oxidation potential of bis-diimine copper(I) complexes is cathodically shifted relative to those of the corresponding ruthenium complexes. The oxidation of copper(I) into copper(II) is accompanied by a severe flattening of the perpendicular geometry of the ligands, in order to evolve towards the more stable planar-like structure. Thus, it is not surprising that the size of the substituents at the 2 and 9 positions of the phenanthroline ligands controls the degree of destabilization of the copper(II) geometry. Bulky substituents anodically shift to the oxidation potential of the CuII/I redox couple. As a result, the CuII/I oxidation potential can be used as a parameter to evaluate the rigidity of the coordination sphere around copper. Moreover, from an electrochemical point of view, this couple is “quasi-reversible”, which indicates that there is an important structural change between the forward and the backward electron transfers at the electrode. It can be therefore concluded that copper(I) complexes are generally powerful reductants in the excited-state (for example E(CuII/I*) = −1.11 V vs SCE for [Cu(dpp)2]+, where dpp stands for 2,9-diphenyl-1,10-phenanthroline) and even more than the extensively used [Ru(bpy)3]2+ (E(RuIII/II*) = −0.85 V vs SCE). In contrast, copper(I) complexes are known to be quite poor photooxidants, as shown by the low E(S*/S−) potential (E = 0.12 V vs SCE for [Cu(dpp)2]+) [16].
4 Heteroleptic bis-diimine copper(I) complexes for application in photoinduced electron transfer
The transduction of light energy into chemical potentials is the holy grail of artificial photosynthesis [1]. A key step towards this goal is to develop molecular systems that transform solar energy into a long-lived charge separated state with high quantum yield. The key principle to achieve a long-lived charge separated state is to perform a long-range charge separation by fractioning it into discrete short distance charge shift steps [18]. It has led to the concept of triads such as the D–S–A systems, in which D and A are electron-donor and acceptor components, respectively, with S being a photosensitizer. Ideally, D and A should be well-separated in distance in order to avoid the fast charge recombination that arises by spatial proximity. Therefore the linear geometry is the best organization to enlarge the distance between them (Fig. 1). Linear and rigid rod-like molecular arrays have been constructed with several photo-sensitizers (S = porphyrin, and ruthenium or osmium complex) in D–S–A triads and in some cases long-lived charge separated states were obtained [12,19]. If transduction of light energy into chemical potential using homoleptic copper(I)-diimine complexes was monitored very early [20], surprisingly, heteroleptic [copper(diimineA)(diimineB)]+ complexes have scarcely been used for such a type of construction, in spite of the potential interest. The relevance of such an endeavor is the longer lifetime of a natural vectorial charge separated state (i.e. MLCT) of the copper(diimine)2 complex compared to an organic sensitizer (porphyrin), the lower cost of copper with respect to ruthenium or osmium and the easy accessibility to a linear geometry with tetrahedral complexes compared to octahedral ones. For this application of heteroleptic bis-diimine copper(I) complexes, the important property is its excited-state lifetime or its emission quantum yield, but these two properties are usually strongly related. A long-lived excited-state gives time to efficiently perform long-range charge transfer with the adjacent donor or acceptor modules.
The first report of a heteroleptic copper(I) complex based dyad Cu-A dates back to late 1990s; in this work by Meyer and co-workers, a methylviologen acceptor is appended to a bipyridine unit (Fig. 3) [21]. The copper(I) ion coordinated by the latter ligand and the coordination sphere is completed by two triphenylphosphine ligands, which are known to allow isolating heteroleptic copper(I) complexes, thanks to their bulkiness. Formation of a charge separated state (lifetime of a few tens ns) was monitored in acetonitrile. This lead was followed by Armaroli, Nierengarten and co-workers, who replaced triphenylphosphine ligands by POP (bis[2-(diphenylphosphino)phenyl] ether) and dppf (1,1′-bis(diphenylphosphino)ferrocene) and a diimine ligand connected to a C60 as the electron acceptor [22]. Sauvage and coworkers have also developed many beautiful interlocked rotaxanes and catenanes built around bis-phenanthroline copper(I) complexes [23]. The latter complex elegantly mimics, to some extent the role of the accessory bacteriochlorophyll in the natural photosynthetic reaction center, in assisting the photoinduced charge separation between a zinc and a gold porphyrin [24]. However, in these systems, the copper complex was not the photoexcited unit and hence these molecules lie outside the frame of the present review. Although very inspiring to the scientific community, no further discussion on these appealing molecules will therefore be made.
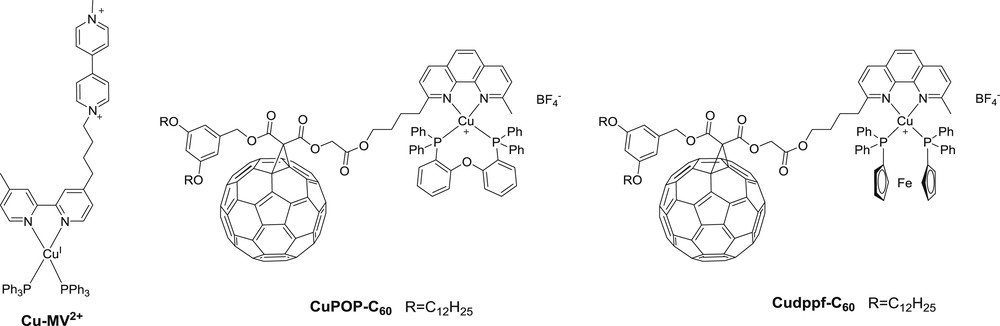
Structures of phosphine-based heteroleptic copper(I) complexe dyads [22].
Similarly, little will be told about molecules 3 and 4 (Fig. 4) because the latter consists of homoleptic bis-diimine copper(I) complexes [25]. In the case of 3, the phenanthroline ligand was modified in α of the chelating nitrogen atoms by one bis-methanofullerene derivative C60 on one side. The resulting homoleptic copper(I) complex underwent charge separation, yielding CuII–C60− when the copper-bis-diimine core was excited, despite a rather weak thermodynamic driving force (ca. −0.13 eV); unfortunately, no lifetime could be retrieved from the data. Importantly, the authors highlighted the great advantage that the MLCT excited state is a charge separated state in nature, favoring the formation of the ultimate charge separated state by lowering the activation barrier of the latter step. The same authors later reported the use of (CuI–C60)2 helicates (eg. 4, Fig. 4) to design photosensitive arrays; bis-phenanthroline ligands were modified with methanolfullerene or bis-methanofullerene. Charge separation upon excitation of the bis-diimine copper(I) core was invoked in each case.
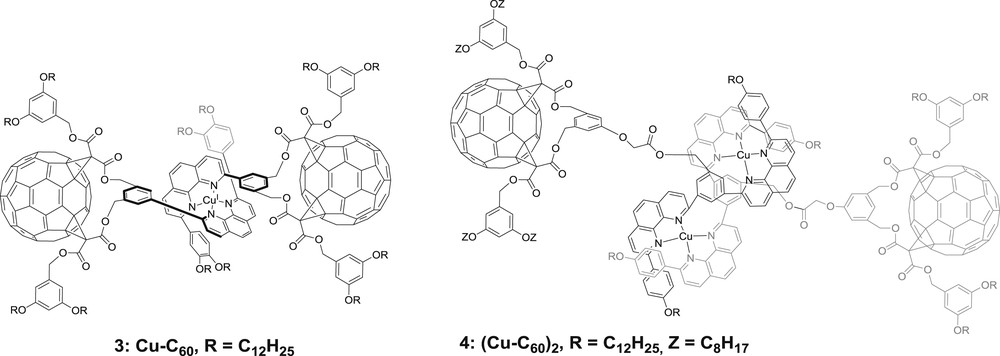
Structures of homoleptic complexes 3 and 4 designed for photo-induced charge separation [25].
The isolation of stable heteroleptic copper(I)-bis-diimine complexes was made possible through the use of rotaxanes and catenanes. In this particular case, the strong affinity of copper(I) ions for the coordinating phenanthroline ligand is employed to isolate those elegant supramolecules. In the case of rotaxane 5 (Fig. 5), a phenanthroline containing macrocycle and a “thread” made of another diimine cavity functionalized by two methanofullerene stoppers are assembled by coordination of a copper(I) ion [26]. The resulting heteroleptic system cannot be unmade thanks to the steric bulk of the C60 stoppers preventing the thread from sliding away through the loop of the macrocycle. Selective excitation in the MLCT of the bis-diimine copper(I) dye was difficult due to the overlapping, weak absorption bands of C60 and Cu(phen)2 units. Nevertheless, photo-induced electron transfer upon excitation in the visible of a dichloromethane solution of 5 was reported, corresponding formally to a CuII–(C60−)C60 species. An estimation of ca. 1.5 eV photonic energy was stored in the system, but a very fast back electron transfer was hypothesized although no spectroscopic proof of the latter fact was given owing to the difficulty to monitor C60− or Cu(II)-bis-diimine spectral features.
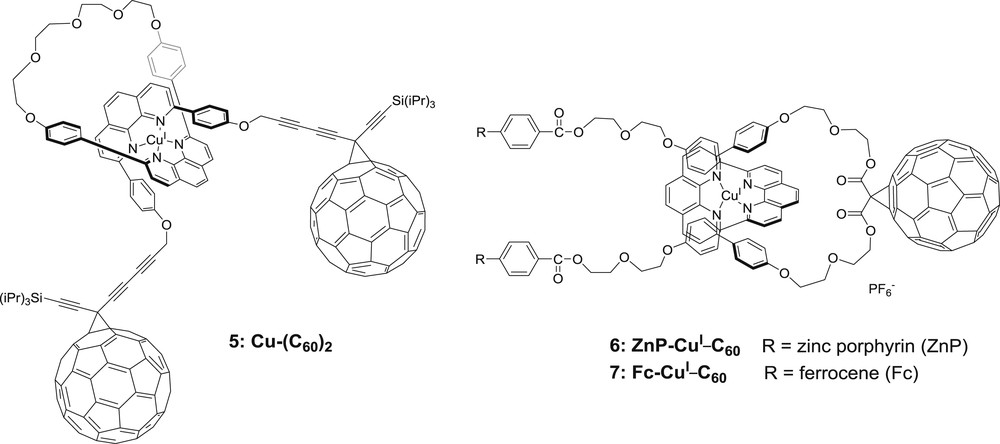
Structures of rotaxanes 5–7 [26,27].
A very interesting series of rotaxanes and catenanes was reported, involving a copper(I)-bis-phenanthroline core connected on one side by a C60 acceptor and on the other side by one or two electron donors [27] (Figs. 5 and 6). In the case of molecule 6, porphyrin units as electron donors yielded new rotaxanes behaving as dyads and triads. The rather high conformational mobility of rotaxanes entailed various kinetic behaviors for the photo-induced processes, depending on the external medium (isotropic, nematic, glassy phases…).
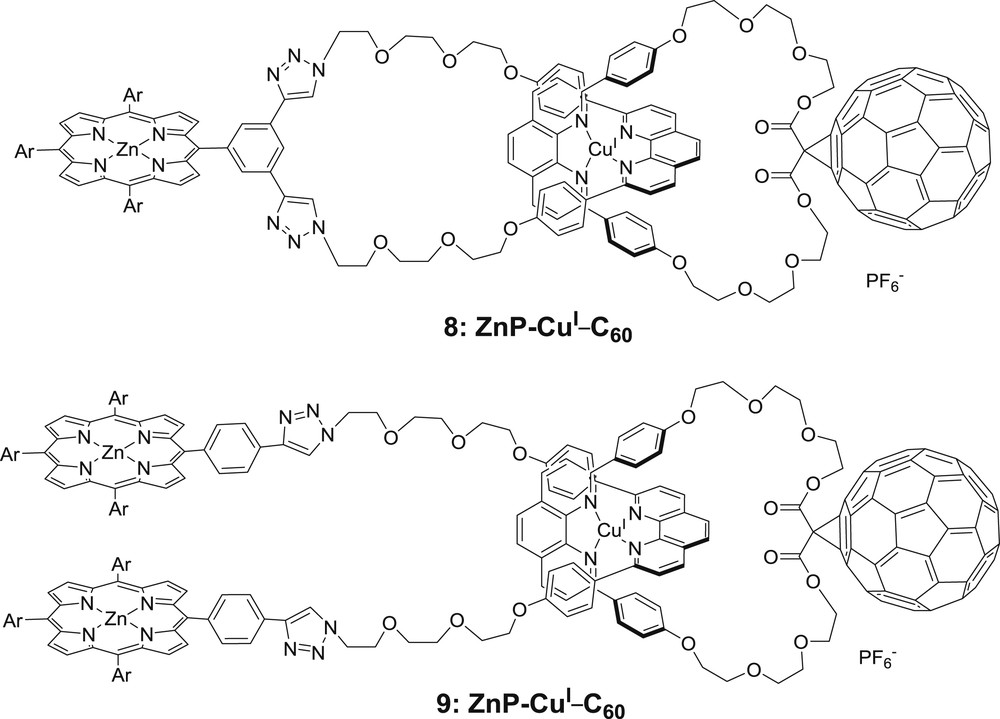
Structures of the catenane 8 and rotaxane 9 [28].
The replacement of porphyrins by two ferrocene units was also realized (7) [27]. The nature of the C60 acceptor (methanofullerene or bis-methanofullerene) as well as the distance between the redox units (increasing the number of glycol links and replacing peptide bonds by triazoles) were varied (Fig. 6). Thermodynamically allowed photo-induced charge separation occurred within these triads, yielding in all cases a reduced fullerene and an oxidized copper(II)-bis-phenanthroline complex (ZnP–CuII–C60−). Let us mention that if the porphyrin moieties were the photo-excited chromophores in this case, a fast energy transfer to the MLCT excited state occurred, and all subsequent photophysical processes were operated by the directly or indirectly excited bis-phenanthroline copper(I) center. The lifetimes of the charge separated states, storing an energy of ca. 1.20 eV, were comprised between 15 and 16 ns in butyronitrile. No charge shift to the ferrocene donors was clearly monitored (the ferrocenium features are difficult to assign in transient absorption spectroscopy), despite the existence of a longer lived state, assigned to 3C60*. The authors invoked too long a distance between the ferrocene units and the photo-oxidized copper(II) center.
To investigate charge separation processes between subunits, a new family of parent catenanes was studied, consisting of interlocked loops, one bearing a fullerene acceptor, and the other a porphyrin donor (zinc, magnesium or free base porphyrins), both loops being held tight thanks to a copper-bis-phenanthroline unit (Fig. 6) [28]. In the case of zinc porphyrins (molecule 8), photoexcitation of the supramolecules lead to the stepwise formation of long-lived, spatially extended charge separated states ZnP+–CuI–C60−. In short, the triplet state of the copper complex converted into a primary charge separated state ZnP–CuII–C60− and a thermodynamically allowed electron shift from the zinc porphyrin to the copper(II) complex yielded the final ZnP+–CuI–C60− charge separated state. Lifetimes were comprised between 0.24 μs for molecule 9 and 1.1 μs for molecule 8; the increased rigidity of catenates vs. rotaxanes was held responsible for this discrepancy.
The same systems were studied, where Zn2+ was replaced by Mg2+ within the coordinating porphyrin macrocycle (MgP) and the case of free base porphyrins (PH2) was explored too [28]. Similar to ZnP derivatives, photo-excitation of the system lead to a primary charge separation state corresponding formally to Porphyrin–CuII–C60−, followed by a secondary charge shift yielding the extended charge separation state exhibiting an oxidized porphyrin moiety and a reduced C60 fragment. However, the efficiency of the formation of the latter was diminished compared to molecule 8, because the excited porphyrin chromophores MgP or PH2 competitively deactivate in favor of the ground state instead of the singlet or triplet MLCT excited state. In addition, the thermodynamic driving forces associated to the formation of the preliminary charge separated state Porphyrin–CuII–C60− are smaller than in the case of 8.
Of course, the synthesis of catenanes or rotaxanes requires a know-how and several steps, including purification of the intermediate molecules. The alleviation of the synthetic burden towards the isolation of heteroleptic copper(I)-bis-diimine dyads and triads is desirable. The counter-productive lability of the coordination sphere did not prevent Elliott and co-workers to study triad 10 (Fig. 7) [6a]. The latter consists of two phenanthroline ligands, one bearing methylviologen (MV2+) acceptors and the other tetramethylphenothiazine (PTZ) donors. By carefully choosing a nonpolar solvent (ortho-difluorobenzene), they managed to isolate a statistical mixture of homoleptic MV2+CuI–MV2+, PTZ–CuI–PTZ and heteroleptic MV2+–CuI–PTZ 10, the latter being predominant. A meticulous analysis of the various contributions from all three species to the transient absorption spectra of the mixture made possible to prove the existence of a photo-induced charge separation state in both the dyad CuI–MV2+ (lifetime of the charge separated state CuII–MV+ τCSS = 43 ns) and the triad MV2+–CuI–PTZ (MV+–CuI–PTZ+, τCSS > 100 ns) where the longer lifetime obtained for the latter was justified by the longer distance between the acceptor and the donor (Fig. 8).
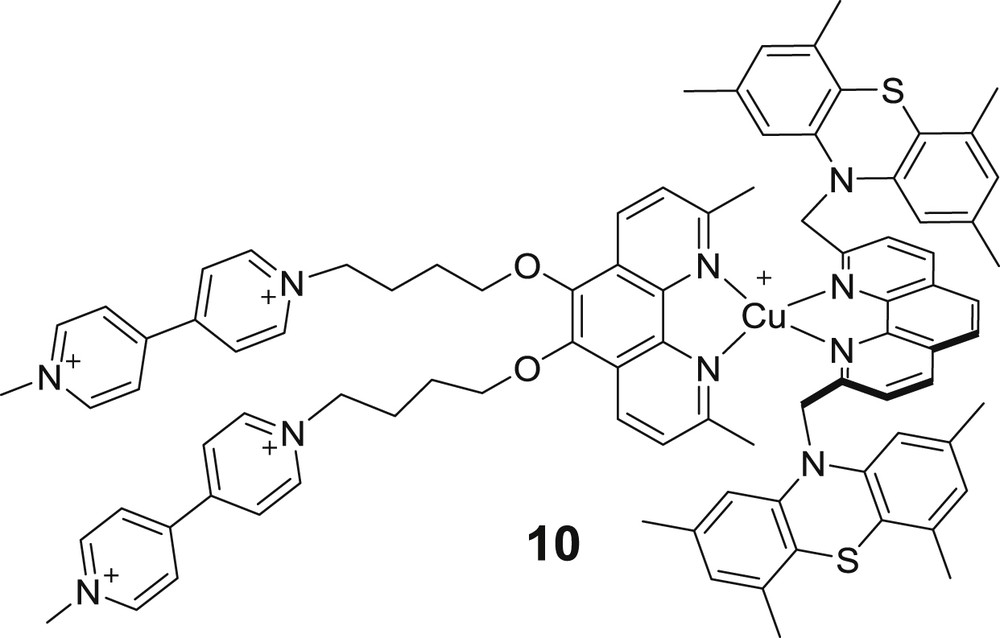
Structure of triad 10 reported by Elliott and co-workers [6a].
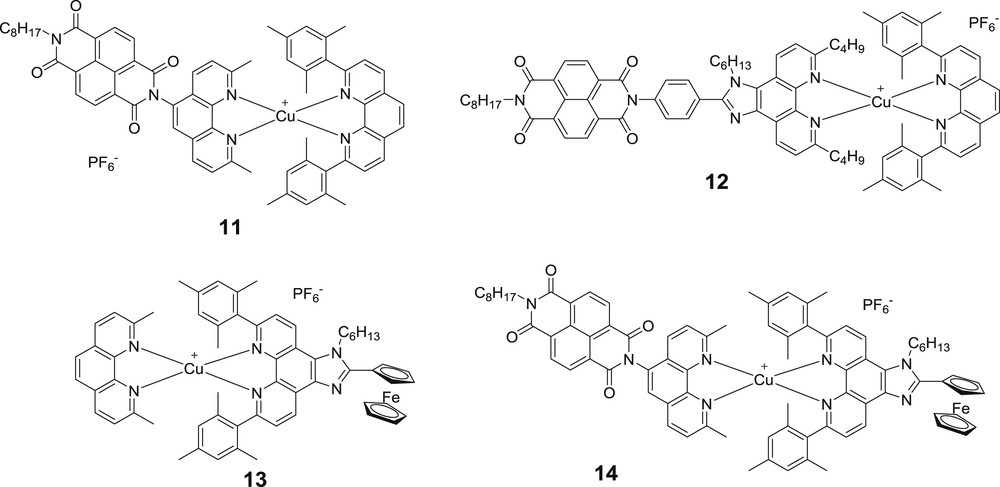
Structure of the dyads and triad 14 prepared by Odobel and co-workers [6b].
Nevertheless, the analysis of the photophysical data was considerably burdened by the presence of not a single but three different species at the same time. Notwithstanding the added workload, it is difficult to imagine a possible application of copper as a valuable substitute for ruthenium in such a case. Application of the HETPHEN synthetic strategy allowed to prepare pure and stable batches of three dyads (11, 12 and 13) and a triad (14), based on the naphtalenediimide (NDI) acceptor and ferrocene donor [6b]. The distance between the NDI unit and the copper(I) center was varied through the use of different organic linkers (see 11 and 12). In dyad 12, the NDI proved to be too remote from the copper(I) chromophore to allow photo-induced electron transfer upon excitation of the latter. A photo-induced charge separated state (NDI−–CuII, τCSS = 2.7 ns) was clearly observed for dyad 12 when the MLCT of the copper(I) center was excited. Appending a ferrocene moiety entails a secondary charge shift to the latter, yielding the extended charge separation state NDI−–CuI–Fc+, as proven by the increased lifetime of 34 ns (Fig. 9). As mentioned above, spectroscopic fingerprints of Fc+ are difficult to monitor by conventional transient absorption spectroscopy and therefore could not be detected. Importantly, the efficiency of the formation of the CSS in the triad is superior to 90%. The analysis of the femtosecond transient absorption spectroscopy results enabled to elucidate the detailed deactivation mechanism of the photoexcited copper complex in triad 10 (Fig. 9). In particular, it was possible to evidence the formation of the charge separation from the unrelaxed singlet 1MLCT, which is sufficiently fast (540 fs) to compete with the flattening process.
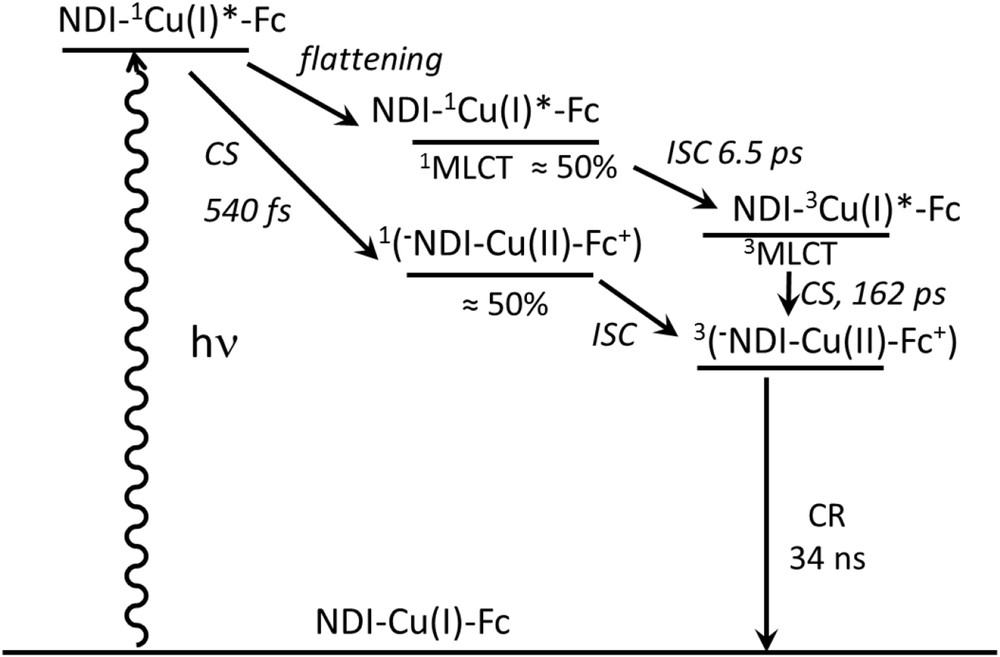
Summary of all the deactivation processes occurring upon light excitation into the MLCT absorption band of the copper complex in the triad 14 [6b].
5 Heteroleptic bis-diimine copper(I) complexes as sensitizers in dye-sensitized solar cells
Dye sensitized solar cells are an attractive photovoltaic technology for electricity production because they can be cheap to manufacture, light weight, flexible, the color of the device can be changed at will and they offer transparency and perform better under low light conditions than any other generations of solar cells [29]. Today, the record efficiency has reached 13% in laboratory with porphyrin derivatives [30], but ruthenium polypyridine complexes remain among the most studied and highly efficient sensitizers used for this application [11b,31].
Upon light absorption (hν), the sensitizer excited-state (S*) becomes a stronger reductant than the ground-state and promotes electron injection into the conduction band (CB) of the semiconductor (eq. (1) and (2)). The ground-state of the sensitizer is regenerated by the redox mediator (M) which is then oxidized into M+ (eq. (3)). The latter diffuses into the solution and shuttles the positive charge to the counter-electrode where it can be reduced back to its original state (eq. (4)). The injected charges migrate to the transparent conducting electrode (TCO) and pass into the external circuit to perform electrical work until they reach the counter-electrode (CE) where they are collected by a oxidized redox mediator (eq. (4)). Summarily, the reactions involved in the operation principle are indicated in Fig. 10.
| S + hν → S∗ | (1) |
| S∗ + SC → S+ + SC− | (2) |
| S+ + M → S + M+ | (3) |
| CE(e−) + M+ → CE + M | (4) |
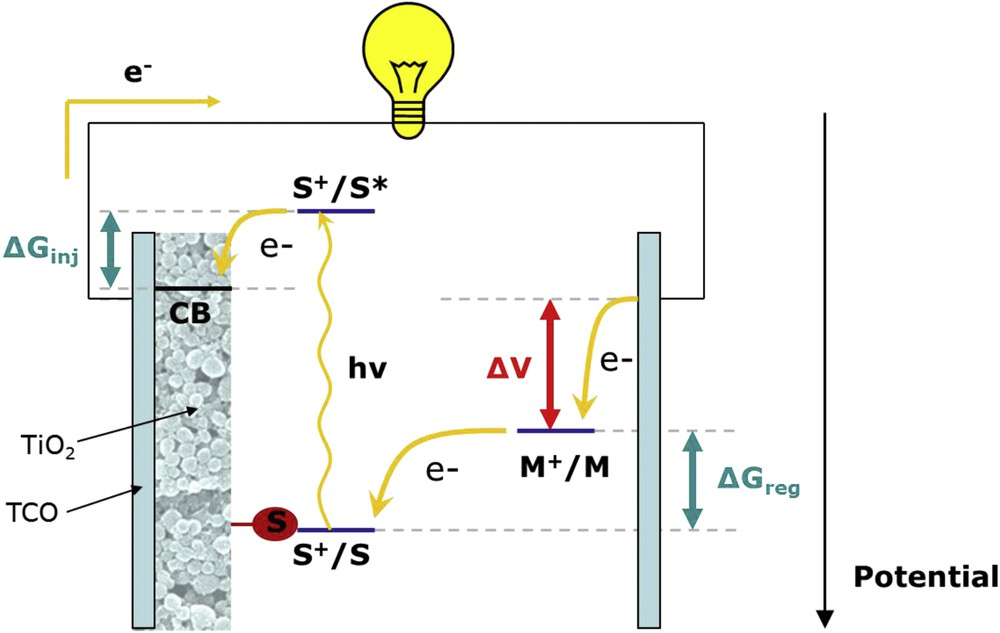
Schematic representation of a DSSC and its working principle. S = sensitizer, TCO = transparent conductive oxide. M = redox mediator.
Another important application of the photoanodes is their utilization as a photocatalytic system in a half-cell for water splitting (oxidation of water into O2) [32]. Implementation of the above photoanode with a photocathode (sensitized NiO electrode for ex.) opens the possibility to develop tandem DSSCs and tandem photocatalytic devices which will necessarily display higher photoconversion efficiencies than single photoelectrodes owing to their potentially larger light collection cross-section.
One distinctive feature of all the efficient sensitizers reported so far is the presence of a strong charge transfer transition with a suitable vectorialization of the charge movement within the excited-state. Indeed, for efficient electron injection into the TiO2 conduction band, the electron density must be shifted from one extremity of the dye to the anchoring group upon light absorption. This characteristic ensures a sizable electronic coupling of the excited state with the wave function of the semiconductor (SC) conduction band (fast injection) and a remote distribution of the positive charge from the SC surface after hole injection (slow charge recombination). These engineering rules are nicely illustrated with efficient ruthenium complexes such as N3 (Fig. 11), which was the champion dye for several decades (giving 10% PCE). In the latter complex, the HOMO orbital is strongly localized on the thiocyanato ligands, exposed to the electrolyte, facilitating thus the oxidation of the redox mediator, while the LUMO is carried by the two anchoring ligands (bipyridine dicarboxylic acid); hence an ultrafast electron injection can occur owing to the proximity with the SC. Heteroleptic bis-diimine copper(I) complexes are particularly amenable sensitizers for DSSCs. The impossible preparation of heteroleptic copper complexes until recently is certainly the major reason why these complexes have been rarely investigated as dyes in Grätzel solar cells (about 30 papers published so far versus more than 2000 publications for ruthenium polypyridine complexes: source WEB of knowledge). The first advantage is the larger extent to which the electronic properties of the sensitizer can be tuned thanks to the presence of two different ligands. Secondly, the unique possibility to arrange two different diimine ligands around the copper(I) center enables to design push-pull complexes in which one ligand will be fonctionnalized by anchor groups to immobilize the dye on the TiO2 surface and facilitate electron injection (it will bear the LUMO) and the other ligand will contain electron releasing groups to carry the positive charge after electron injection (it will bear the HOMO) [33]. This molecular design, abundantly used with ruthenium sensitizers, is certainly one of the reasons for their successes in DSSCs and probably represents the most promising approach to develop efficient bis-diimine copper(I) complexes for DSSCs (Fig. 11).
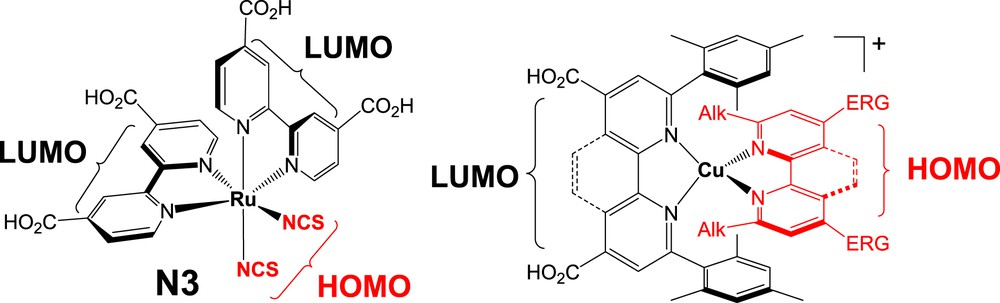
Illustration of the push-pull character of an efficient ruthenium sensitizer (N3) and the general structure of heteroleptic bis-diimine copper(I) complexes for DSSCs. Alk = alkyl chain, ERG = electron releasing group.
The first investigation of bis-diimine copper complexes as sensitizers of wide bandgap semiconductors started in 1983, but naturally with some homoleptic bis-phenanthroline complexes 15–17 designed by Sauvage and co-workers and tested on the ZnO photoanode by Alonso Vante and co-workers (Fig. 12) [34]. The complexes were free of anchoring groups and therefore the measured photocurrent was low, but this study provides the first demonstration that this class of complexes could inject electrons in n-type SCs until a relatively low energy wavelength (700 nm). Few years later, another homoleptic bis-phenanthroline copper(I) complex 17 was investigated by the same teams [35]. It carries carboxylic anchoring groups and it was tested on both ZnO and TiO2. The performances were still low, but the devices far from being optimized as the redox mediator was the quinone/hydroquinone couple [35]. Interestingly, a recent photophysical study by X-ray transient absorption spectroscopy of this complex grafted on TiO2 showed that an ultrafast electron injection can occur from the singlet 1MLCT excited state before intersystem crossing (ISC) to the triplet state [36]. Another important contribution came in 2002, with the work of Sakati and co-workers, who prepared the first DSSC with the bis(tetramethyl-bipyridine-dicarboxylic acid) copper(I) complex 19 [37]. Under AM 1.5 and using the classical iodide/triiodide electrolyte, this complex yields 2.5% photoconversion efficiency (PCE) (Voc = 640 mV, Jsc = 2.9 mA/cm2) with a circa 30% maximum IPCE around 480 nm.
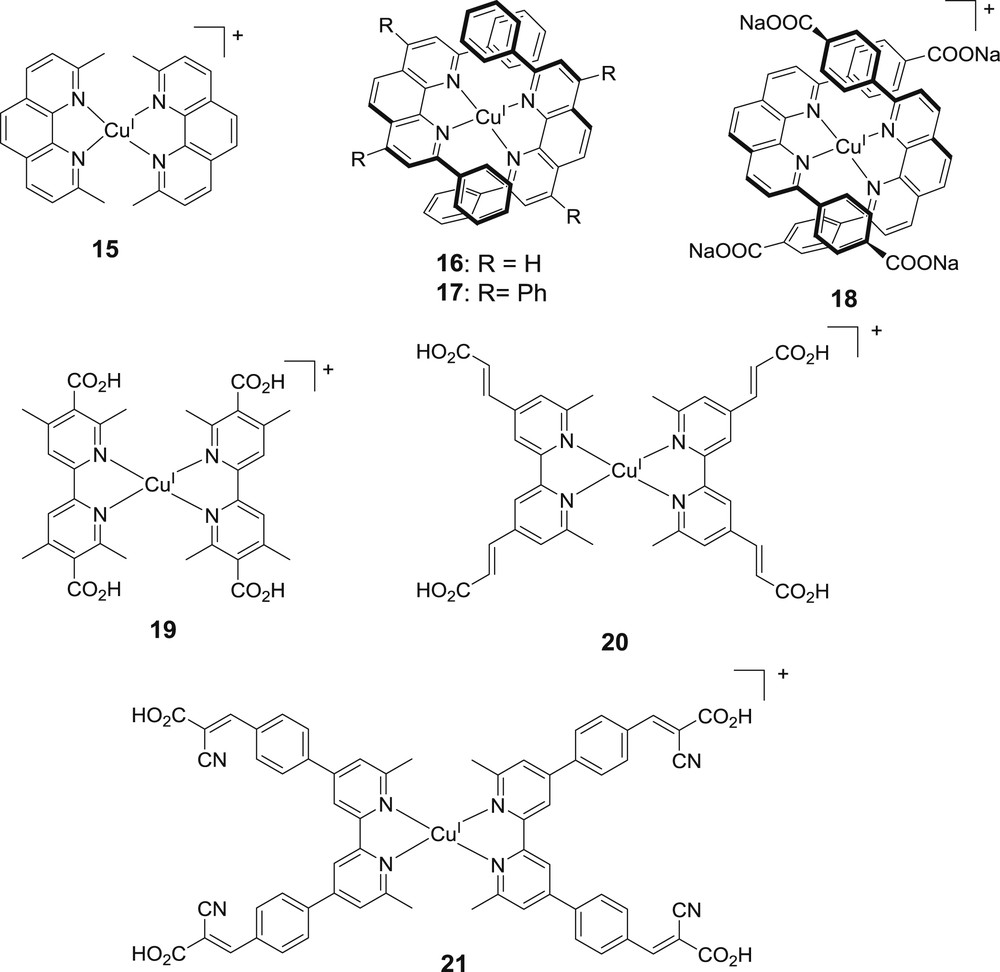
Structures of some homoleptic bis-phenanthroline copper(I) complexes investigated in DSSCs.
Another breakthrough was reported by Bessho and co-workers in 2008 with the bis(6,6′-dimethyl-bipyridine-bis-4,4′-vinylcarboxylic acid) copper(I) complex 20 [38]. A 2.3% PCE (Voc = 556 mV, Jsc = 5.9 mA/cm2, ff = 70%) was achieved thanks to the red shifted MLCT transition of this complex (ɛ = 6700 M−1 cm−1 at 450 nm). A similar homoleptic bis-bipyridine copper(I), with phenyl acrylic acid substituents on the bpy (complex 21 in Fig. 12), was reported in 2012 and it exhibits the same overall PCE (2.3%) [39].
Although these series of homoleptic bis-diimine copper(I) complexes display encouraging photovoltaic performances in DSSCs, they were far from those measured for ruthenium complexes such as the reference N719 dye (9.7% under the same conditions reported in the work of Bessho and co-workers [38]). A new and fruitful direction could come from the design of heteroleptic bis-diimine copper(I) complexes, whose photoredox and absorption properties could be optimized to a much larger extent. This assumption was first supported by quantum chemical calculations published by Guo and co-workers [40]. These calculations showed that heteroleptic bis-diimine copper(I) complexes theoretically exhibit suitable properties to act as efficient sensitizers in DSSCs and they could potentially achieve similar PCEs as those of ruthenium complexes. In 2009, Constable and co-workers reported an interesting and straightforward strategy to immobilize heteroleptic bis-diimine copper(I) complexes on TiO2. It consists of a two-step procedure starting with the initial attachment to the TiO2 surface of a diimine ligand functionalized with anchoring groups such as the dimethyl bipyridine dicarboxylic acid 22 (Fig. 13). In a second step, the as-coated TiO2 electrode was dipped in a solution of a homoleptic bis-diimine copper(I) complex such as 23. The well-known ligand exchange reaction around the copper(I) center with the diimine ligand leads to the formation of the heteroleptic complex at the surface of TiO2 comprising the anchoring ligand and the ligand coming from the homoleptic complex in solution (Fig. 13). Recently, it had been shown that the conditions to run this reaction (particularly the concentration of the precursor homoleptic complex) are crucial to achieve the best performances [41]. This methodology was applied to a large panoply of different anchoring ligands and ancillary ligands (Fig. 14). Interestingly, Kelvin probe force microscopy study revealed that, upon chemisorption on the TiO2 surface, the dipole moment of the positively charged copper(I) complex (A2)Cu(27) is inversely oriented as compared to that of the benchmark ruthenium complex N719 (N719 is the same dye as N3 shown in Fig. 11 but in which two carboxylic acid groups of the latter are neutralized by tetrabutylammonium hydroxide) as it points away from the surface [42]. As a consequence, the CB band bending is also inverted with the copper complex and this explains the lower Voc generally measured with the copper diimine complexes than with ruthenium sensitizers. Very recently, neutral copper(I) sensitizers were prepared and investigated in DSSCs using dipyrromethene as ancillary ligands (for example: (A1)Cu(32) Fig. 14). Unfortunately, the photovoltaic performances and particularly the Voc were relatively low owing to a low regeneration driving force (PCE = 0.41% see Table 1) [43]. Indeed the anionic ligand cathodically shifts the oxidation potential of the CuII/I couple and makes the complex a powerful reductant but a poor oxidant in the oxidation sate + II.
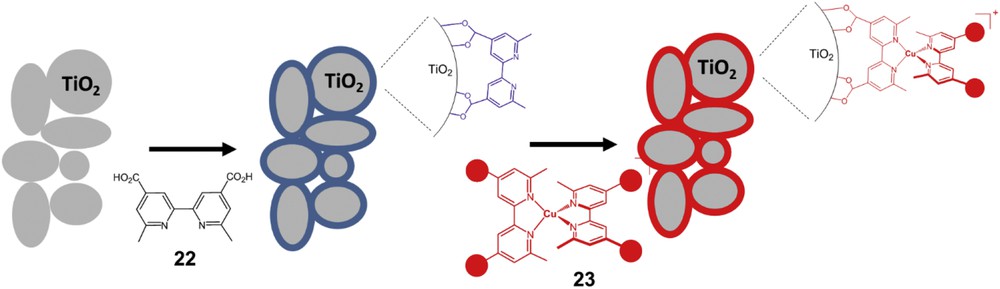
Schematic representation of the two step preparation of heteroleptic bis-diimine copper(I) complexes on the nanocrystalline TiO2 electrode.
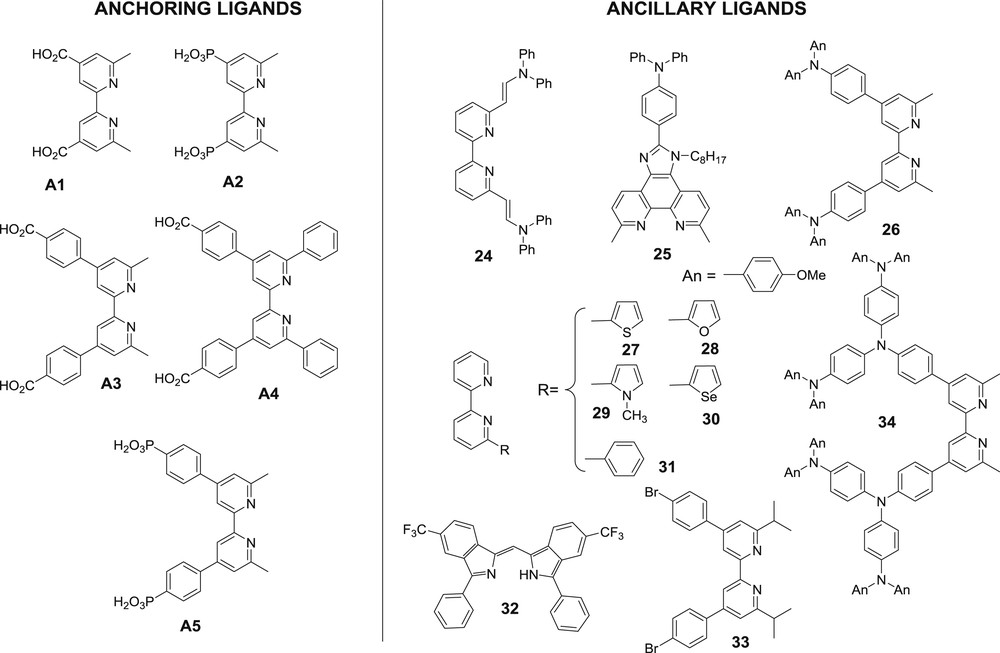
Structures of the anchoring (left) and ancillary ligands (right) used to form heteroleptic bis-diimine copper(I) sensitizers on the TiO2 surface.
Summary of the metrics of the DSSCs prepared with a selection of heteroleptic bis-diimine copper(I) sensitizers prepared according to the procedure shown in Fig. 3 and recorded under simulated AM1.5 (1000 W/m2). The ligand legend is shown in Fig. 14. Voc = open circuit voltage, Jsc = short circuit current density, ff = fill factor and PCE = photoconversion efficiency.
| Complexes | Electrolyte | Voc mV | Jsc mA/cm2 | ff % | PCE % | Ref. |
| (A1)Cu(24) | I3−/I− | 482 | 1 | 65 | 0.47 | [44] |
| (A2)Cu(24) | I3−/I− | 609 | 5 | 60 | 1.95 | [44] |
| (A3)Cu(24) | I3−/I− | 555 | 5 | 60 | 1.70 | [44] |
| (A4)Cu(24) | I3−/I− | 555 | 1 | 63 | 0.51 | [44] |
| (A1)Cu(28) | I3−/I− | 451 | 1 | 62 | 0.40 | [45] |
| (A2)Cu(28) | I3−/I− | 561 | 4 | 60 | 1.51 | [45] |
| (A3)Cu(28) | I3−/I− | 506 | 5 | 54 | 1.25 | [45] |
| (A5)Cu(33) | I3−/I− | 576 | 6.91 | 72 | 2.87 | [46] |
| (A2)Cu(26) | I3−/I− | 593 | 9.55 | 67 | 3.77 | [47] |
| (A2)Cu(34) | I3−/I− | 580 | 6.62 | 65 | 2.51 | [47] |
| (A5)Cu(25) | I3−/I− | 612 | 2.73 | 70 | 1.18 | [48] |
| (A5)Cu(25) | CoIII/II | 575 | 5.11 | 71 | 2.02 | [48] |
| (A1)Cu(32) | I3−/I− | 520 | 1.21 | 64 | 0.41 | [43] |
One interesting advantage of this approach lies in the fact that the screening of a large variety of ligand combinations can be quickly made just by immobilizing a given anchoring ligand on TiO2 and then exposing it to various homoleptic copper(I) complexes. Thus, many different heteroleptic bis-diimine copper(I) complexes could be quickly prepared in a simple manner. In contrast, the intrinsic physico-chemical properties of the as-obtained heteroleptic bis-diimine copper(I) complexes (redox potentials, spectroscopic properties) cannot be easily measured. This can be a drawback to establish structure/properties relationships, which are crucial to the rational design highly performing sensitizers. Also, the dying conditions are more difficult to optimize as more parameters entail this process (immobilization of the anchoring ligand and formation of the complex in a second step). The photoconversion efficiencies measured on nanocrytalline TiO2 films under AM 1.5 (100 W/m2) of a selection of heteroleptic bis-diimine copper(I) sensitizers are collected in Table 1.
From the many combinations of heteroleptic bis-diimine copper(I) complexes investigated in TiO2 based DSSCs, one can observe that the best anchoring ligands are those substituted with phosphonic acid (ligands A2 & A5). This could be rationalized by the fact that these complexes could be neutral in spite of the positive charge brought by Cu(I), thanks to the potential bis deprotonation of the phosphonic acid moieties, which can neutralize the positive charge. Indeed, it has often been seen that positively charged sensitizers display low PCEs in DSSCs. This is certainly the consequence of large spacing between the immobilized dyes on the TiO2 surface (owing to electrostatic repulsions) and to the higher triiodide concentration near the surface of TiO2 resulting from ion paring with the dyes. Among the anchoring ligands containing the carboxylic acid moieties, the most performing one is usually A3. The most efficient ancillary ligands are those which contain electron releasing substituents such as ligands 24–26 and 34, in agreement with the postulating engineering rule mentioned above. These substituents have two beneficial features. First, they raise the HOMO level on the copper center and consequently red-shift the MLCT absorption band. Secondly, they can accept a significant amount of the spin density of the hole after electron injection in TiO2 and this retards the charge recombination with the injected electron in the TiO2 conduction band. The strategy of Constable to make heteroleptic copper(I) complexes directly on TiO2 films was used by Elliot and co-workers with the complexes 35–37 (Fig. 15) [49]. These complexes proved to be compatible with the tris(bis-tertbutylbipyridine) cobalt(III/II) redox mediator. The highest photovoltaic performances were measured with the dyad 37, whose phenothiazine moieties (PTZ) play the role of secondary electron donors and scavenge the holes formed on copper after electron injection. However, the overall photovoltaic performances are low (Voc = 570 mV, Jsc = 0.54 mA/cm2, ff = 21% with complex 37) probably because of the feeble MLCT extinction coefficient and the low diffusion constant of the bulky redox mediator. The photophysical study of the complex 37 immobilized on TiO2 indicates that the hole transfer to PTZ units indeed occurs with a rate constant faster than (7 ns)−1 and is formed within a 80% quantum yield. It recombines with a time constant of about 100 μs. The utilization of a cobalt electrolyte with a bis-diimine copper(I) sensitizer was also successfully reported by Constable and co-workers with the complex (A5)Cu(25) whose performances are higher than those delivered with the conventional iodide/triiodide electrolyte (Table 1) [48]. It is worth noting that the complex (A5)Cu(25) can be used with a much less encumbered Co(bpy)33/2+ redox shuttle, whose diffusion constant is much higher than that of the tris(bis-tertbutylbipyridine) cobalt complex and permits to theoretically reach higher Jsc. Moreover, cobalt electrolytes are less corrosive, absorb much less visible light and enable to reach higher photovoltage than that of iodide/triiodide. Collectively, these results are encouraging in view of using bis-diimine copper(I) complexes in DSSCs.
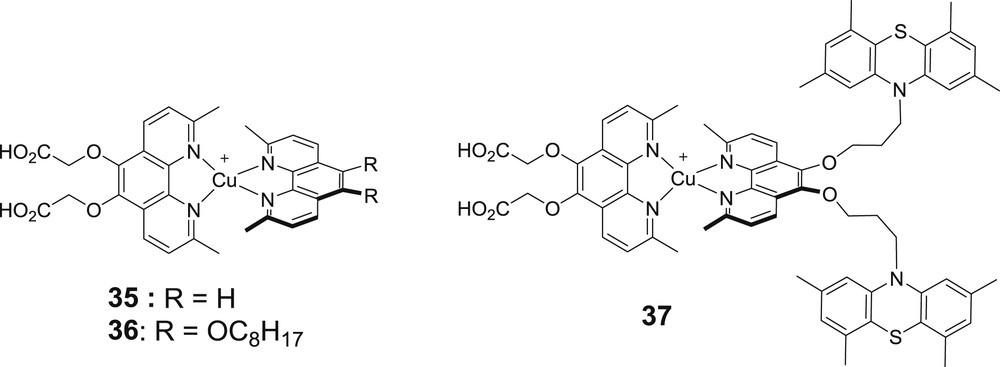
Structures of the three heteroleptic bis-diimine copper(I) sensitizers studied by Elliot and co-workers [49].
It was only in 2012, that the first stable and isolated heteroleptic bis-diimine copper(I) complexes designed for DSSCs were reported by the group of Odobel (Fig. 16) [33a]. The HETPHEN concept was implemented for the development of heteroleptic bis-diimine copper(I) complexes for this specific application. Hence, the sterically encumbered ligand 6,6′-dimesityl-phenanthroline 1 was associated with bisquinoline dicarboxylic acid, which serves as the anchoring group. The latter is known to exhibit a low lying LUMO orbital, which is perfectly suited to accept the electron in the MLCT excited state and to induce a bathochromic shift of the latter. The resulting complexes exhibit a broad MLCT transition spreading over a wide wavelength window over 700 nm, but at the expense of a rather low extinction coefficient limiting the absorbance of the TiO2 photoanode. On the other side, a trisarylamine moiety was appended in the ancillary ligand (complex 39 Fig. 16) in order to slow down charge recombination. The photovoltaic performances of these complexes are relatively modest (0.71% and 0.25% for 38 and 39 respectively) certainly because the LUMO on the bisquinoline dicarboxylic acid is too low lying relative to the TiO2 conduction band position, imposing thus a weak injection driving force. The feeble light harvesting efficiency of the TiO2 electrodes is also another weakness of these systems at the origin of the low photocurrent density.
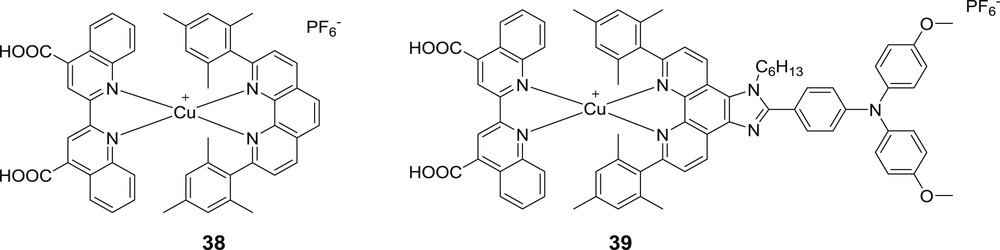
First series of heteroleptic bis-diimine copper(I) sensitizers developed by the group of Odobel [33a].
A real improvement came with a new series of heteroleptic complexes bearing chromogenic ancillary ligands. It had been demonstrated earlier that the introduction of π-conjugated moieties on the bipyridine (styryl group) increasingly enhances the extinction coefficient of the copper complexes [50]. This strategy was naturally applied to copper complexes for DSSCs and resulted in highly absorbing dyes (Fig. 17).
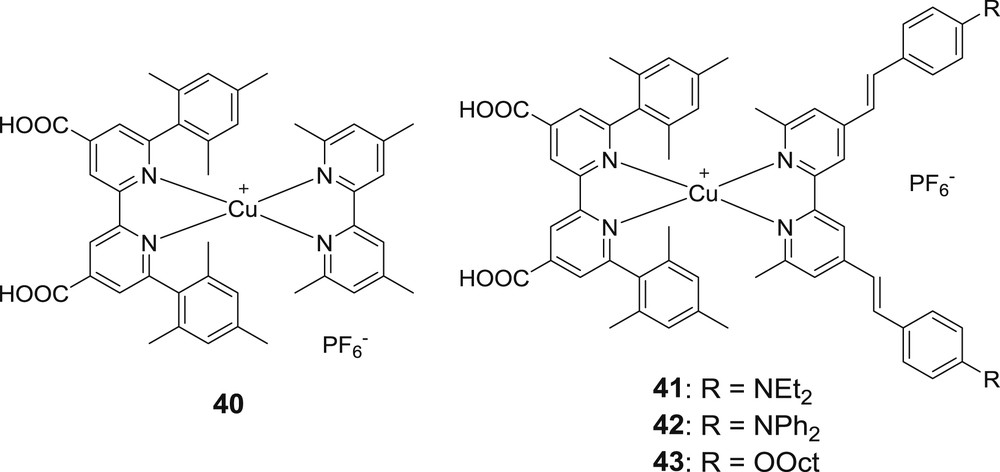
Second series of heteroleptic bis-diimine copper(I) sensitizers developed by the group of Odobel [33b].
The photovoltaic performances of these dyes are increased when deoxycholic acid is used, because it diminishes their deleterious aggregation tendency probably induced by the organic moieties on the ancillary ligands. The two best dyes are 41 and 42, which contain amine substituents on the styryl moieties as they display respectively 4.66% (Voc = 605 mV, Jsc = 10.86 mA/cm2 and ff = 71%) and 4.42% photoconversion efficiencies under AM1.5 (1000 W/m2). It is worth noting that dye 41 reaches the highest PCE ever reported with bis-diimine copper(I) sensitizers and underscores the benefit of push-pull like structures which are only possible with heteroleptic complexes. Interestingly, the photoactivity on these complexes in TiO2 based DSSCs spans over 700 nm, but the maximum IPCE reaches only 50%, pointing that there is room for improvement, namely by increasing the electron injection quantum yield (Fig. 18).
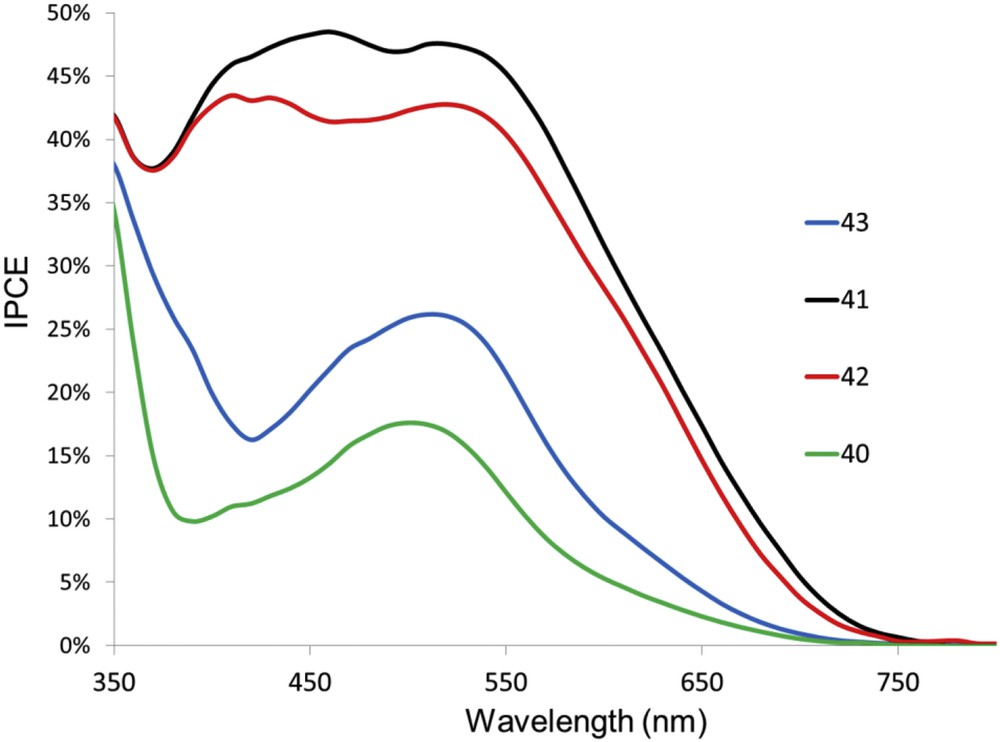
Photoaction spectra of the dyes 40–43 in a TiO2 based DSSC with I3−/I− as the redox shuttle.
6 Conclusions and perspectives
Bis-diimine copper(I) complexes are valuable dyes to be used for solar energy conversion schemes first because they exhibit MLCT transition in the visible spectrum. Furthermore, copper is a relatively abundant element on earth and consequently quite affordable. The latter condition is fundamental to envision the materials that will be used for large scale conversion of solar energy into fuels or electricity. The attractive properties of copper complexes have driven an increasing interest with the clear objective to replace the highly used and quite well performing polypyridine ruthenium, platinum and iridium complexes for these applications. The main limitations of these copper(I) complexes stem from their usually low extinction coefficient on the MLCT transition, the quite short lived excited state and the difficulty to prepare stable heteroleptic complexes with diimine ligands. The extinction coefficient can be importantly enhanced by expanding the π-conjugated length on the ligands, which become intrinsically highly absorbing and enable to increase the distance of the charge transfer. The excited-state lifetime of the MLCT can certainly be enhanced by freezing the movement of the ligands around copper(I) via bulky substituents placed at suitable positions. Great success has already been demonstrated with tert-butyl substituents or with bidentate phosphines, which achieve room temperature lifetimes over μs timescale. Interestingly, the encumbered ligand used for the HETPHEN strategy can play this role. The systems presented in this review highlighted the great benefit of heteroleptic copper complexes brought by the HETPHEN synthetic strategy. The possibility to assemble two different ligands around copper enables to tune the properties of the dye with a much larger extent than what two identical ligands can do. Moreover, it gives the possibility to enhance the vectoralisation of the electron transfer and to enhance the number of functional groups that can be introduced on the systems. The heteroleptic copper(I) complexes presented in this article demonstrate their real superiority over homoleptic homologs for long range photoinduced charge separation and as sensitizers in DSSCs. In addition, they can also be advantageously used for other applications such as organic photovoltaics, photocatalysis, catalysis and light emitting diodes. The above examples are only preliminary results and we can foresee that many potential new developments will come with heteroleptic complexes, because the degree of freedom to tune the properties is very large. Indeed, the bulky substituents at the α position of nitrogen were essentially mesityl, but many other encumbered groups could be envisioned and they can carry specific properties. The diimine ligands used so far were only bipyridine and phenanthroline but other imine systems are possible and could be even simpler to prepare. Overall, the use of heteroleptic copper(I) complexes is certainly at its early age and the successes presented in this review can certainly spark the developments of new systems with intriguing and valuable properties.
Acknowledgments
The Agence Nationale de Recherche (ANR) is gratefully acknowledged for the financial support through the ANR Blanc “HeteroCop” (ANR-09-BLAN-0183-01). LUMOMAT and COST Action (CM1202 PERSPECT-H2O) are also acknowledged for support of the research.