

1 Introduction
Microbial glycolipids represent a group of structurally diverse low molecular weight compounds generally produced by fermentation processes. Depending on their microbial origin and structural characteristics, they are classified into four main groups including rhamnolipids, sophorolipids, trehalose lipids and mannosylerythritol lipids (Fig. 1). Glycolipid structures are composed of a hydrophilic head linked to a hydrophobic tail. The hydrophilic moiety is typically a mono- or disaccharide unit while the hydrophobic tail is derived from a fatty acid side chain. This amphiphilic structure gives glycolipids interesting surfactant properties, thereby allowing them to reduce surface and interfacial tension between liquids, solids and gases and to form micelles and emulsions in liquid systems [1].
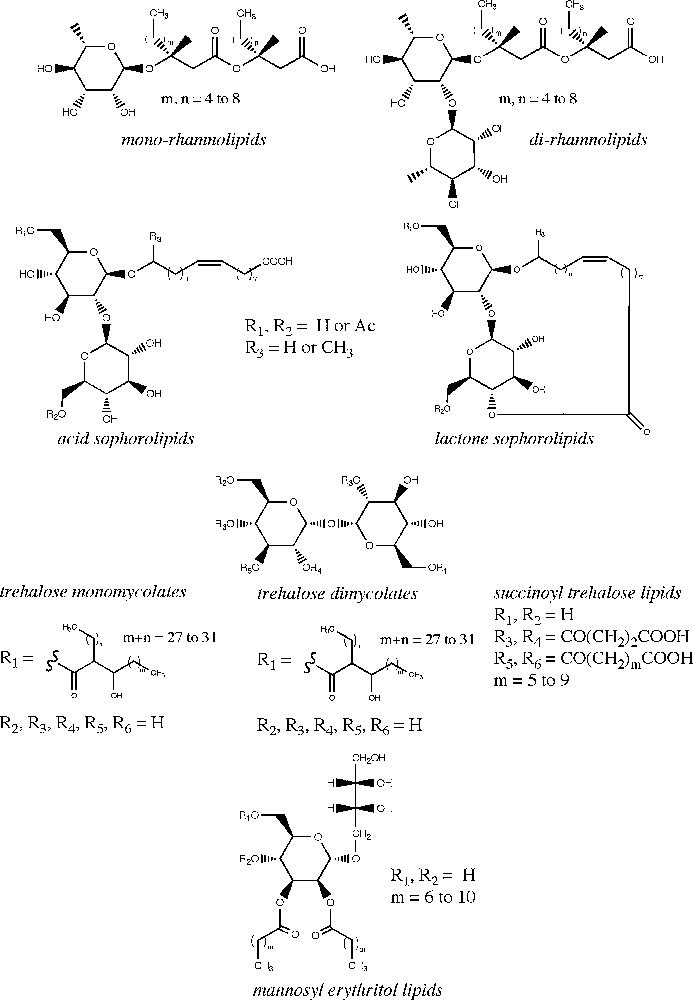
Microbial glycolipid structures.
Worldwide surfactant production is about 10 million tons per year [2], and the majority of these compounds are petroleum derived. Microbial glycolipids produced from renewable substrates are an attractive and environmentally friendly alternative to petroleum surfactants because of their biodegradability and low toxicity. Glycolipids are also potentially interesting candidates for different environmental applications such as bioremediation and oil recovery from soil [3,4]. For example, rhamnolipids are able to scavenge organic compounds, which are sorbed to soil particles as well as mobilize contaminants such as heavy metals, oils and other toxic pollutants by pseudosolubilisation or emulsification during soil washing treatments [5,6]. In addition to their environmental potential, glycolipids have also found a range of applications in the cosmetic, detergent, food, and pharmaceutical industries [7]. Mixtures of rhamnolipids and sophorolipids are able to form microemulsions in different vegetable oils that are applicable to biofuel production, hard surface cleansers and drug delivery [8]. Glycolipids are also becoming interesting alternatives to synthetic drugs due to their antimicrobial and antiviral activities [9–13].
Despite these promising physicochemical and biological properties, the commercialization of glycolipids as microbial biosurfactants has been hampered due to high production costs. Another major difficulty arises from the complexity of the glycolipid mixtures, which are naturally produced by yeast or bacteria. Their structural diversity in fermentation media limits the understanding of individual mechanisms of action and limits their valorization in cosmetic or health-related areas. Efforts are currently being made to improve fermentation and downstream processes to recover glycolipids in higher quantities, with better purity and efficient production costs [14,15]. Thus, the optimization of extraction and purification methods to obtain glycolipids either as selected mixtures or in pure form remains an important challenge.
This paper presents an overview of the different strategies developed in the last several years to obtain highly concentrated glycolipid extracts or to individually characterize these compounds. In particular we will focus on solid support-free liquid-liquid separation techniques such as centrifugal partition chromatography (CPC) or counter-current chromatography (CCC). These separation techniques have shown promising perspectives for the preparative-scale separation and purification of lipids and related compounds [16], but their application to glycolipids and more generally to natural surfactants remains scarce. The main advantages and limitations of CPC and CCC methods regarding glycolipid investigation and valorization will be discussed.
2 The main classes of glycolipids
2.1 Rhamnolipids
Rhamnolipids are mainly produced by Pseudomonas aeruginosa and Burkholderia sp. [17–19] and are by far the best studied glycolipids due to their fungicide properties and promising applications in the agricultural industry [20,21]. More than 60 different rhamnolipid structures have been identified. They comprise one or two L-rhamnose units linked through a β-glycoside bond to up to three hydroxy fatty acid groups (Fig. 1). The fatty acid chain length varies from 8 to 14 carbon atoms with or without unsaturation. In bacteria cells, rhamnolipids play a crucial role in the uptake and degradation of insoluble substrates and in biofilm development [19]. They possess strong surface active properties interesting for the large-scale production of biosurfactants and are also used to enhance the biodegradation of toxic organic soil contaminants [22]. Rhamnolipids have also demonstrated significant antiviral and antifungal activities against herpes simplex virus types I and II and phytopathogenic fungi, respectively [11–13]. Due to their effect on bacterial cell surface structures, rhamnolipids also display antibacterial activities [23,24].
2.2 Sophorolipids
Sophorolipids are synthesized by a number of nonpathogenic yeasts such as Candida bombicola, Candida apicola or Thodotorula bogoriensis [25]. They are released in the extracellular medium to act as extracellular carbon storage material for the regulation of high osmotic strength produced from high sugar concentrations [26]. Globally, sophorolipids lower the surface tension of water from 73 mN/m to about 30–40 mN/m, with a critical micelle concentration of 40 to 100 mg/L. They show excellent skin compatibility and thus are employed in various cosmetic formulations. Sophorolipids have also demonstrated interesting antimicrobial activity against plant pathogenic fungi [27], a capacity to inhibit the human immunodeficiency virus [10] and more recently a potential for liver cancer treatment [28]. Their structure consists of one sophorose unit (a glucose disaccharide with an unusual β-1,2 bond) which is β-linked to a hydroxy fatty acid containing 16 to 18 carbon atoms and one or more unsaturations [29]. Sophorose can be acetylated at the 6′- and/or 6′′ position and the hydroxy fatty acid can be linked to the sophorose unit at the terminal or subterminal position (Fig. 1). The carboxylic end of the fatty acid is either free or esterified at the 4′′ position (lactone form). The physicochemical and biological properties of sophorolipids are significantly influenced by the distribution of the lactone vs. acid forms produced in the fermentative broth. In general, lactone sophorolipids are more efficient in reducing surface tension and are better antimicrobial agents, whereas acidic sophorolipids display better foaming properties. Acetyl groups can also lower the hydrophilicity of sophorolipids and enhance their antiviral and cytokine stimulating effects [10].
2.3 Trehalose lipids
Trehalose lipids are produced by a variety of microorganisms including Rhodococcus erythropolis [30], Mycobacterium, Nocardia and Corynebacterium strains [31]. Their structures vary in the number and length (from 20 to 90 carbon atoms) of the esterified fatty acid chain. Trehalose is a glucose disaccharide with an α, α-1, 1-glycoside bond (Fig. 1). Similarly to rhamnolipids, trehalose lipids can be used as additives in environmental remediation to enhance the solubility of hydrophobic soil contaminants and stimulate the biodegradation of hydrocarbons [6]. They also exhibit biological activities, more particularly trehalose 6,6′-dimycolate which possess antitumor, antiviral and antimicrobial activities [32,33]. Succinoyl trehalose lipids have been reported to inhibit growth and induce differentiation in human promyelocytic leukaemia cell lines depending on the structure of the hydrophobic moiety [34,35]. Certain trehalose lipids however, particularly those produced from the Rhodococcus genus, are often bound to the cell envelope, what negatively affects their production yield and industrial recovery [6].
2.4 Mannosylerythritol lipids
Together with sophorolipids, mannosylerythritol lipids are the only surfactants produced in large quantities by yeasts. Mannosylerythritol lipids are mainly synthesized by Pseudozyma sp. and in a lesser extent by Schizonella melanogramma and Ustilago sp. The structure of mannosylerythritol lipids strongly depends on their microbial origin. They contain a 4-O-β-D-mannopyranosyl-meso-erythritol as the hydrophilic group and a fatty acid and/or an acetyl group as the hydrophobic moiety (Fig. 1). The structural variations of mannosylerythritol lipids are mainly due to the number and position of the acetyl groups on mannose or erythritol and to the fatty acid chain length and degree of saturation [30]. There is an increasing interest in mannosylerythritol lipids because of their promising pharmaceutical applications [36,37]. The self-assembling properties of these lipids enhance their efficiency in gene transfection [30,38]. They also present antitumor and differentiation inducing activities against human leukemia cells [35]. In addition to their excellent surface activity, these compounds can be recovered in high yields (> 100 g/L) thus becoming economically attractive for industrial purposes.
3 Microbial glycolipid valorization
Several major challenges must be overcome to make the use of microbial glycolipids more profitable. First, their production costs have to be significantly reduced. Large-scale production must be optimized in order to economically compete with chemical surfactants. Particular efforts have been made to find more appropriate and cheaper substrates among oils, alkanes or fatty acids and their corresponding esters as carbon sources [39,40]. In an attempt to reduce substrate costs while keeping the environmentally friendly character of glycolipids, inexpensive vegetable oils are still often used as a carbon source for glycolipid production [41–45]. Other aspects focusing on the development of over-producing strains, improvement of cultural factors, fermentation conditions and downstream processes are currently being investigated [46,47].
The use of microbial glycolipids in cosmetic and health-related areas remains limited due to the lack of information regarding the biological role of the individual species and their toxicity toward human systems. Efficient separation and purification tools are thus required to examine each compound individually in an attempt to understand their specific biological role, with a particular focus on their antimicrobial and antitumor mechanisms of action. This is becoming more and more feasible with the help of recent developments in preparative-scale purification techniques.
3.1 Downstream processes: an overview of glycolipid extraction methods
The extraction of microbial glycolipids requires a preliminary separation of the microorganisms from the fermentation media. (Fig. 2) At an industrial scale, bacteria or yeast cells are classically removed by centrifugation. As glycolipids are generally heavier than water, it is also possible to just decant them from the fermentation media after heating or pH adjustment if necessary. Another alternative is to add ethanol in order to dissolve the glycolipids and then remove the cells by centrifugation. This process was recently described for the extraction of sophorolipid mixtures [48]. Other methods based on precipitation are also commonly used to remove glycolipids from the fermentation media. Acidification or salting-out of the media makes glycolipids less soluble in the aqueous phases. After precipitation, they can be easily collected by centrifugation. The precipitation of rhamnolipids, for instance, is generally performed after acidification of the media [49] or after the addition of salts such as aluminum sulfate [50]. Acid precipitation has also been employed for the extraction of succinoyl trehalose lipids [51]. Diafiltration followed by isopropanol precipitation was proposed to isolate trehalose lipids from Rhodococcus sp. strains [52]. All of these centrifugation and precipitation methods are very convenient when working with large volumes. However, the resulting crude extracts often remain contaminated by lipids or carbohydrate compounds. Solvent extraction is a more efficient alternative to more cleanly isolate the glycolipid mixtures. Polar organic solvents such as chloroform, methanol or ethyl acetate are the most widely used. For instance the extraction of sophorolipids is classically performed by using ethyl acetate [53]. An additional extraction with apolar solvents such as n-hexane, pentane or methyl tert-butyl ether can also be useful to remove the residual lipid substrates, which are co-extracted with sophorolipids [42,54]. For the extraction of trehalose lipids, the preferred solvents are either a mixture of chloroform/methanol (2:1, v/v), methyl tert-butyl ether or a mixture of ethyl acetate/methanol (8:1, v/v) [55,56]. Rhamnolipid extraction is typically carried out by using ethyl acetate or a mixture of chloroform/methanol (2:1, v/v) [50,57]. The extraction yield can be improved if a preliminary acidification step is performed to reduce solubility in water.
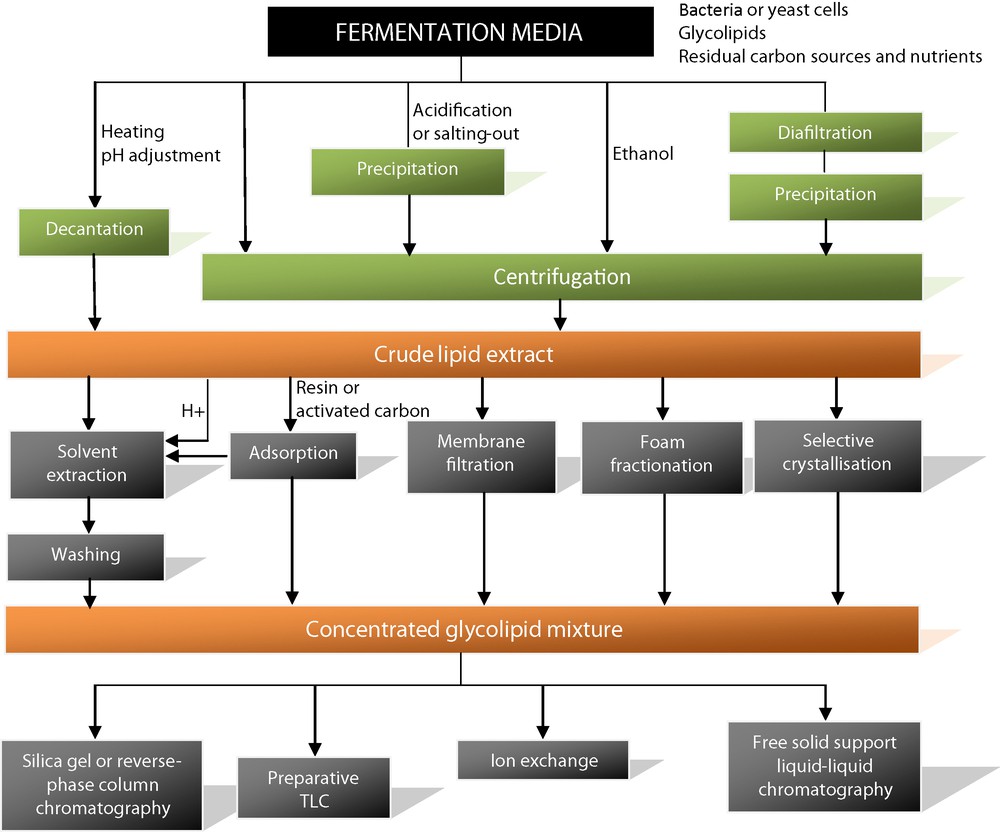
Downstream processes: glycolipid extraction and purification methods.
Adsorption techniques represent another interesting alternative to solvent extraction. This is a continuous process, which consumes only small amounts of solvent. Adsorption methods on resins or activated carbon have been reported for rhamnolipids [23,58]. Adsorption on Amberlite XAD followed by solvent extraction and heating was reported for the recovery of mannosylerythritol lipids with high purity [59]. Membrane filtration [60] or foam fractionation [61] are also useful methods for the extraction of microbial glycolipids after cell separation. Selective crystallization can be applied after precipitation or extraction of glycolipids from culture broths. The lactone forms of sophorolipids crystallize, for example, in phthalate and phosphate buffers, whereas the acidic forms remain soluble in water [62].
3.2 Downstream processes: an overview of glycolipid separation methods (Fig. 2)
Despite many investigations, the separation of individual glycolipids remains difficult due to the strong structural similarities within each class. The molecular species often vary only in the number of sugars, length of the fatty acid chain or degree of saturation. A number of chromatographic methods have been reported for the separation of individual glycolipids after their removal from the fermentation media. Silica-gel or preparative reversed phase column chromatography, for instance, have been described for the purification of sophorolipids and rhamnolipids [63,64]. The separation of the polar sophorolipid acid structures from the less polar lactone forms can be easily achieved, but the purification of the highly diverse individual structures within each group remains quite complex [29]. Another method for rhamnolipids was performed on preparative silica-gel TLC plates with a solvent mixture of chloroform/methanol (9:1 v/v) and subsequently a mixture of chloroform/methanol/water/acetic acid (65:15:2:0.1 v/v) to distinguish the mono- from the disaccharide polar head groups [65]. Silica gel column chromatography was also used to purify mannosylerythritol lipids by gradient elution of a mixture of chloroform/acetone [59,66]. For the separation of trehalose lipids, several solvent systems have been proposed, but the one most extensively used is a mixture of chloroform/methanol (2:1 v/v) [67]. Although these methods allow the purification of milligram quantities of individual compounds, glycolipid purification remains laborious because these molecules generally represent a minor part of the crude fermentation media extracts. This is particularly true for trehalose lipids, which are produced at low concentration in a fermentation media, which often contains a large excess of n-alkanes used as substrates. Other separation methods based on the ionization properties of glycolipids, for example the negative charge of rhamnolipids at high pH values, have also been proposed [50,68]. Ion-exchange columns can be applied to separate glycolipids from larger quantities of culture broths. Other methods based on the specific polarity of the different glycolipid species appear interesting for the fractionation of glycolipids from crude extracts of fermentation medium. Recently, a purification method using colloidal magnetic nanoparticles was applied to different rhamnolipids produced from P. aeruginosa [69]. It can be seen from the abundance and variety of purification and separation techniques found in the literature that current methods are not yet suitable for the industrial-scale purification of microbial glycolipids.
4 Solid support-free liquid-liquid separation systems: an original alternative for the separation and purification of glycolipids
4.1 The technology of support-free liquid-liquid separation
Droplet Counter-Current Chromatography (DCCC), CCC and CPC refer to particular types of support-free liquid-liquid chromatographic systems where solutes are separated on the basis of their partition coefficients between two immiscible solvent systems [70]. These techniques are an excellent alternative to circumvent the problems associated with solid phase adsorbents and to preserve the chemical integrity of mixtures subjected to fractionation.
DCCC relies on the continuous pumping of droplets of a mobile phase through an immiscible stationary phase [71]. Typical DCCC instruments are made of 50–500 vertical thin glass columns interconnected by Teflon tubing. In the 1980s, DCCC was presented as a good strategy for the initial fractionation of crude plant extracts [72]. However, even if the initial sample amount can reach the gram scale for preparative isolation, the separation resolution remains critical and DCCC systems only work at low flow rates (<1 mL/min), what results in poor process productivity. Support-free liquid-liquid separation technologies were initially developed by Martin and Synge in 1946 [73]. CCC was then introduced by Ito in 1970 [74,75] and CPC by Nunogaki in 1982 [76]. In CCC, hydrodynamic instruments are used where the separation takes place in a so-called “multilayer coil” made of a long piece of continuous tubing wrapped around a holder in multiple layers (Fig. 3). The resulting multiple coils can be connected in series to increase the total volume of the instrument. The coil is submitted to a centrifugal force field and rotates around its own axis while simultaneously rotating around the central axis of the system. This results in the retention of the stationary phase and partitioning of the analytes between the two phases. In contrast, CPC uses hydrostatic instruments, which are made of a spinning rotor, comprised of a series of interconnected cells engraved circumferentially on stacked disks in rotation around a single axis. In CPC, the centrifugal force field applied to maintain one phase stationary inside the column remains constant in intensity and direction. In both systems, the mobile phase is pumped through the stationary phase and the equilibrium between both phases depends on the intensity of the centrifugal force (rotation speed), the flow rate of the mobile phase and the physicochemical properties of the biphasic solvent system. These liquid-liquid chromatography techniques are very interesting in terms of selectivity, sample loading capacity and scale-up ability [70,77]. In contrast to other chromatographic techniques, their main advantages are the absence of irreversible adsorption due to the lack of solid support and the risk of analyte degradation. They also allow total recovery of injected samples (up to 100%), and consume low solvent quantities. CPC and High Speed CCC (HSCCC) are the two variants of support-free liquid-liquid chromatographic systems being used today. One of the key points for an efficient CCC or CPC separation relies on the choice of a suitable solvent system, which dissolves and efficiently partitions the analytes between both phases while displaying a sufficient stability at the hydrodynamic equilibrium. When the biphasic solvent system has been optimized, the pumping mode is then chosen. In CPC, when the lower phase is selected as the stationary phase, the experiment is performed in the “ascending mode”. When the lower phase is selected as the mobile phase, the experiment is performed in the “descending mode”. Another interesting aspect of these systems is the ability to operate in dual mode by switching the flow direction and switching the mobile and stationary phases [78]. In addition to the classical elution mode (isocratic or gradient), other development strategies based on displacement methods can be applied. Depending on the nature of the target compound, displacement by pH-zone refining can be a suitable development mode for the separation and purification of compounds whose electrical charge depends on the pH value, while ion-exchange displacement methods are devoted to the purification of ionic solutes. The column capacity of support-free liquid-liquid separation instruments ranges from 200 to 1000 mL for laboratory-scale R&D applications. Pilot and industrial instruments for large-scale applications with column capacity up to 25 L are progressively appearing on the market. Advances in CPC and CCC instrument technologies are currently focused on the improvement of design, scaling-up capacities and operating conditions to guarantee industrial applications with high productivity.
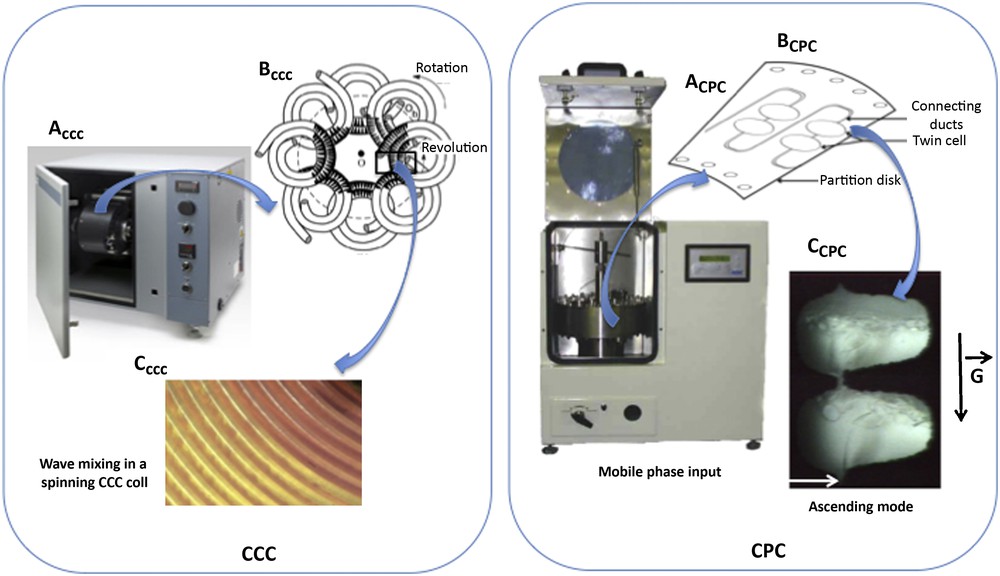
CCC and CPC support-free liquid-liquid separation systems.
4.2 Application of centrifugal partition chromatography and counter-current chromatography to the purification of lipid-related compounds
DCCC, CPC and CCC have been widely used for the extraction and purification of natural products including polyphenols, alkaloids and terpenoids of plant origin [79–82]. (Table 1) In the last few years, CPC and CCC methods have also emerged as promising tools to purify complex lipid mixtures including fatty acids, phospholipids and sphingolipid-derived compounds [16]. Table 1 gives some examples of DCCC, CCC or CPC methods reported for the separation of lipids and related compounds. In the earliest studies, DCCC experiments were generally achieved by eluting two solvent systems of different polarity step by step in the elution mode. This strategy was applied to the fractionation of a human serum lipid extract into neutral lipids, fatty acids and phospholipids [83], or to the fractionation of mouse and whale brain gangliosides into sialosylgangliosides or cerebroside sulfuric esters having the same carbohydrate moiety but different fatty acids and long chain base composition [84]. At that time, the methods were not sufficiently selective to achieve separation between fatty acids having different chain length or degree of unsaturation. Later, CCC and CPC were preferentially optimized to separate polyunsaturated from saturated fatty acid derivatives. In 1988 for instance, ethyl esters of C18:0, C18:1, C18:2, and C18:3 fatty acids were successfully separated from a Borago oil extract by CPC [85]. The ethyl esters of C18:2 and C18:3 were separated in a first elution step in the ascending mode while C18:1 was recovered after switching the system to the descending mode. Lastly, γ-linoleic acid (C18:3) was recovered with 98.3% purity. In another work conducted on an oil extract of the marine phytoplankton Skeletonema costatum, a semi-preparative scale CCC method was developed to eliminate all of the saturated fatty acids in a first elution step and subsequently isolate the mixture of biologically interesting C16:3, C18:4, C20:5 and C22:6 polyunsaturated fatty acids [86]. CPC was also used to separate different classes of glycerol derivatives including mono-acylglycerides, digalactosyl-monoglycerides, monogalactosyl-diglycerides and digalactosyl-diglycerides present in a wheat gluten oil extract for their valorization in cosmetic formulations [87].
Application of CCC and CPC methods for the purification of lipid compounds.
| Method | Sample loading (mass injected) | Solvent system (proportions) | Elution mode | Flow rate (mL/min) | Rotation (rpm) | Reference |
| DCCC of neutral lipids, fatty acids, and phospholipids | Human serum lipid extract (nd) | n-heptane/n-butanol/CHCl3/MeOH:60% AA (3:2:2:3:5 v/v) n-heptane/benzene/CHCl3/ethanol/60% AA (2:3:1:4:3 v/v) | Ascending | nd | nd | Konobu et al., 1976 [83] |
| DCCC of mono- and di-sialosylgangliosides | Crude Glycolipid extract of mouse brain (150 mg) | CHCl3/MeOH/H2O/n-propanol (50:60:40:5 v/v) | Ascending | nd | nd | Otsuka et al., 1981 [84] |
| DCCC of mouse brain sialoylgangliosides | Crude Glycolipid extract of mouse brain (150 mg) | CHCl3/MeOH/0.5% CaCl2/n-propanol (50:60:40:6 v/v) | Ascending | nd | nd | Otsuka et al., 1981 [84] |
| DCCC of polysialosylgangliosides, cerebroside sulfuric esters and neutral cerebrosides | Crude Glycolipid extract of whale brain (150 mg) | CHCl3/methanol/H2O/n-propanol (50:60:40:2 v/v) | Ascending | nd | nd | Otsuka et al., 1981 [84] |
| CPC of unsaturated fatty acid ethyl esters | Borago oil extract (nd) | n-hexane/ACN (1:1 v/v) | 1) Ascending 2) Descending | Murayama et al., 1988 [85] | ||
| Two-step CCC of polyunsaturated fatty acids (143 mL column) | Oil extract of the micro-algae Skeletonema costatum (2.4 g) | n-heptane/ACN 3%/water (first step) n-heptane/MeOH/H2O (second step) (500:415:85 v/v) | Descending | 2 | 850 | Bousquet and Le Goffic, 1995 [86] |
| Two-step CCC of polyunsaturated fatty acids (143 mL column) | Oil extracts of the micro-algae Skeletonema costatum and Isochrysis galbana (nd) | n-heptane/ACN (nd) | Descending | 3 | 720 | Bousquet et al., 2002 |
| CPC of phosphatidylcholine | Crude phospholipid extract of squid (nd) | n-heptane/ethyl acetate/ACN (1/0.65/1 v/v) | 1) Ascending 2) Descending | Baudimant et al., 1996 | ||
| HSCCC of phospholipids (165 mL column) | Hen egg yolk extract (0.75 g) | CHCl3/n-heptane/n-butanol/MeOH/60% AA (2:3:2:3:5 v/v) | Ascending | 1 | 1000 | Shibusawa et al., 2005 [89] |
| CPC of glycerol derivatives including glycosylglycerides and monoacylglycerides (250 mL column) | Wheat gluten oil extract (460 mg) | n-heptane/ethyl acetate/acetone/MeOH/H2O (2:2:2:1:1 v/v) | 1) Ascending 2) Descending | 5 | 1500 | Zeches-Hanrot et al., 2006 [87] |
| CPC of glycerol derivatives including glycosylglycerides and monoacylglycerides (250 mL column) | Wheat gluten oil extract (820 mg) | n-heptane/ethyl acetate/acetone/MeOH/H2O (2:2:2:1:1 v/v) | 1) Ascending 2) Descending | 5 | 1800 | Zeches-Hanrot et al., 2006 [87] |
| CPC of glycerol derivatives including glycosylglycerides and monoacylglycerides (250 mL column) | Wheat gluten oil extract (680 mg) | n-heptane/ACN/n-butanol (60:38:12 v/v) | Ascending | 5 | 1200 | Zeches-Hanrot et al., 2006 [87] |
| CPC of glycerol derivatives including glycosylglycerides and monoacylglycerides (250 mL column) | Wheat gluten oil extract (450 mg) | Hexamethyldisiloxane/ACN/tetrahydrofuran/H2O (9:5:2:0.4 v/v) | Ascending | 5 | 1800 | Zeches-Hanrot et al., 2006 [87] |
| CPC of neutral glycolipids (240 mL column) | 3.2 L of Vero cell culture after incubation with 50 μM n-dodecyl-β-lactoside | CHCl3/MeOH/H2O (5:6:4 v/v) | Descending | 4 | 700 | Kato et al., 2008 [90] |
| CPC of acidic glycolipids (240 mL column) | 1) 3.2 L of Vero cell culture after incubation with 50 μM n-dodecyl-β-lactoside 2) rat brain extract (nd) | n-butanol/ethanol/1% AA (4:1:5 v/v) | Descending | 4 | 1200 | Kato et al., 2008 [90] |
| CPC of mono- and dirhamnolipids (205 mL column) | Rhamnolipid commercial mixture (484 mg) | n-heptane/ethyl acetate/MeOH/H2O (1:1:1:1 v/v) | Ascending | 8 | 1300 | Varnier et al., 2009 [13] |
| CPC of acidic gangliosides (240 mL column) | Rat brain lipid extract pre-purified on a strong anion exchange cartridge (nd) | n-butanol/MtBE/ACN/H2O (3:1:1:5 v/v/v/v) | Descending | 4 | 1200 | Kato and Hatanaka, 2008 [91] |
| HSCCC of acidic gangliosides (320 mL column) | Rat brain lipid extract pre-purified on anion exchange cartridge (nd) | n-butanol/MtBE/ACN/H2O (3:1:1:5 v/v) | Descending | 2 | 1000 | Kato and Hatanaka, 2008 [91] |
| CPC of acidic gangliosides (240 mL column) | Rat brain lipid extract without pre-purification (nd) | n-butanol/MtBE/ACN/H2O (3:1:1:5 v/v) | Ascending | 4 | 1200 | Kato and Hatanaka, 2008 [91] |
| CPC of α-galactosylceramide analogues (250 mL column) | Organic synthesis reaction media after filtration and drying (8 mg) | n-heptane/dichloromethane/ACN/MeOH/H2O (10:5:5:5:1 v/v) | Ascending | 6 | 1300 | Haudrechy et al., 2010 [92] |
Other complex lipid classes such as phospholipids, rhamnolipids or gangliosides have also been successfully separated by CCC or CPC techniques, with sample quantities ranging from milligrams to grams. For example, mono- and di-rhamnolipids were isolated from a crude commercial rhamnolipid mixture by CPC in a classical elution mode [13]. As shown in Fig. 4, the main selectivity criterion during this separation was guided by the number of sugar units, the monorhamnosides being eluted before the di-rhamnosides. Within both groups, the compounds were organized according to their fatty acid chain length.
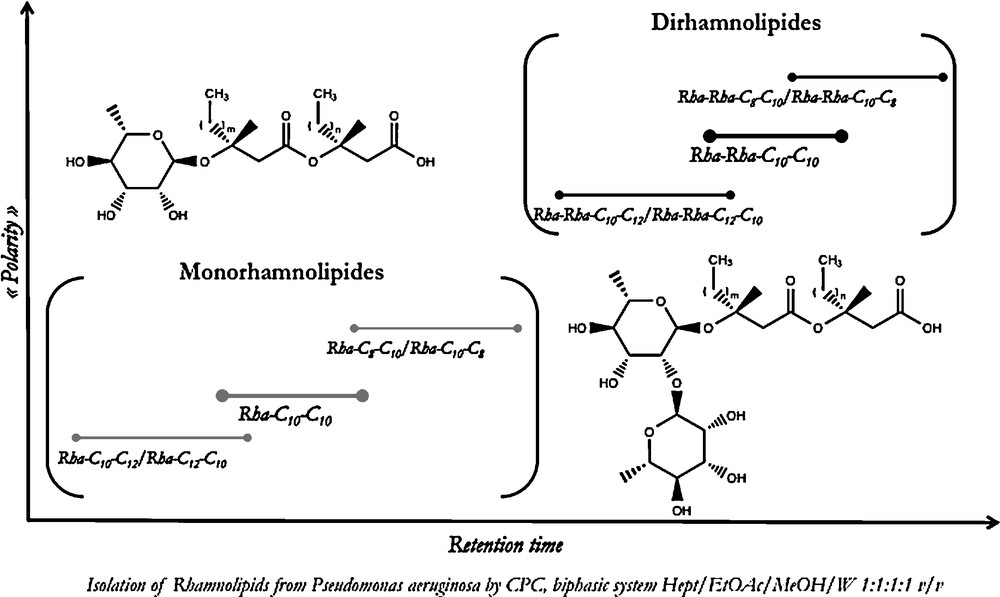
Isolation of rhamnolipids from Pseudomonas aeruginosa by CPC.
In another work, phosphatidylcholine was isolated from a crude lipid extract of squid [88]. More recently, the fractionation of phosphatidylcholine and phosphatidylethanolamine was achieved by high-speedCCC starting from a hen egg yolk extract [89]. Gangliosides and acidic sialylated glycosphingolipids have also been successfully separated from neutral glycolipid analogues starting from a rat brain extract. In this work, a two-step CPC elution method was optimized either by increasing the polarity of the organic phase or by acidifying the solvent system [90]. The acidification resulted in a better solubility of gangliosides in the organic stationary phase, thus leading to better retention of these compounds inside the column and in their step-wise selective elution. More recently, the same research group developed a CPC procedure to separate individual ganglioside analogues from a lipid extract of rat brain [91]. A series of C-glycoside analogues of α-galactosylceramides were also recently separated by CPC [92].
4.3 Centrifugal partition chromatography and counter-current chromatography perspectives for the preparative-scale purification of microbial glycolipids
The data presented in Table 1 demonstrates that support-free liquid-liquid separation techniques provide a wide range of efficient procedures for the purification of lipid compounds, including not only simple lipids, fatty acids or glycerol derivatives but also more complex compounds like phospholipids or glycolipids. The great potential of these strategies relies on the diversity of possible solvent combinations covering a large range of polarities and solubility constants, as well as the easy and simple use of these methods. Depending on the biphasic solvent system, flow rate and rotation speed conditions, CCC and CPC techniques are able to achieve the separation of polar and non-polar natural lipids, glycolipids having different carbohydrate head groups or of fatty acid derivatives differing in their degree of unsaturation or chain length. These techniques also display clear advantages over classical liquid-solid chromatographic techniques in terms of column capacity, sample recovery, solvent consumption and irreversible adsorption risk.
However, the major hurdle in the large-scale purification of microbial glycolipids remains the structural similarities between individual species. Most importantly, the inherent surfactant character of glycolipids appears incompatible with an optimal equilibrium between the two immiscible phases inside the columns. Generally, the polar solvent used in CPC or CCC systems display a high viscosity and a low interfacial tension, which can lead to poor retention of the stationary phase and to a low separation efficiency when introducing amphiphilic species into the system. In Table 1, a variety of solvent systems have been presented as suitable for the separation of complex lipids. Generally, the organic phase contains a non-polar solvent, most often n-heptane, which allows washing of the residual fatty acids present in the initial crude oil extract. The presence of a more polar solvent in the organic phase such as ethyl acetate, chloroform or n-butanol allows the separation of amphiphilic structures differing in their sugar head groups or between fatty acids differing in their degree of unsaturation The aqueous phase is often a highly polar mixture of methanol and water which permits the capture or elution (depending on the elution mode) of the residual carbohydrate substrates or maintains the highly polar compounds inside the aqueous phase. The acidification or salting out of the solvent system can also orientate the solubility of the analytes in a targeted way. At present, CPC and CCC studies have only been performed on the milligram to gram scale of crude lipid extracts with flow rates ranging from 1 mL/min to 8 mL/min (Table 1). It is not yet clear if sample loading and flow rate limitations are directly related to the physicochemical properties of the lipid compounds under examination, or if the CCC or CPC methods reported so far have not totally investigated these aspects. It would be interesting to examine in detail the effect of an increased flow rate or sample loading on the stationary phase retention and on glycolipid separation efficiency.
5 Conclusion
In spite of a long history of glycolipid production from microorganisms such as P. aeruginosa, C. bombicola, and R. erythropolis, research in this area remains dynamic, in particular because of the recent exponential development of white biotechnologies. Although the production of glycolipids in mass markets such as biosurfactants is viable, the development of these molecules in areas with higher added value is hampered by the difficulty of their isolation in pure form. Nevertheless, interesting biological activity has been demonstrated for many glycolipids in the framework of antimicrobial or antitumoral agents. Effective separation and purification tools are thus required to obtain each compound individually and to undertake structure activity relationship studies. The support-free liquid-liquid chromatographic techniques such as countercurrent chromatography and centrifugal partition chromatography appear to be efficient and promising for glycolipid purification mainly because of their preparative character and their ability to purify complex mixtures of homologous compounds.
Acknowledgements
The authors thank the “Région Champagne-Ardenne”, “Agro-industrie Recherches et Développement” and Soliance for financial support. The authors thank also Laurence Grare, Emilien Mathiot and Anthony Bresin from Agro-industrie Recherches et Développement for providing rhamnolipid extracts and for fruitful discussions.