

1 Introduction
The layered solids herein studied are obtained by the assembling of the planar anionic block [Ni(CN)4]2− through a transition metal ion (T2+) which links neighboring blocks at their N ends. In the as-synthesized material, the available coordination sites on the T metal centers found at the layer plane are occupied by coordinated water molecules which, in turn, serve to stabilize additional water molecules in the interlayer region through hydrogen bonding interactions. From this fact, the layer–layer distance and the material crystal structure depend on the amount of water molecules found in that region and on the resulting hydrogen bonding interactions among them. This suggests that in such a system of layered solids, the water content modifies the host structure resulting in different phases. This possibility is explored in this study where for a given T metal three different phases were found. The crystal structure of these phases was resolved and refined from X-ray powder diffraction (XRD) data. The structural information was complemented with infrared (IR), UV–vis, Raman and thermogravimetric (TG) data.
Within this family of layered solids, structural information is available [1–5] for some T metals. For T = Fe, Co and Cd, a phase containing six water molecules per formula unit, T[Ni(CN)4]·6H2O, which crystallizes with an orthorhombic unit cell (Pnma space group) has been documented and their crystal structures refined [1–4]. For T = Ni, the existence of three different phases, named K, L0 and L1, has been reported [5], all of them crystallizing with an orthorhombic unit cell of different cell edges. To the best of our knowledge, the crystal structure of these three phases remains unknown. No additional information is available about the members of the studied series of layered compounds.
The existence of available coordination sites on the layer plane for this series of solids has stimulated the incorporation of pillar spacers between neighboring layers to obtain 3D porous materials of tailored channels dimension and geometry [6], which have been evaluated for hydrogen storage [7,8]. With pyrazine as pillar molecule, the obtained solids show unique structural features at room and low temperatures [9–11]. Related to the ability of the iron(2 + ) atom to form both high and low spin complexes, the series Fe1−xTx[M(CN)4].pyz where T = Co, Ni and M = Ni, Pd, Pt, has been intensively studied in order to understand the nature of its spin-crossover behavior, particularly the pronounced hysteresis loop that there is observed [12–16]. In the case of monodentate NH3 or bidentate α,ω-diaminoalkanes as auxiliary ligands coordinated to the transition metal ion (T2+), the layered materials belong to a family of compounds known as Hofmann-type clathrates with a high ability for the organic molecules sequestering [17]. The herein reported structural study for the series T[Ni(CN)4]·xH2O with T = Mn, Co, Ni was stimulated by the renewed interest on molecular materials, their properties and potential applications.
2 Experimental
The materials under study were prepared by mixing aqueous solutions (0.01 M) of K2[Ni(CN)4] and the sulfate of the T metals involved (Mn2+, Co2+, Ni2+). The resulting precipitates were aged for 24 h within the mother liquor and then isolated by centrifugation. The solid fraction was repeatedly washed with distilled water to remove all the accompanying ions and then air-dried until constant weight. Details on the conditions used to obtain the different phases are discussed below. The nature of the obtained powders as tetracyanonickelate (II) salts was corroborated from IR and Raman spectra. The metal atomic ratio in the obtained powders was established from X-ray dispersed-energy spectroscopy (EDS) analyses. The materials thermal stability and their hydration degree were evaluated recording TG curves. All the reagents used were analytical grade from Sigma-Aldrich.
The TG curves were run using a high resolution TA Instrument (Hi-ResTM) thermo-gravimetric analyzer TGA Q 5000. IR spectra were collected by the KBr pressed disk technique using an FT spectrophotometer (Spectrum One from Perkin-Elmer). Raman spectra were collected in the 3500–100 cm−1 frequency range, an Almega XR Dispersive Raman spectrometer equipped with an Olympus microscope (BX51) was used to obtain the Raman spectra. The excitation source was 532 nm radiation from a Nd:YVO4 laser (frequency-doubled) and the incident power at the sample was of ∼ 2.5 mW. UV–vis spectra were recorded with a Perkin-Elmer spectrometer using the integration sphere method.
The XRD powder patterns were recorded in the Bragg–Brentano geometry using CuKα radiation and a D8 Advance diffractometer (from Bruker). High-resolution XRD patterns were collected at XPD-10B beamline of the LNLS synchrotron radiation facility (Campinas, Brazil). The experimental details are available from Supplementary information. The crystal structures were solved ab initio by direct methods using the program SHELXS [18] from extracted intensities according to the Le Bail method [19]. Physical considerations and information from the remaining techniques were used in order to select the appropriate structural models to be refined, and then to check the obtained structure. The structural refinement from these XRD powder patterns was performed with the Rietveld method using the FullProf program [20] and pseudo-Voigt peak shape function. Peak profiles were calculated within 10 times the full width at half maximum (FWHM). The background was modeled by a third-order polynomial. The interatomic C–N and Ni-C distances were constrained to take values within certain limits considering results from single crystal studies of Fe, Co and Cd tetracyanonickelates [1–4].
3 Results and discussion
3.1 Nature of the solids to be studied
Without exception, a 1:1 T:Ni metals ratio was found from the recorded EDS spectra. In addition, the obtained IR and Raman spectra show the set of ν(CN), ν(NiC) and δ(NiCN) vibrations characteristic of metal tetracyanonickelate (II) [9–11]. Fig. 1 shows typical IR spectra for the studied series where such vibrations are observed. The formation of the –CN–T–NC– coordination bond is easily sensed by the shift for the ν(CN) vibration, from 2123 cm−1 in K2[Ni(CN)4] to 2152, 2160 and 2168 cm−1, for T = Mn, Co, Ni, respectively. In these spectra, the presence of both coordinated and weakly bonded water molecules is sensed by the appearance of narrow and broad ν(OH) stretching bands above and below 3500 cm−1, respectively. For the δ(HOH) vibration, two bands or a broad one are observed corresponding to two types of crystal water molecules. From the recorded XRD powder patterns (Fig. 2), two orthorhombic and a tetragonal phases were identified; their unit cell parameters and space group are available from Table 1. According to their unit cell edges, two of these three phases (the orthorhombic ones) coincide with those proposed for T = Ni [5]. The labels of K, L0 and L1 given for the Ni phases [5] will be maintained for the analogues of Mn and Co. The amount of water molecules within neighboring layers was determined from the recorded TG curves. Fig. 3 shows representative TG curves for the considered series of solids; K, L0 and L1 phases crystallize with four, six and three water molecules per formula unit. The evolution on heating of these water molecules depends on the T metal involved and on the phases formed. This last behavior suggests that the crystal structure is related to the interactions between water molecules (discussed below). The recorded UV–vis spectra complete the required information on the nature of the studied series of solids. Fig. 4 shows the spectra for the sub-series Ni(H2O)2[Ni(CN)4]·xH2O. In these spectra, peaks below 300 nm denote the presence of a Ni atom with square planar geometry; meanwhile peaks above 300 nm are associated with a Ni atom in a pseudo-octahedral coordination [5]. According to this information, water molecules are only coordinated to the bridge metal (T), but they show slight electronic differences, nonetheless. Regarding the same composition, visible region denotes the main differences among the three different external metallic centers. From Supplementary information, a comparison of UV–vis spectra for the hexa-aquo composition T[Ni(CN)4]·6H2O, for M = Ni, Co, Mn is available. In the UV region, all spectra display charge transfer bands previously assigned to the common fragment [Ni(CN)4]2− [5]. In accordance with a high spin configuration d5 in an octahedral environment, transitions for Mn2+ are spin and orbital forbidden, which results in no bands within the visible region. A d7 configuration for Co2+ has three spin-allowed transitions; two of them are visible in the spectrum and are within the normal range for reported cobalt compounds in octahedral environment. The first transition (ν1) is a weak and broad band in the near IR at 1092 nm, assigned to the 4T2g ←4T1g(F) transition. The more energetic 4T1g (P)←4T1g(F) transition (ν3) appears at 494 nm [21]. The ν2 transition appears at slightly higher energy, usually as a shoulder for that intense band. The absorption spectrum for octahedral nickel center (Ni2+,d8) is discussed below (Section 3.4).
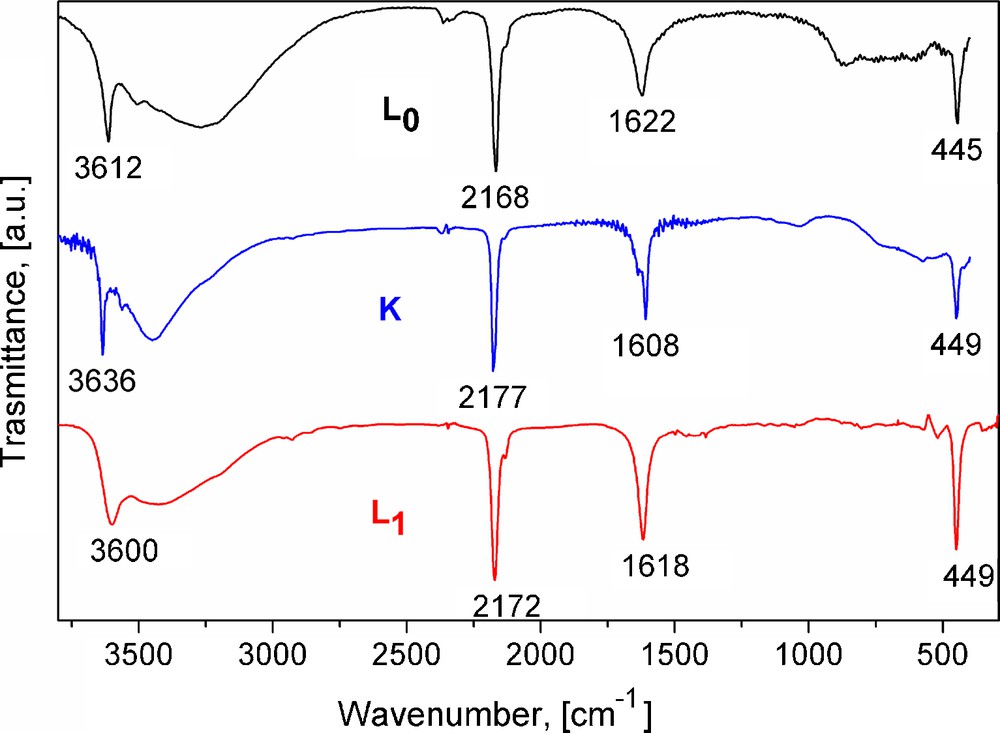
IR spectra for L0, K and L1 phases of Ni(H2O)2[Ni(CN)4]·xH2O. In the IR spectra, the structural differences for the three orthorhombic phases are appreciated. Analogue spectra were obtained for T = Mn, Co.
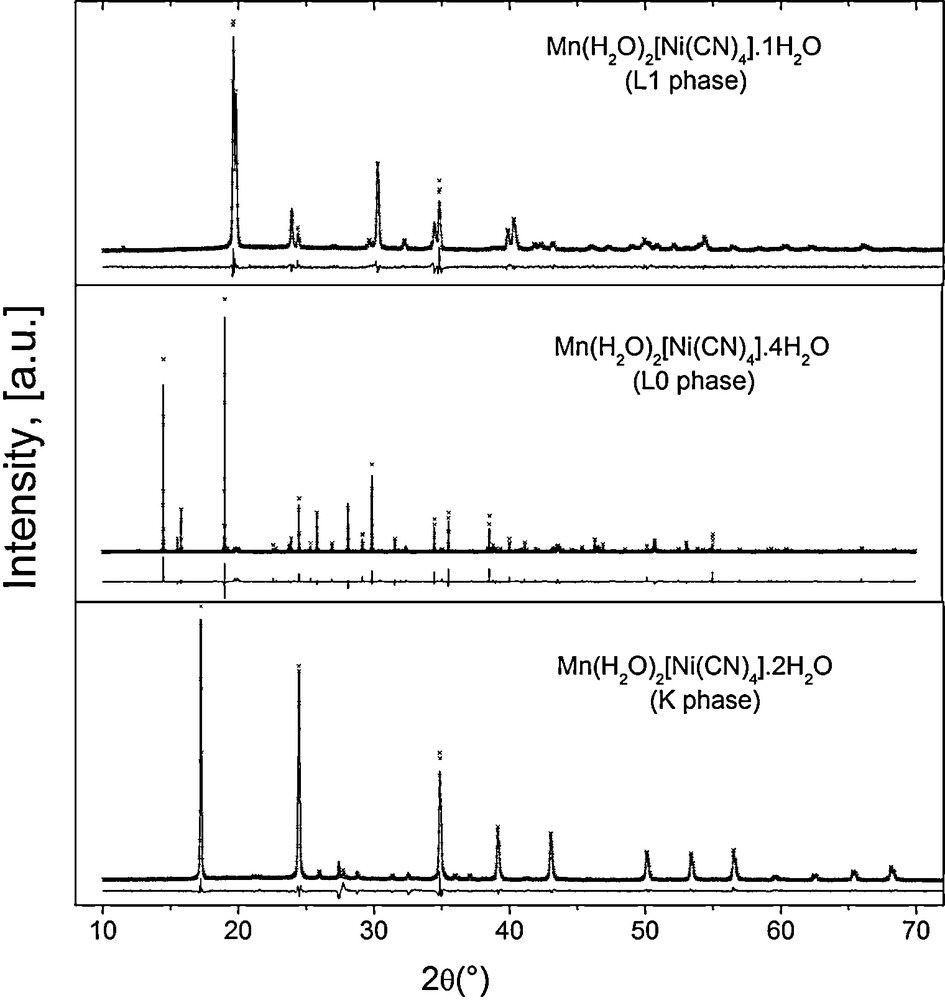
XRD powder patterns for Mn(H2O)2[Ni(CN)4]·xH2O, corresponding to K, L0, and L1 phases. Analogue patterns were obtained for T = Co, Ni.
Unit cell edges (in Å), cell volume (Å3) per formula unit (VZ) and space group for the series T(H2O)2[Ni(CN)4]·xH2O with T = Mn, Co, Ni.
| Phase | Mn | Co | Ni |
| K | a = b = 7.279(2) | a = b = 7.182(3) | a = b = 7.145(2) |
| c = 10.291(2) | c = 10.139(2) | c = 10.116(2) | |
| VZ = 272.63 | VZ = 261.50 | VZ = 258.21 | |
| P4/mmm | P4/mmm | P4/mmm | |
| L0 | a = 12.304(4) | a = 12.195(2) | a = 12.207(4) |
| b = 14.125 (6) | b = 13.885(3) | b = 13.860(5) | |
| c = 7.308(6) | c = 7.143(3) | c = 7.124(3) | |
| VZ = 317.52 | VZ = 302.38 | VZ = 301.32 | |
| Pnma | Pnma | Pnma | |
| L1 | a = 7.303(2) | a = 7.115(2) | a = 7.091(4) |
| b = 14.539(2) | b = 14.264(3) | b =14.135(3) | |
| c = 9.044(2) | c = 8.898(1) | c = 8.876(5) | |
| VZ = 240.07 | VZ = 225.76 | VZ = 222.26 | |
| Imma | Imma | Imma |
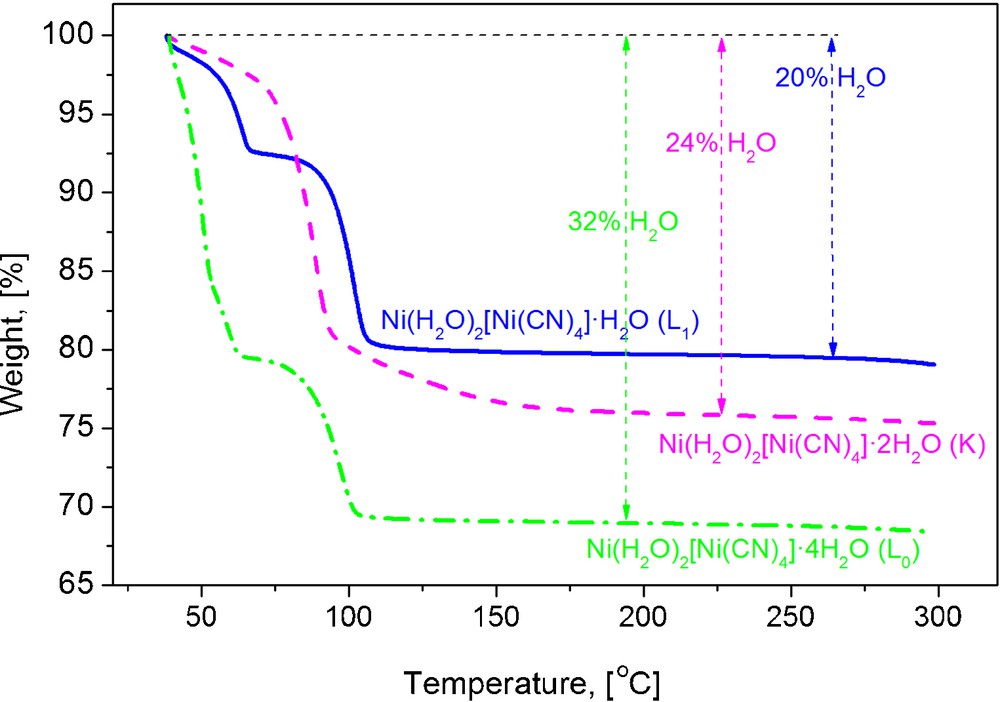
TG curves for Ni(H2O)2[Ni(CN)4]·xH2O. The weight loss for the dehydration region corresponds to the evolution of four, six and three water molecules per formula unit; for the phases K, L0 and L1, respectively.
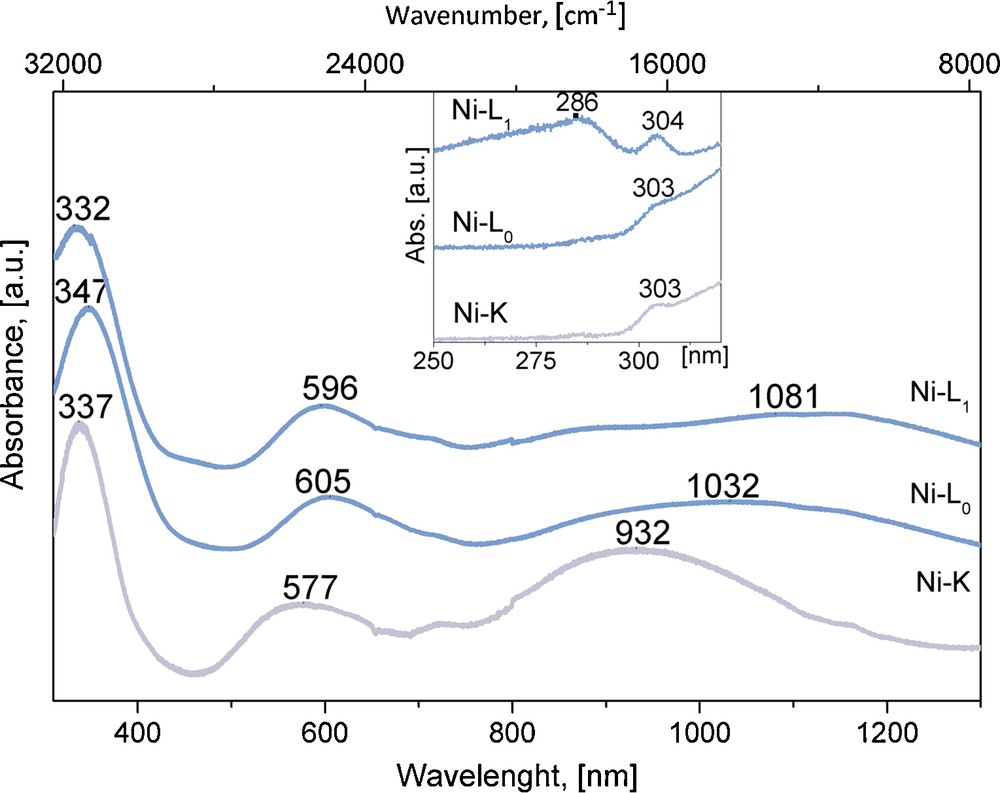
UV–vis spectra for Ni(H2O)2[Ni(CN)4]·xH2O. These spectra correspond to square and octahedral coordination for Ni and T metals, respectively.
3.2 Crystal structures
The crystal structure for K, L0 and L1 phases were refined in the space groups P4/mmm, Pnma, Imma, respectively. The refined atomic positions and occupation and thermal factors, and the calculated interatomic distances and bond angles are available from Supplementary information. This structural information was also deposited at ICSD database with file numbers indicated below. The relevant interatomic distances and bond angles are summarized in Table 2. For K phase, the structure is formed by flat T(H2O)2[Ni(CN)4] layers; the T–N–C angle is 180° (Fig. 5). The two additional water molecules per formula unit are found linked to the coordinated ones. The oxygen–oxygen (OC–OHB) distance between these types of water molecules is, as average, 3.60 Å (Table 2), a value characteristic of weak hydrogen bonding type interactions [22]. These weak hydrogen bonding bridges link coordinated water molecules from neighboring layers contributing to the solid stability.
Relevant bond distances and angles for the series of layer solids under study.
| Phase | OC–OHB1 (Å) | OC−OHB2 (Å) | T–N–C angle (°) |
| K (Mn) | 3.64(2) | – | 180 |
| L0 (Mn) | 2.78(5) | 2.89(4) | 166.0(3) |
| L1 (Mn) | 3.19(2) | – | 169.8(4) |
| K (Co) | 3.60(3) | – | 180 |
| L0 (Co) | 2.79(5) | 2.82(4) | 169.2(2) |
| L1 (Co) | 3.19(2) | – | 174.9(4) |
| K (Ni) | 3.57(2) | – | 180 |
| L0 (Ni) | 2.53(2) | 2.91(1) | 167.4(2) |
| L1 (Ni) | 3.16(4) | – | 171.8(4) |

Framework for K phase. Coordinated water molecules from neighboring layers remains linked through aquo bridges (hydrogen bonding interactions).
For L0 phase, the layers T(H2O)2[Ni(CN)4] adopt a wave-shaped conformation related to the alternating linking of their coordinated water molecules by non-coordinated waters (Fig. 6). The oxygen-oxygen distance, OC–OHB1, corresponding to these aquo bridges is, as average, 2.71 Å (Table 2). Relatively weakest aquo bridges, <OC–OHB2> = 2.87 Å, are found which are linking coordinated water molecules from neighboring layers (Fig. 6, Table 2). These last aquo-bridges maintain the layers together. The layers stacking along the c axis appears as in-phase waves. This phase (L0) has two additional weakly bonded water molecules stabilized within the interlayer region through weak hydrogen bonding interactions. The weakly bonded water molecules found in the interlayer region adopt a 2D ice-like structure of edge-shearing hexagons formed of three connected oxygen atoms (Supplementary information). Such 2D ice-like network has been already observed for the cadmium analogue, Cd(H2O)2[Ni(CN)4]·4H2O [4].
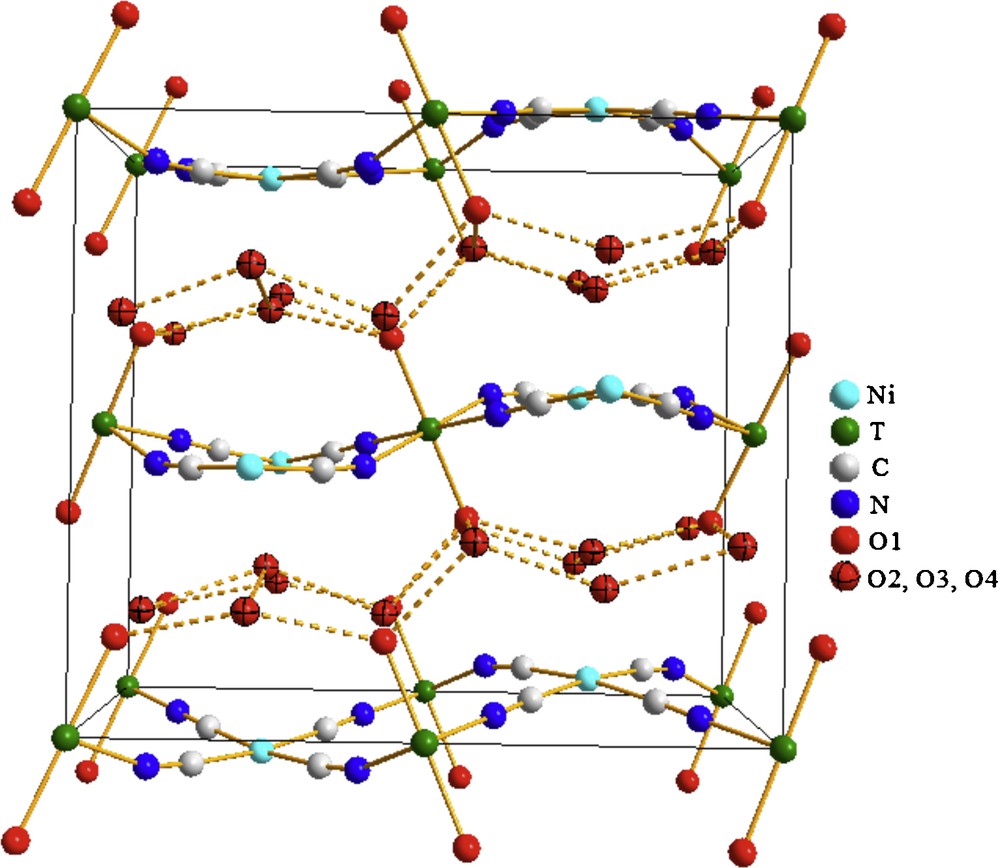
Framework for L0 phase. Coordinated water molecules from a same layer and from neighboring layers remain linked through aquo bridges (hydrogen bonding). Along the c axis, the layers are stacked according to in-phase waves.
For L1 phase, T(H2O)2[Ni(CN)4]·H2O, the non-coordinated water molecule is found forming bridges among coordinated waters from a same layer. These bridges appear alternated at both sides of a given layer (Fig. 7). This leads to a weave-shaped layers configuration, similar to that already observed for L0 phase but, in order to minimize the free volume for the interlayer region the layers adopt an out-of-phase weaves configuration when they are observed along the c axis. The average OC–OHB distance for the aquo bridges is 3.15 Å, significantly larger than the value observed for the analogous interaction in L0 phase (2.71 Å) (Table 2). The 2D ice-like network observed in L0 phase polarizes the negative charge of OHB atoms in the bridges and this induces a higher ability for their protons for the hydrogen bonding interactions with the coordinated water molecules. This results into the observed smaller OC–OHB distance for L0 phase.

Framework for L1 phase. Coordinated water molecules from different layers remain linked through aquo bridges. Along the c axis, the layers are stacked according to out-of-phase waves in order to minimize the interlayer region free space.
The cell volume per formula unit for the three phase follows the order Mn > Co > Ni (Table 2). That order corresponds to a negative correlation with the metal polarizing power (Ze/r2): Mn (3.282) < Co (3.652) < Ni (3.858) [23]. Such behavior finds explanation in the metal interaction with its coordinated water molecules. A highly polarizing metal, e.g. Ni, subtracts a high amount of charge from the oxygen atom of coordinated waters and this enhances the ability of their protons to form hydrogen bridges with non-coordinated water molecules reducing the OC–OHB distance, which is effectively observed from the refined crystal structures (Table 2). The net result of such an induced effect is the observed negative correlation for the cell volume; an increase for the metal polarizing power leads to a more compact unit cell. The role of the metal polarizing power on the coordination bond with the water molecules and on the network of aquo bridges is also sensed by the material dehydration temperature and the IR and Raman spectra (discussed below).
3.3 Thermal behavior on heating
L0 and L1 show an analogue behavior on heating. The non-coordinated water molecules evolve below 60 °C and then up to 100 °C the coordinated ones are also liberated (Fig. 3). The obtained anhydrous solid then remains stable up to above 300 °C when the decomposition of the T[Ni(CN)4] layers is detected. The inflexion observed at 50 °C in the TG curve of L0 reveals the existence of two slightly different types of weakly bonded water molecules; those in the 2D-ice like structure and the ones forming aquo bridges between coordinated water molecules.
The behavior on heating for K phase is quite different; both coordinated and non-coordinated water molecules evolve together in a single step. It seems, the disrupting of the weak aquo bridges between coordinated water molecules destabilizes the framework and this favors the material dehydration as a continuous process. For about 95 °C, practically all the crystal water has abandoned the solid and then a smooth weight loss up to 150° is observed which is probably related to the loss of water molecules trapped in the collapsed layered network. According to the weight loss profile for L0 and L1 phases, the aquo bridges between coordinated water molecules from different layers are more stable than those that are linking water molecules from a same layer. Probably the thermal excitation of the lattice vibrational states for these phases favors the water bridges disrupting at relatively low temperature because the presence of these bridges are forcing the layers to adopt the wave-shaped configuration. It seems, for flat layers of K phase, the excitation of vibration states has a less pronounced effect on the flexible lateral aquo bridges that are connecting the coordinated water molecules along the c axis (Fig. 5). This could also explain the evolution of both non-coordinated and coordinated water molecules for K phase in a single step.
For different metals (Mn, Co, Ni) and a same phase, the temperature of evolution of both weakly bonded and coordinated water molecules follows the order Ni > Co > Mn (Supplementary information). This is related to the above-discussed role of metal polarizing power on the strength of the metal-water bonding and its induced effect on the network of hydrogen bonding interactions.
The network of aquo bridges formed in the interlayer region has a decisive role in the long range crystalline order observed in the studied series of 2D solids; if the crystal water is removed a solid amorphous to XRD results. When the solid is rehydrated, the long-range order is only partially restored.
3.4 Nature of the observed fine structure in the IR, Raman and UV–vis spectra
The frequency for the ν(CN) vibrations observed in IR and Raman spectra is an indirect sensor for the metal interaction with its coordinated water molecules and also for the bridge formation between water molecules. The metal T bonding to the CN ligand at the N end takes place through the ligand 5σ orbital which has certain anti-bonding character for the C≡N bond. From this fact, an increase for the metal (T) polarizing power leads to an increase for ν(CN) frequency. When the metal subtracts charge from a ligand like the water molecule, a decrease for the ν(CN) frequency is observed. Likewise, a variation in the electron density on the oxygen atom of the coordinated water molecule, by its participation in hydrogen bonding interactions, for instance, is sensed as a small but perceptible variation for the frequency of the ν(CN) vibration. Such mechanism explains the frequency difference observed for the ν(CN) vibration in K, L0 and L1 phases (Fig. 1). The higher frequency corresponds to K phase where the non-coordinated water molecules participate of relatively weak hydrogen bonding interactions with the coordinated ones. The low ν(CN) frequency observed for L0 is related to the 2d-ice like network linked to the coordinated water molecules. An analogue behavior is observed from the Raman spectra (Supplementary information).
UV–vis spectra also show a fine structure; for a same T metal the absorption bands are observed at slightly different wavelength (Fig. 4). In the UV region, all spectra display charge transfer bands previously assigned to the common fragment [Ni(CN)4]2− [5]. It is in the visible region where marked differences arise, as a result of quite distinct environment at the pseudo-octahedral NiN4(OH2)2 chromophore. Among the three spin-allowed transitions, the most sensitive one is the 3T2g←3TA2g(F) transition; which in the ligand field approach for octahedral complexes, corresponds to the 10Dq parameter. This transition senses the energetic difference between basal and the first metal excited states. Under this model, the 3T2g←3TA2g(F) transition from K phase at 932 nm corresponds to the phase with the highest gap, followed by Lo (1032 nm); the smaller gap is found for L1 (1081 nm). However, hydration degree stands not as the key factor to the energetic levels distribution found in this series. Rigidity in layers seems to be preponderant feature in determining the possible excited states, since K phase display a narrower band at lower wavelength. Related to the wave-shaped conformation adopted by the layer in Lo and L1 phases, transitions to excited states are possible in a greater extent. Broader bands assigned to the 3T2g←3TA2g(F) transition suggest a greater orbital mixing than in K phase, which in turn decreases the energy gap. This hypothesis is supported by the average distances for octahedral T center, (T–N and T–O) previously discussed, which show that the K phase presents the most distorted octahedron and thus the maximum gap; while L1 phase shows the most regular octahedron among this series and thus the minimum gap. The remaining two bands in the visible region are assigned to the 3T1g(F)← 3TA2g(F) and 3T1g(P)←3TA2g(F) spin-allowed transitions for an octahedral metal center. Their behavior shows no clear trend.
3.5 Formation conditions for the different phases
The K phase was only obtained under hydrothermal conditions at about 70 °C and heating time of about 72 h. This agrees with the above mentioned probably temperature role to disrupt the aquo bridges between coordinated water molecules from a same layer (phases L0 and L1). L1 phase is obtained by precipitation at room temperature and then the solid washed and dried without prolonged aging. L0 modification is formed also by precipitation but under stirring and then aging the precipitate for a week within the mother liquor. It seems, the stirring and aging processes favor the incorporation of a greater amount of water molecules into the interlayer region and the 2D ice-like network formation. This phase (L0) was detected in powders obtained from the non-stirred precipitate aged for weeks within the mother liquor. From these facts, K and L0 appear to be the high and room temperature thermodynamic products, respectively; while L1 is an intermediate kinetic product.
4 Conclusions
For the series T(H2O)2[Ni(CN)4]·xH2O with T = Mn, Co, Ni, the formation of three structural modifications was observed, two of them correspond to room and high temperature thermodynamic products, T(H2O)2[Ni(CN)4]·4H2O (L0, Pnma) and T(H2O)2[Ni(CN)4]·2H2O (K, P4/mmm), respectively; and an intermediate room temperature kinetic product, T(H2O)2[Ni(CN)4]·H2O (L1, Imma). The crystal structure for these three phases was solved and then refined from XRD powder patterns using the Rietveld method. The refined crystal structures were supported by the recorded TG curves and IR, Raman, and UV–vis spectra. All the spectral signals show a fine structure related to the nature of the aquo bridges formed between the water molecules found in the interlayer region.
Acknowledgements
This study was partially supported by the Projects SEP-CONACyT-2009-01-129048 and 2010-01-155413. The authors thank Dr. N. Barba-Behrens for the experimental facility used in the UV–vis spectra recording.
Appendix A Supplementary data
Structural information derived from the crystal structures refinement has also been deposited at ICSD Fachinformationszentrum Karlsruhe (FIZ) (E-mail: crysdatafiz-karlsruhe.de) with ICSD file numbers: 422110: Mn(H2O)2[Ni(CN)4]·2H2O; 422112: Mn(H2O)2[Ni(CN)4]·4H2O; 422115: Mn(H2O)2[Ni(CN)4]·H2O; 422109: Co(H2O)2[Ni(CN)4]·2H2O; 422114: Co(H2O)2[Ni(CN)4]·H2O; 422111: Ni(H2O)2[Ni(CN)4]·2H2O; 422113: Ni(H2O)2[Ni(CN)4]·4H2O; 422116: Ni(H2O)2[Ni(CN)4]·H2O.