

1 Introduction
Noncovalent interactions such as hydrogen bonding, π–π interactions, Van der Waals contacts and other weak forces play crucial roles in several fields, such as supramolecular chemistry, molecular recognition, biochemistry and materials science [1–3]. Many research efforts have focused on systematically studying a variety of noncovalent forces to observe the interesting cooperative effects in the formation and function of supramolecular architectures. As is well known, O − H···O/N, C − H···O/N, C − H···π interactions and aromatic π-stacking interactions are used to construct a range of supramolecular architectures, because of their strength, selectivity and directionality [4]. More importantly, highly ordered molecular assemblies into chains, sheets and networks as efficient strategies in achieving desired supramolecular architectures are of current interest [5,6].
Schiff-base compounds have been extensively investigated over the past few decades, and therefore attracted considerable attention due to their intriguing properties and the potential applications, such as catalysis, magnetics, nonlinear optics and drug design [7–11]. The bidentate Schiff-base ligands with —CR = N–N = CR— acceptor groups have been reported to generate supramolecular systems with interesting host-guest chemistry [12]. The bidentate and multidentate Schiff-base ligands with terminal pyridyl groups have also been investigated to generate coordination polymers with novel network patterns, and the multidentate Schiff-base ligands have contributed to one-dimensional metal-organic nanometer tube which are further linked together through weak C − H···X bonds [13]. Considering the ligand structure features, salicylaldazines are suitable candidates for investigating the possibilities for intramolecular hydrogen bonding and intramolecular proton transfer process between two groups, due to the high degree of conjugation and the presence of OH groups close to the azine nitrogens [14]. Our research interest has focused on investigating the nature of noncovalent interactions in determining the supramolecular structures of new benzaldazine derivatives and clarifying the relationship of the corresponding structure and fluorescent properties. Herein we wish to report synthesis and characterization of three compounds 1–3 with the general formula Ar1(CH = N–N = HC)Ar2 (Ar1 = Ar2 = 2-OH-3,5-tBu2C6H2 (1), Ar1 = Ar2 = 2-BrC6H4 (2), Ar1 = ortho-C6H4(NHC6H3-Me2-2,6), Ar2 = 2-FC6H4 (3)). The luminescent properties of these benzaldazine derivatives 1–3 were studied both in solution and in the solid state. The effect of the functional groups on molecular packing and fluorescent properties in the solid state and solution with different solvents was also investigated.
2 Experimental
2.1 General
All solvents and reagents were commercially obtained and used as received. 3,5-Di-tert-butyl-2-hydroxybenzaldehyde was synthesized according to the literature method [15]. 1H NMR and 13C NMR spectra were measured using a Varian Mercury-300 NMR spectrometer. IR spectra were recorded on a Nicolet Impact 410 FTIR spectrometer using KBr pellets. The elemental analyses were performed on a Perkin-Elmer 2400 analyzer. UV–vis absorption spectra were recorded on an UV-3100 spectrophotometer. Fluorescent measurements were carried out on a RF-5301PC. All melting points were determined by an X-5 micr-melting point apparatus and are uncorrected.
2.2 Synthesis of 3,5-di-tBu-2-OHC6H4(CH = N–N = HC)C6H4-OH-2-tBu-di-5,3 (1)
3,5-di-tert-butyl-2-hydroxybenzaldehyde (0.40 g, 2.00 mmol) was added to hydrazine hydrate (0.05 g, 1.00 mmol) in 5 mL ethanol. The resulting bright yellowish mixture was refluxed for 2 h. Upon cooling the reaction mixture to room temperature, yellow solids were precipitated. The obtained solid product was washed with cold ethanol, then dried in vacuo to give the desired product in excellent yield. Yield: 0.45 g (96%). M.p. 219.2–221.4 °C. Anal. Calcd. for C30H44N2O2 (464.68): C 77.54, H 9.54, N 6.03. Found: C 77.48, H 9.62, N 6.10. 1H NMR (300 MHz, CDCl3): δ 11.89 (s, 2H), 8.76 (s, 2H), 7.46 (d, J = 2.0 Hz, 2H), 7.17 (d, J = 2.0 Hz, 2H), 1.47 (s, 18H), 1.32 (s, 18H) ppm. 13C NMR (75 MHz, CDCl3): δ 165.2, 156.8, 141.3, 136.9, 128.2, 126.9, 116.7, 31.4, 29.4 ppm. IR (KBr, cm−1): υ 2960, 1623, 1592, 1439, 1390, 1362, 1251, 1201, 1173, 963, 717.
2.3 Synthesis of ortho-C6H4Br(CH = N–N = HC)C6H4Br-ortho (2)
Similarly, 2-bromo-benzaldehyde (0.37 g, 2.00 mmol) and hydrazine hydrate (0.05 g, 1.00 mmol) in ethanol afforded compound 2 as yellow solid. Yield: 0.34 g (94%). M.p. 173.2–173.4 °C. Anal. Calcd. for C14H10N2Br2 (366.05): C 45.94, H 2.75, N 7.65. Found: C 45.98, H 2.82, N 7.54. 1H NMR (300 MHz, CDCl3): δ 9.02 (s, 2H), 8.23 (d, J = 1.8 Hz, 2H), 8.20 (d, J = 1.8 Hz, 2H), 7.42–7.29 (m, 4H) ppm. 13C NMR (75 MHz, CDCl3): δ 161.4, 133.2, 132.7, 132.4, 128.6, 127.6, 125.8 ppm. IR (KBr, cm−1): υ 1613, 1559, 1431, 1316, 1270, 1024, 953, 751, 640, 439.
2.4 Synthesis of ortho-C6H4F(CH = N–N = HC)C6H4(NHC6H3-Me2-2,6)-ortho (3)
Compound ortho-C6H4F(CH = N–N = HC)C6H4F-ortho was synthesized by the similar procedure as compound 1. Hydrazine hydrate (0.05 g, 1.00 mmol) and 2-fluoro-benzaldehyde (0.25 g, 2.00 mmol) in ethanol afforded compound ortho-C6H4F(CH = N–N = HC)C6H4F-ortho as yellow solid. Yield: 0.24 g (98%). M.p. 132.3–132.8 °C. Anal. Calcd. for C14H10N2F2 (244.24): C 68.85, H 4.13, N 11.47. Found: C 68.94, H 4.25, N 11.30. 1H NMR (300 MHz, CDCl3): δ 8.93 (s, 2H), 8.13 (t, J = 6.9 Hz, 2H), 7.48–7.42 (m, 2H), 7.23–7.10 (m, 4H) ppm. 13C NMR (75 MHz, CDCl3): δ 158.8, 155.4, 150.6, 150.6, 127.8, 127.7, 122.5, 122.5, 119.3, 119.2, 116.6, 116.4, 110.9, 110.6 ppm. IR (KBr, cm−1): υ 1627, 1483, 1457, 1233, 1096, 959, 814, 753, 653, 462.
A solution of n-BuLi (1.40 mL, 1.90 mmol) in n-hexane was added to a solution of 2,6-dimethylaniline (0.22 mL, 1.80 mmol) in THF (20 mL) at 0 °C. The mixture was allowed to warm to room temperature and stirred for additional 2 h. The resulting solution was transferred into a solution of ortho-C6H4F(CH = N-N = HC)C6H4F-ortho (0.40 g, 1.60 mmol) in THF (10 mL) at 25 °C. After stirring for 2 h, the reaction was quenched with H2O (20 mL). The layers were separated and the water layer was washed with Et2O (2 × 30 mL). The combined organic phase was dried over anhydrous MgSO4 for 2 h, and filtered. The organic phase was evaporated to dryness to give the crude product as brown oil. The crude product was purified by column chromatography on silica gel with ethyl acetate/petroleum ether (1:5 in volume) as eluent to give the pure product as yellowish crystals. Yield: 0.48 g (85%). M.p.: 117.6–118.3 °C. Anal. Calcd. for C22H20N3F (345.41): C 76.50, H 5.84, N 12.17. Found: C 76.42, H 5.90, N 12.13. 1H NMR (300 MHz, CDCl3): δ 9.80 (s, 1H), 8.89 (s, 1H), 8.86 (s, 1H), 8.13 (t, J = 6.6 Hz, 1H), 7.50–7.33 (m, 2H), 7.24–7.05 (m, 6H), 6.72 (t, J = 7.4 Hz, 1H), 6.26 (d, J = 8.3 Hz, 1H), 2.24 (s, 6H) ppm. 13C NMR (75 MHz, CDCl3): δ 166.1, 153.8, 136.5, 134.4, 132.4, 132.2, 132.1, 128.2, 127.4, 127.3, 126.2, 124.2, 124.1, 115.8, 115.8, 115.6, 114.9, 111.6, 18.2. IR (KBr, cm−1): υ 2922, 1622, 1576, 1456, 1318, 1198, 1099, 768, 754, 465.
2.5 X-ray structure determinations of 1, 2 and 3
The block-shaped single yellow crystals for X-ray analysis of 1 and 2 were obtained from a tetrahydrofuran solution and diethyl ether solution upon cooling to 0 °C, respectively. Single crystals of 3 suitable for X-ray structural analysis were obtained from slow evaporation of a methanol solution. Diffraction data were collected at 226(2) K (for 1) or at 293 K (for 2 and 3) on a Bruker Smart diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) for all compounds. The structures were solved by direct methods [16] and refined by full-matrix least-squares on F2. All non-hydrogen atoms were refined anisotropically and the hydrogen atoms were included in idealized position. All calculations were performed using the SHELXTL [17] crystallographic software packages. Details of the crystal data, data collections, and structure refinements are summarized in Table 1 (1, 2 and 3). CCDC −845948 (1), −845949 (2) and −845950 (3) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/deposit [or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336 033; Email: deposit@ccdc cam ac uk].
Crystal data and structure refinement for compounds 1, 2 and 3.
| Data | 1 | 2 | 3 |
| Formula | C30 H44 N2 O2 | C14 H10 Br2 N2 | C22H20FN3 |
| Fw | 464.67 | 366.06 | 345.41 |
| Temperature/K | 226(2) | 293(2) | 293(2) |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group | P–1 | P2(1)/c | P–1 |
| a/Å | 6.4048(8) | 6.8769(14) | 7.4180(15) |
| b/Å | 10.3535(12) | 4.0470(8) | 10.139(2) |
| c/Å | 11.3096(14) | 23.621(5) | 12.916(3) |
| α/° | 81.512(3) | 90.00 | 106.19 (3) |
| β/° | 86.313(2) | 90.96(3) | 98.12(3) |
| γ/° | 72.688(2) | 90.00 | 93.82(3) |
| Volume (Å3) | 707.98(15) | 657.3(2) | 917.8(3) |
| Z | 1 | 2 | 2 |
| Dcalcd (Mg m−3) | 1.090 | 1.850 | 1.250 |
| F(000) | 254 | 356 | 364 |
| θ range for data collection | 1.82° to 26.03° | 2.40° to 27.48° | 3.00° to 27.48° |
| Limiting indices | −7 ≤ h ≤ 5, −12 ≤ k ≤ 9, −13 ≤ l ≤ 12 | −8 ≤ h ≤ 8, −4 ≤ k ≤ 5, −30 ≤ l ≤ 30 | −9 ≤ h ≤ 9, −13 ≤ k ≤ 13, −16 ≤ l ≤ 16 |
| Data/restraints/parameters | 2705/0/161 | 1505/0/83 | 4166/0/241 |
| Goodness-of-fit on F2 | 1.007 | 1.174 | 1.058 |
| Final R indices [I > 2σ(I)] | R1a = 0.0660, wR2b = 0.1443 | R1a = 0.0276, wR2b = 0.0804 | R1a = 0.0450, wR2b = 0.1201 |
| R indices (all data) | R1a = 0.1293, wR2b = 0.1739 | R1a = 0.0385, wR2b = 0.1051 | R1a = 0.0774, wR2b = 0.1354 |
| Largest diff. peak and hole/e.A−3 | 0.227 and-0.164 | 0.335 and-0.327 | 0.155 and-0.150 |
a R1 = ∑||Fo|–|Fc||/∑|Fo|.
b wR2 = [∑[w (Fo2–Fc2)2]/∑[w (Fo2)2]]1/2.
3 Results and discussion
3.1 Synthesis of compounds 1–3
Compounds 1–2 Ar(CH = N–N = HC)Ar (Ar1 = Ar2 = 2-OH-3,5-tBu2C6H2 (1), Ar1 = Ar2 = 2-BrC6H4 (2)) were known and readily synthesized by the condensation reaction of hydrazine hydrate with 2.0 equivalent of 3,5-diterbutylsalicylaldehyde and ortho-bromobenzaldehyde, respectively, according to the previous literature [18] (Scheme 1). Compounds 1–2 were characterized by 1H NMR, 13C NMR, IR spectroscopy along with elemental analysis. The imino –N = CH– protons of compounds 1–2 exhibit resonances at 8.76 and 9.02 ppm, respectively, while the resonances for the corresponding imino -N = CH- carbon atoms are observed at 165.2 and 161.4 ppm, respectively. Compound 3 ortho-C6H4(NHC6H3-Me2-2,6)(CH = N–N = HC)C6H4F-ortho was synthesized via a nucleophilic substitution reaction of ortho-C6H4F(CH = N-N = HC)C6H4F-ortho with the lithium salt of 2,6-dimethylaniline according to a modified literature procedure [19]. The compound was also characterized by 1H NMR, 13C NMR, IR spectroscopy along with elemental analysis. The 1H NMR spectrum of 3 exhibits two singlets with resonances at 8.89 and 8.86 ppm for two –N = CH– protons, while the resonances for the corresponding imino carbon atoms appear at 166.1 and 153.8 ppm. The lower field signal at 9.80 ppm is assigned to the NH proton in 3.
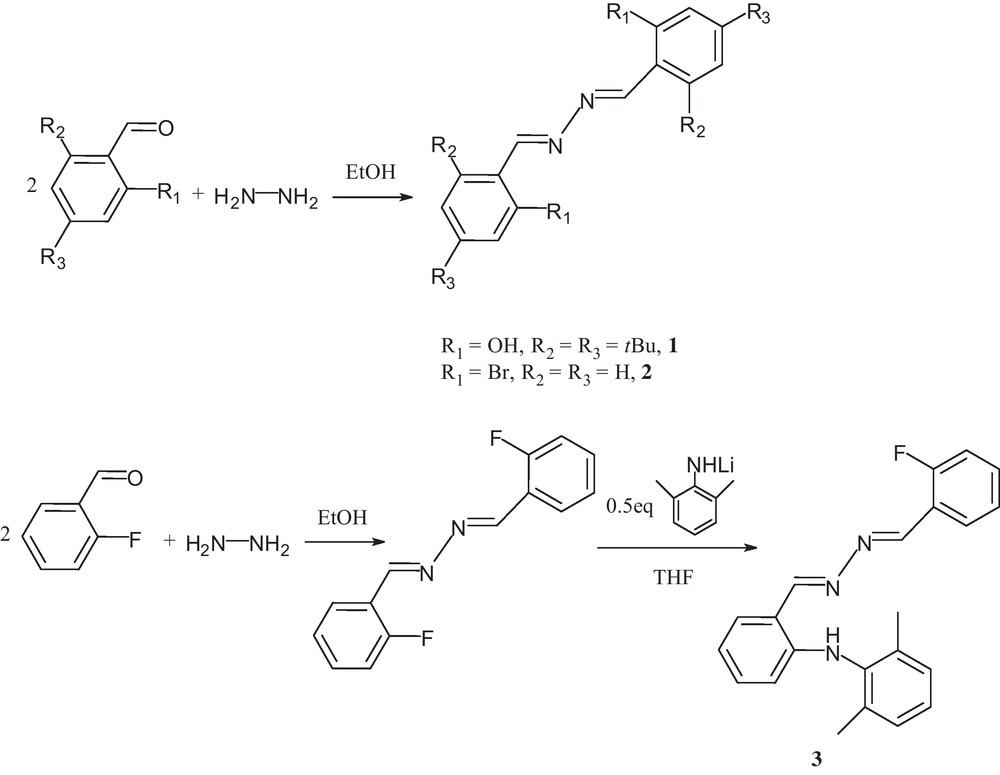
Synthetic route for compounds 1–3.
3.2 Crystal structures
The molecular structures of the compounds 1, 2 and 3 were determined by X-ray crystallographic analysis. The crystallographic data and refinement parameters are presented in Table 1. The ORTEP drawings of molecular structures and packing of 1, 2 and 3 are shown in Figs. 1–3, respectively. The molecule of compound 1 has crystallographic twofold rotation symmetry (Fig. 1(a)). The asymmetric unit of the compound is composed of one-half of the molecule. The imino groups in compound 1 are nearly coplanar with the two benzene rings (N1A–N1–C7–C2: 178.551(224)°, C7A–N1A–N1–C7: 180.000(234)°, C3–C2–C7–N1: 175.000(242)°, C1–C2–C7–N1: 2.679(399)°, with similar torsion angles in comparison with compound ortho-OHC6H4(CH = N–N = HC)C6H4OH-ortho [14,18b,20]. The two benzene rings are also in trans configuration with respect to C7–N1 and C7A–N1A bonds relative to N1–N1A. The intramolecular O–H···N hydrogen bonds form a six-membered ring, generating a S(6) ring motifs [21]. The intramolecular and intermolecular C–H···O interactions are also observed (Table 2), which are typical for Schiff bases derived from salicylaldehydes [22]. Additionally, the π–π stacking interactions play an important role in forming slightly staggered and parallel column structure (J-aggregation structure, Fig. 1(b)). The molecule of compound 2 has also crystallographic twofold rotation symmetry (Fig. 2(a)). The two benzyl rings are coplanar and are in transconfiguration with C7–N1 and C7A–N1A bonds relative to N1–N1A with the torsion angles being −174.5° (C1–C2–C7–N1), −178.8° (C2–C7–N1–N1A), 180.0° (C7–N1–N1A–C7A), 178.8° (N1–N1A–C7A–C2A), respectively. The molecules are linked via intermolecular Br···Br interactions (Br···Br = 3.7714 (10) Å) into sheet running parallel to the b axis (Fig. 2(b)). In compound 3, the intramolecular N–H···N hydrogen bond forms a six-membered ring, generating a S(6) ring motif. (Fig. 3(a)) The two benzyl rings bonded to the imino carbons are nearly copolanar and are in trans configuration with C15–N2 and C16–N3 bonds relative to N2–N3 and nearly copolanar with the torsion angles being 167.4° (C3–C2–C15–N2), −10° (C1–C2–C15–N2), −177.3° (C2–C15–N2–N3), 168.8° (C15–N2–N3–C16), 179.0° (N2–N3–C16–C17), 14.1° (N3–C16–C17–C22) and −164.4° (N3–C16–C17–C18), respectively. The dihedral angles between the 2,6-dimethylphenyl ring and the other two phenyl ring are 68.3° and 61.6°, respectively. The crystal structure of 3 is consolidated by intramolecular N − H···N hydrogen bonds, intramolecular C − H···F interactions and weak intermolecular C − H···F interactions [3.275(2) Å, 111.9°, (x + 1, y, z)] arising from the interactions between the fluoro atom and the two hydrogen atoms of methyl, which further form an infinite zig-zag one-dimension chain. Moreover, Two adjacent chains are linked together by intermolecular C − H···N interactions [3.630(2) Å, 135.8°, (x + 1, y, z)] (Fig. 3(b)).
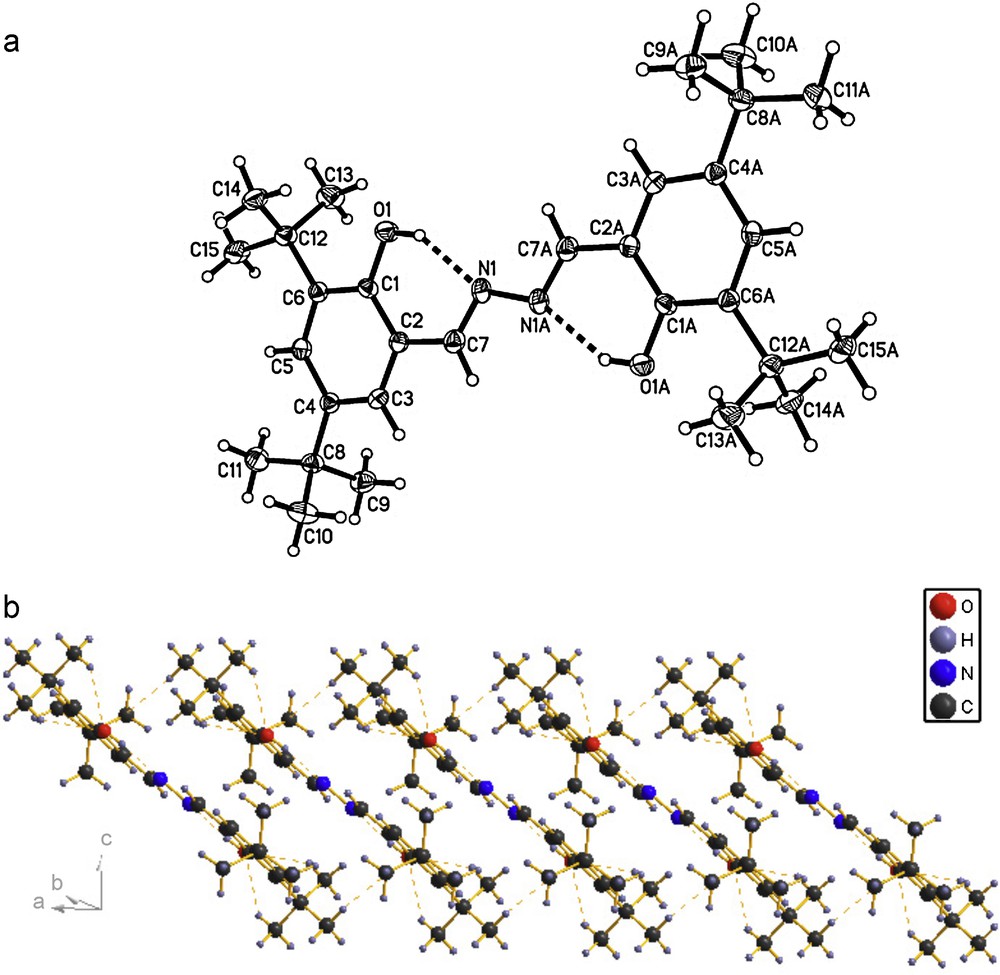
(a) Molecular structure of compound 1 (hydrogen bonds are indicated by dashed lines. The thermal ellipsoids are drawn at 30% probability levels). Selected bond distances (Å) and angles (°): O(1)–H(1) = 0.8200, N(1)–C(7) = 1.274(3), N(1)–N(1)#1 = 1.409(4), O(1)–C(1) = 1.359(3); C(1)–O(1)–H(1) = 109.5, C(7)–N(1)–N(1)#1 = 113.2(3), O(1)–C(1)–C(6) = 119.4(2), N(1)–C(7)–C(2) = 123.9(3), N(1)–C(7)–H(7) = 118.1; (b) The packing of compound 1 (hydrogen bonds are indicated by dashed lines.).
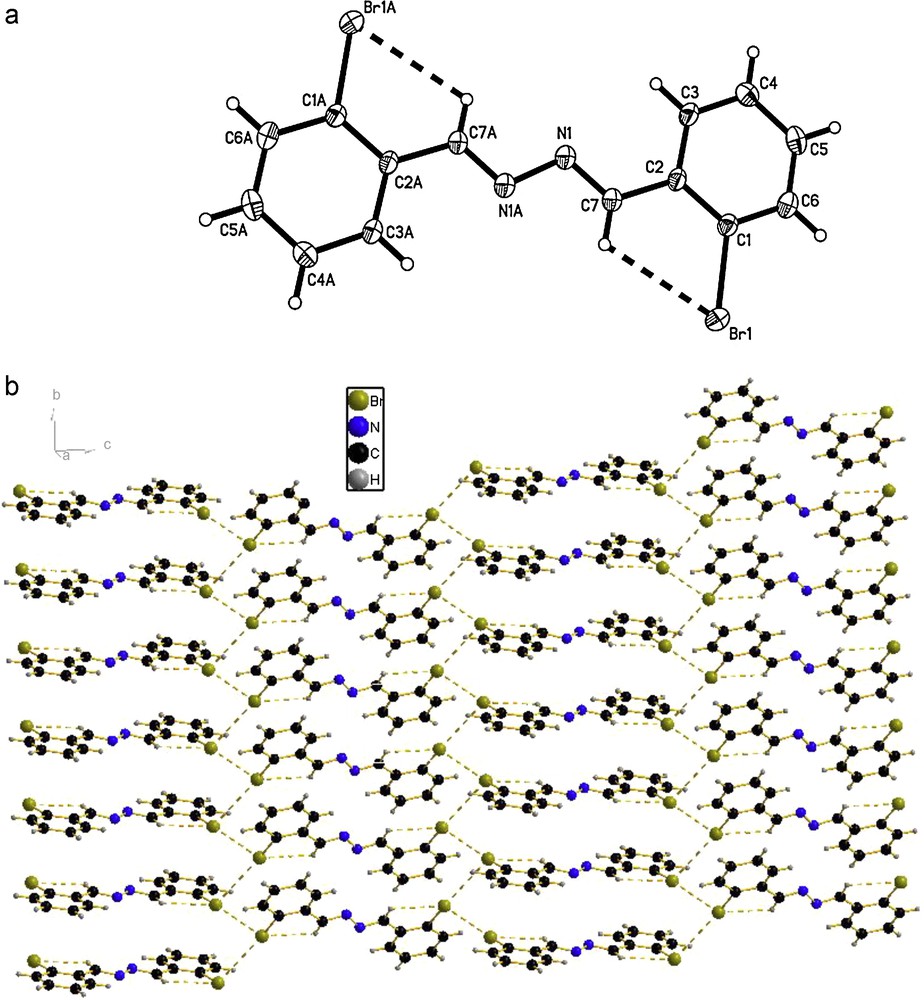
(a) Molecular structure of compound 2 (the thermal ellipsoids are drawn at 30% probability levels). Selected bond distances (Å) and angles (°): Br(1)–C(1) = 1.903(3), N(1)–C(7) = 1.278(4), N(1)–N(1)#1 = 1.419(6); C(7)–N(1)–N(1)#1 = 111.8(4), C(6)–C(1)–Br(1) = 117.0(3), N(1)–C(7)–C(2) = 119.7(3); (b) The packing of compound 2. (C − H···Br and Br···Br interactions are indicated by dashed lines.).
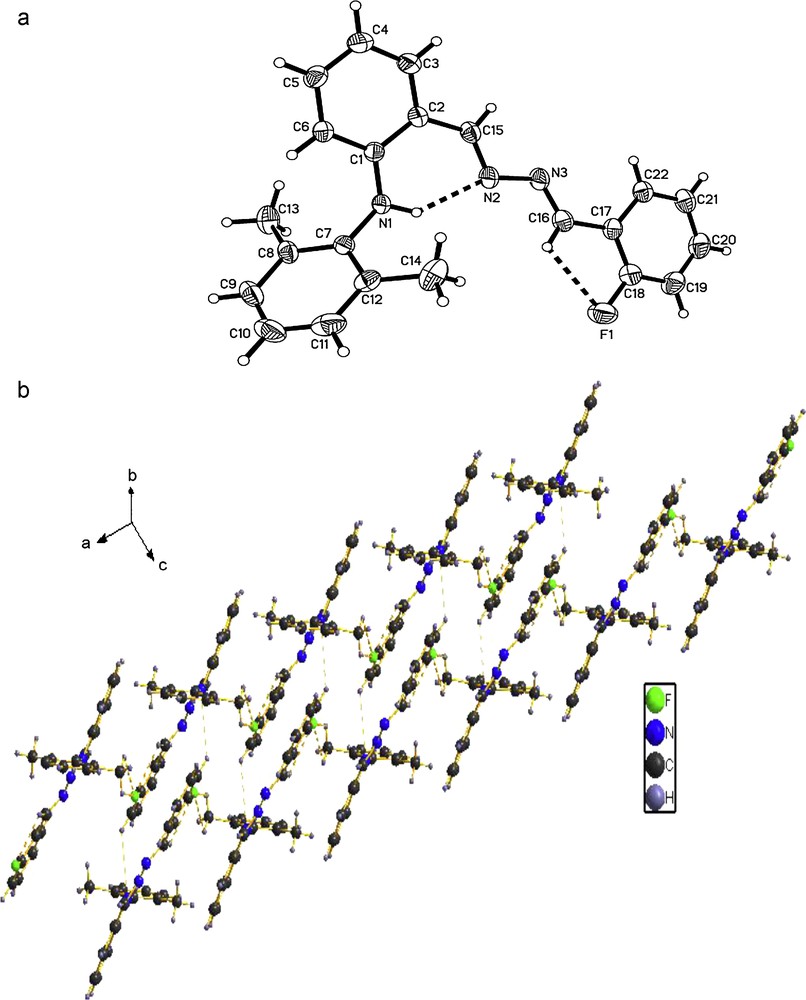
(a) Molecular structure of compound 3 (hydrogen bonds are indicated by dashed lines. The thermal ellipsoids are drawn at 30% probability levels.). Selected bond distances (Å) and angles (°): F(1)–C(18) = 1.3558(18), N(1)–C(1) = 1.3767(18), N(1)–C(7) = 1.4260(17), N(1)–H(1) = 0.860(17), N(2)–N(3) = 1.4047(18); C(1)–N(1)–C(7) = 124.37(12), C(1)–N(1)–H(1) = 114.1(10), C(15)–N(2)–N(3) = 111.99(12), F(1)–C(18)–C(19) = 117.92(15); (b) The packing of compound 3. (C − H···N and C − H···F interactions are indicated by dashed lines.).
Hydrogen-bond geometries for 1, 2, and 3.
| Structure | D–H···A | d(D–H)(Å) | d(H···A)(Å) | d(D···A)(Å) | <(DHA) (deg) |
| 1a #1 | C(13)–H(13A)···O(1) | 0.96 | 2.34 | 2.990(3) | 124.1 |
| C(14)–H(14 C)···O(1) | 0.96 | 2.35 | 3.010(3) | 125.6 | |
| O(1)–H(1)···N(1) | 0.82 | 1.92 | 2.650(3) | 148.2 | |
| C(15)–H(15 C)···O(1)#2 | 0.96 | 2.99 | 3.865(4) | 152.2 | |
| 2b #1 | C(7)–H(7)···Br(7) | 0.93 | 2.7824(7) | 3.1973(34) | 108.192(205) |
| 3c | N(1)–H(1)···N(2) | 0.860(17) | 2.065(16) | 2.766(2) | 138.1(13) |
| C(16)–H(16)···F(1) | 0.93 | 2.51 | 2.783(2) | 97.3 | |
| C(19)–H(19)···N(1)#1 | 0.93 | 2.90 | 3.630(2) | 135.8 | |
| C(13)–H(13A)···F(1)#2 | 0.96 | 2.79 | 3.275(2) | 111.9 | |
| C(13)–H(13B)···F(1)#2 | 0.96 | 2.79 | 3.275(2) | 111.9 |
a #1–x, −y + 1, –z; #2 x + 1, y, z.
b #1–x, −y + 2, −z.
c #1–x + 1, –y + 2, –z + 1; #2 x + 1, y, z.
3.3 UV–vis absorption and photoluminescence spectra
The photophysical properties of compounds 1–3 were investigated by UV–vis and photoluminescence (PL) spectroscopy in solutions at room temperature. The obtained spectral data are summarized in Supporting Information Table ST-1. The UV–vis spectra of all compounds exhibited two to four broad absorption bands with strong absorption bands at λmax = 310, 298, 298 nm, respectively. The absorption maxima are in the range of 279–404 nm.
All compounds 1–3 produce bright fluorescence in the solid state with emission maxima of 579, 469 and 496 nm, respectively. The emission maxima of compounds 2 and 3 are blue-shifted about 27 nm and 83 nm compared with the emission maximum of compound 1, respectively. This is due to the difference of the substituents in the aromatic ring that might affect the conjugated extent and π-stacking interactions of these compounds [23]. The emission behaviors of compounds 1−3 in different solvents (1-chlorobenzene, methylene chloride (CH2Cl2), tetrahydrofuran (THF), ethanol (EtOH), dimethylformamide [DMF]) have been observed. The emission color of compound 1 in solution can be tuned from blue to green with increasing polarity of the solvent (433–569 nm), while the emission color of compound 2–3 can be slightly tuned in the blue region with increasing polarity of the solvent (404–437 nm for 2, 461–480 nm for 3). In addition, the emission maxima of compounds 1–3 in solution are blue-shifted compared with the ones of the corresponding compounds in solid, similar to that in previous observations [24]. The red shift of the emission maxima from solution to solid state is consistent with the lower energy emission for the latter which is likely resulted from π–π stacking interactions of the molecules in solid state. The typical emission spectra of compound 1 in different solvents and the solid state are shown in Fig. 4. From the fluorescence spectra of compound 1 in different solvents, it is observed that the emission maxima shift to longer wavelength with increasing solvent polarity, indicating that the molecules are significantly solvated in the excited state, resulting in great differences of dipole moments of the molecules. [25–28] In addition, the emission spectra shows red shift in 1-chlorobenzene as compared to that in other solvents, due to the fact that the intermolecular interactions between the aromatic hydrocarbon solvent and the aromatic organic molecular is responsible for the red-shifted emission [29].

Emission spectra of compound 1 in different solvents and the solid state.
4 Conclusions
Three benzaldazine compounds 1–3 with the general formula Ar1(CH = N–N = HC)Ar2 (Ar1 = Ar2 = 2-OH-3,5-tBu2C6H2 (1), Ar1 = Ar2 = 2-BrC6H4 (2), Ar1 = ortho-C6H4(NHC6H3-Me2-2,6), Ar2 = C6H4F-2 (3)) have been prepared by the reaction of hydrazine with different aromatic aldehydes. All compounds were characterized by elemental analysis, 1H NMR, 13C NMR, IR spectroscopy and single-crystal X-ray crystallography. The different supramolecular structures were obtained through different weak interactions (C − H···O, O − H···N and π–π interactions for 1; C − H···Br and Br···Br interactions for 2; C − H···F and C − H···N interactions for 3). Compound 1 shows obvious solvent-dependent fluorescent properties and blue-to-green emission with increasing the solvent polarity. Compounds 2–3 show blue photoluminescence in different solvents. Our work will be useful for the construction of a variety of new transition metal complexes and coordination polymers with novel structures and also provide promising potential for blue-emissive materials.
Acknowledgement
This work was supported by the National Natural Science Foundation of China (Nos. 21074043 and 21004026). One of the authors (Dr. Q. Su) is grateful for the support from Jilin University.