

1 Introduction
Energy consumption has been dramatically increasing in recent decades with a rapid advancement in technology and the swift increase in population worldwide. However, fossil fuels are still the main source of energy. Unfortunately, overconsumption of these fuels has amplified the atmospheric content of carbon dioxide, resulting in severe climate change and global warming. Hydrogen is one of the best energy carriers to satisfy the increasing demand for an efficient and clean energy supply [1]. However, convenient storage and safe transport of hydrogen are the widely accepted technological barriers in “Hydrogen Economy”. This mainly originates from the low density of H2 which makes it extremely difficult to store in compressed or liquefied form [2]. Hence, there is currently an immense interest in developing new and efficient hydrogen storage materials for on-board applications [3].
Exploring interaction between atomic hydrogen and adsorbing surfaces is rather crucial in understanding many important technological processes such as hydrogen storage and transport, fuel cell research, etc. [4,5]. Several theoretical researches for hydrogen-chemisorbed nanostructured materials (e.g. nanotubes, nanoclusters, etc.) have been reported. Ma et al. [6] experimentally found that boron nitride nanotubes (BNNTs) may be used to store hydrogen and they also found that about 70% of the adsorbed hydrogen is chemisorbed. Wu et al. have studied the adsorption of hydrogen on B12N12 nanocluster, showing an exothermic and favorable process [7]; and recently we have investigated H atom adsorption on B12P12 nanocluster and shown that the H atoms prefer to be adsorbed atop the adjacent B and P atoms with the Gibbs free energies of −1.18 and −0.80 eV, respectively [8].
Zinc oxide (ZnO) is a wurtzite-type semiconductive material with a wide band gap. Thermal stability, irradiation resistance, and flexibility to form different nanostructures are the advantages which expedite its potential wide application. Recently, research efforts have been devoted on (XY)n nanostructures such as nanoclusters, nanohorns, nanotubes, and nanowires [9–17]. Concretely, the ZnO nanocluster has been widely studied both theoretically and experimentally [13,14]. Recently we have shown that Zn12O12 nanoclusters may be used as a catalyst in cleavage of Cl2 molecule [12]. In the present work, the interaction of hydrogen with an alternate fullerene-like structure of Zn12O12 cluster has been theoretically investigated in terms of energetic, geometric, and electronic properties.
2 Computational methods
Spin-unrestricted B3LYP hybrid density functional theory (DFT) method within LANL2DZ all electron basis sets were used for the optimization, density of state (DOS), frontier molecular orbitals (FMO) analyses, and energy calculations. The B3LYP has been demonstrated to be a reliable and commonly used level of theory in study of different nanostructures [18–23]. With the optimized structures, binding energy (Ebin) of the H atom on the pure cluster has been obtained using the following equation:
| (1) |
Calculated binding energies (Ebin, eV), HOMO energies (EHOMO), LUMO energies (ELUMO), Fermi level energies (EFL) and HOMO-LUMO energy gap (Eg) of systems (see Fig. 4 for system symbols).
| System | Ebin | QT (e)a | EHOMO | EFL | ELUMO | Eg | ΔEg (%)b |
| Zn12O12 | – | – | −6.84 | −4.82 | −2.80 | 4.04 | – |
| Zn12O12Hc | −1.97 | 0.39 | −3.81d | −3.85 | −2.96 | 0.85 | 79.0 |
| A | −2.49 | 0.11 | −6.55 | −4.65 | −2.76 | 3.79 | 6.2 |
| B | −2.40 | 0.11 | −6.63 | −4.69 | −2.75 | 3.88 | 3.9 |
| C | −2.34 | 0.18 | −5.44 | −4.30 | −3.16 | 2.28 | 43.5 |
| D | −2.09 | 0.39 | −4.13 | −3.17 | −2.22 | 1.91 | 52.7 |
| E | −2.32 | 0.38 | −4.51 | −3.58 | −2.66 | 1.85 | 54.2 |
a Q is defined as the average of total Mulliken charge on the H atoms.
b The change of HOMO-LUMO gap of Zn12O12 after adsorption.
c See Fig. 3.
d Energy of SOMO.
3 Results and discussion
3.1 Geometry of Zn12O12
Optimized structure and geometric parameters of Zn12O12 nanocluster have been shown in Fig. 1. In a structural view, the cluster has been made of six squares and eight hexagons. Structurally, there are two individual ZnO bonds; one is shared by two six-membered rings, 6-MR (B66 bond), and the other between a 4- and a 6-MR (B64 bond). The B64 bond (1.98 Å) is slightly longer than B66 bond (1.91 Å). The values of ZnOZn angle in 6-MR and 4-MR are 116.1° and 88.9° and those of OZnO are 123.5° and 90.7°, respectively (Fig. 2). The obtained geometrical parameters concur with the previous results [25–27]. Reber et al. [25], using a density functional formalism that incorporates exchange and correlation effects within the generalized gradient approximation of Perdew, Burke and Ernzerhof, have shown that the lengths of B66 and B64 bonds are about 1.88 and 1.97 Å, respectively. Yong et al. [26], based on a DFT method, have shown that the Zn12O12 has two types of ZnO bonds, a longer one (1.981 Å) in rhombuses and the other shorter one (1.880 Å) connecting the neighboring rhombuses. They have also demonstrated that the bond angle in the rhombuses is acute at O (86.4°) and obtuse at Zn (91.0°). The corresponding angles in the hexagons have been reported to be 108.2 and 129.4°, respectively. As shown by the molecular electrostatic potential (MEP) map of the Zn12O12 in Fig. 2, Zn atoms are positively charged (blue colors) while the O atoms are negatively charged (red colors) in ZnO bond, indicating ionic character of these bonds.
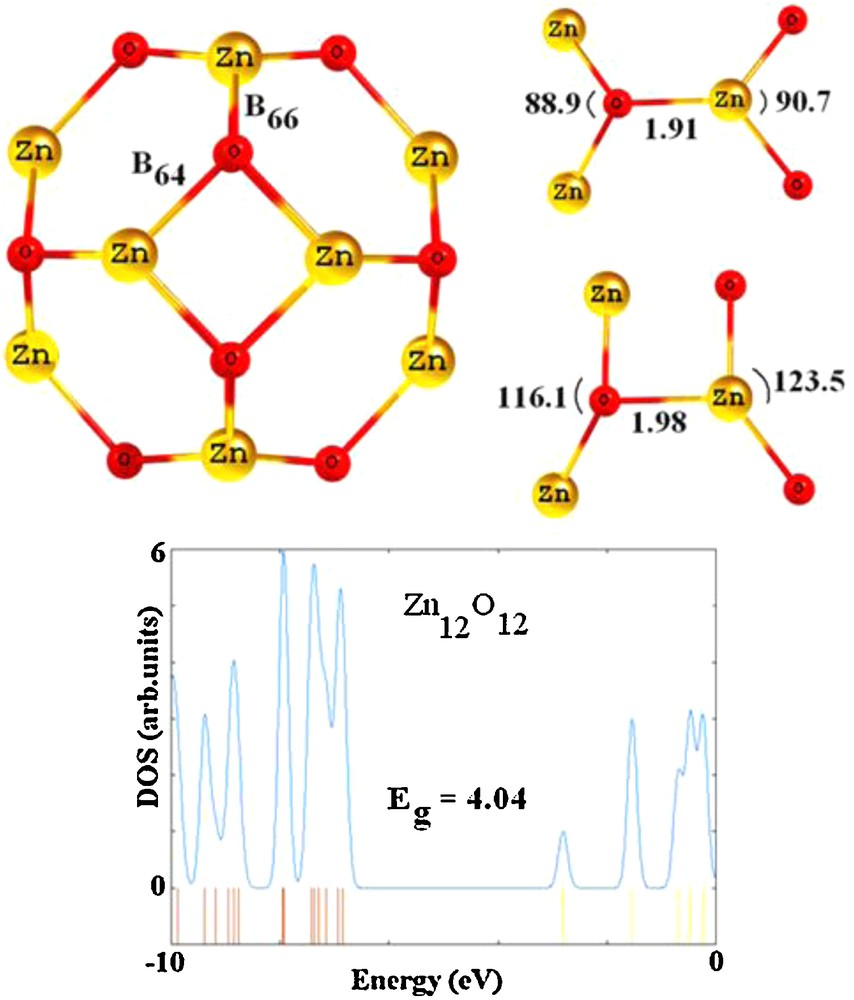
Geometrical parameters of optimized Zn12O12 nanocluster. Distances are in angstrom (Å) and the angles in degrees.

(a): calculated molecular electrostatic potential surface (MEP) of the Zn12O12 cluster. The red and blue colors are referred to the higher and lower electron density, respectively; (b): singly occupied molecular orbital (SOMO) profile of Zn12O12H.
3.2 Atomic hydrogen adsorption
There are two individual sites for the adsorption of an H atom on the cluster; on top of either a Zn or O atom. Therefore, to find the adsorption behavior of an H atom on the cluster, the H atom was initially put on these sites and then relax optimizations were performed. The results (Table 1) show that the adsorption process is site selective and the H atom prefers to be adsorbed on top of an O atom with Ebin of –1.97 eV. In this configuration (Fig. 3), the length of newly formed HO is about 0.98 Å, indicating that the interaction is very strong. The oxygen atom has an unoccupied p orbital that it prefers to accept electrons from the hydrogen, while the zinc atom has fully occupied d orbitals. In other words, in the bare cluster, some charges are transferred from the Zn atoms to the O atoms in the cluster surface; when an H is placed atop an O atom of the cluster, it competes with the Zn atoms for releasing the electron to the O. Based on the Mulliken charge analysis, we have found that the charge on oxygen and zinc atoms of the Zn12O12 cluster is about −0.982 e and +0.982 e, respectively. When one hydrogen atom is added to oxygen, its charge is reduced to −0.895 e, indicating that the H atom acts as an electron donor agent. The H chemisorption on Zn12O12 also induces a local structural deformation. In the stable configuration of Zn12O12H, the ZnO bond lengths and angles involving the H-adsorbed O atom (2.06 and 2.17 Å, 111.9° and 84.7°, respectively) are different from those in the pristine Zn12O12 cluster. The results show that when an H atom is put on the Zn atom of cluster, it shifts to the top of O atom during the relax optimization.
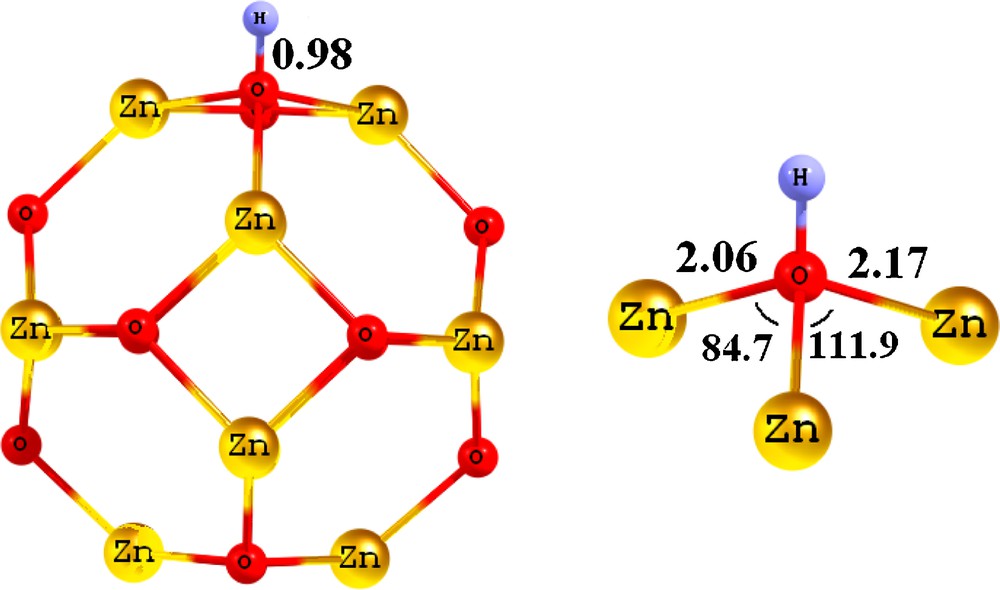
Geometrical parameters of the optimized Zn12O12H nanocluster. Distances are in angstrom (Å) and the angles in degrees.
In the next step, we have investigated the simultaneous adsorption of two H atoms on the cluster, exploring how the prior chemisorption of an H atom influences the adsorption properties of the second one. To this end, we have considered five possible adsorption configurations as shown in Fig. 4, namely, A, B, C, D and E. Then, we have found that the 1, 2 addition on the B64 bond is the most favorable adsorption process (configuration A). The Ebin of this process has been calculated to be about −2.49 eV which is a little larger than that of the case in which two H atoms have been adsorbed on the B66 bond (−2.40 eV, B).

Geometrical parameters of the optimized Zn12O12H2 nanoclusters. Distances are in angstrom (Å).
It is may be due to the fact that the B64 bond is shared between 4- and 6-MRs but the B66 between two 6-MRs; therefore, the B64 bond is thermodynamically favorable site for the adsorption of the H atoms because of more strain in 4-MR compared to that in the 6-MR. The energetically favorable B64 bond breakage upon the adsorption process can be ascribed to the release of strain energy of the 4-MR. Based on the NBO analysis, in configuration A, the B64 bond is broken after the adsorption process and two new bonds are formed, namely, HO and HZn with bond lengths of 0.98 and 1.65 Å, respectively. Similarly, in configuration B, the B66 is broken and length of newly formed HO and HZn is about 0.98 and 1.64 Å, indicating that the interaction is very strong and also the B66 bond length increases (2.99 Å) after 2H adsorption.
The Ebin values of the 1,3 addition of two H atoms atop the oxygen atoms of 6-MR (−2.32 eV, E) and 4-MR (−2.09 eV, D) are smaller than those atop an O and a Zn atom in configurations A and B. Interestingly, when two H atoms are simultaneously adsorbed on two atoms of the cluster, the absolute value of the Ebin per atom is much more negative than that of the case in which only one atom is adsorbed. The relative stability of the configurations is A > B > E > D > C.
To rationalize these phenomena, we have performed frontier molecular orbital analysis (FMO) on the H-Zn12O12 complex. The results show that it has a singly occupied molecular orbital (SOMO) which mainly spreads on the Zn atoms of cluster (Fig. 2b). An H atom also has a SOMO; therefore, it is expected that the second H atom strongly prefers to be interacted with the SOMO of cluster (Zn atom) rather than the O atoms. In other words, the H atom and the cluster act like radicals which react together via SOMO/SOMO interaction. As a result, the configurations A, B, and C are energetically more favorable than the configurations D and E.
3.3 Electronic properties
Subsequently, we have studied the influence of the H adsorption on the electronic properties of the cluster. Density of state (DOS) plot in Fig. 1 shows that the pristine Zn12O12 is a semiconductor with a wide HOMO/LUMO energy gap (Eg) of 4.04 eV which is somewhat larger than that of bulk ZnO (3.4 eV) [28]. It should be noted that here, the Eg also stands for SOMO/LUMO energy gaps for open shell system of H-Zn12O12. When one H atom is adsorbed on the Zn12O12, having an approximately strong interaction, the DOSs of the cluster are significantly changed near the Fermi level and a new impurity state is appeared at the energy level of −3.81 eV. As a result, the Fermi level is noticeably shifted from −4.82 eV in free cluster to −3.85 eV in the H/Zn12O12; therefore, Eg of the cluster is dramatically reduced from 4.04 to 0.81 eV. The results suggest that the semiconductive cluster has been transformed to an n-type one ascribed to the large charge transfer from the hydrogen atom to the cluster. It is noteworthy to mention that the canonical assumption for Fermi level is that in a molecule (at T = 0 K), it lies approximately in the middle of the Eg. The change of Fermi level of a semiconductor alters its field emission current. Since nanoclusters are one of the promising candidates for field emitters, the present results are of great importance. It seems that the H adsorption will facilitate the electron emission from the cluster by making the HOMO unstable.
The results in Table 1 and DOS plots in Fig. 5 show that like the case of one H adsorption, the Fermi level of Zn12O12 is shifted to higher energies in all cases of simultaneous adsorption of two H atoms; but this shift is significantly higher in the case of two H atoms co-adsorption on two O atoms of either 4-MR or 6-MR (configurations D and E). Eg of the cluster is also reduced in all of the cases but its decrement is significant in the configurations D and E. This phenomenon may be explained by the results of Mulliken charge analysis, indicating that similar to the case of one H adsorption, in these configurations a large charge has been transferred (Table 1) from the hydrogen atoms to the cluster which transforms the Zn12O12 cluster from intrinsic semiconductor to an extrinsic n-type one with narrower Eg. The results indicate that the electronic properties of Zn12O12 are very sensitive towards the adsorption of H atoms and also the electrical conductivity of the cluster may alter due to changes of Eg.

Calculated density of states (DOS) for H-adsorbed Zn12O12 complexes.
However, we believe that the increment of Fermi level energy upon the H adsorption may lead to a decrement in the work function that is important in field emission applications. The work function is calculated using the standard procedure by taking the potential energy difference between the vacuum level and the Fermi level, which is the minimum energy required for one electron to be removed from the Fermi level to the vacuum. The decrement in the work function indicates that the field emission properties of the cluster are enhanced upon the adsorption of H atoms. All of the above calculations and findings suggest that the H adsorption has an advantageous effect on the field emission properties of Zn12O12.
3.4 Endohedral adsorption
We have also investigated the adsorption of an H atom inside the cluster. There are two individual sites for the adsorption of an H atom inside the cluster; atop of either a Zn or O atom, similar to the exohedral adsorption. Based on the calculations, it was found that the H atom strongly prefers to be adsorbed on the O atom with Ebin of −0.99 eV as shown in Fig. 6. In the obtained configuration, the length of newly formed HO is about 1.01 Å. The OZn bonds adjacent the adsorbing oxygen atom are significantly elongated upon the H adsorption and also the adsorbing atom is somewhat moved inward. However, these findings reveal that the endohedral adsorption of an H atom on the cluster is much less energetically favorable than the exohedral one. It can be rationalized by the fact that when an H atom is placed on the O atom, it tends to form sp3 hybridization which can easily occur in the case of exohedral adsorption. It is worthy to mention that when the H atom was located adjacent to a Zn atom it was shifted on a neighboring O atom during the relax optimization.
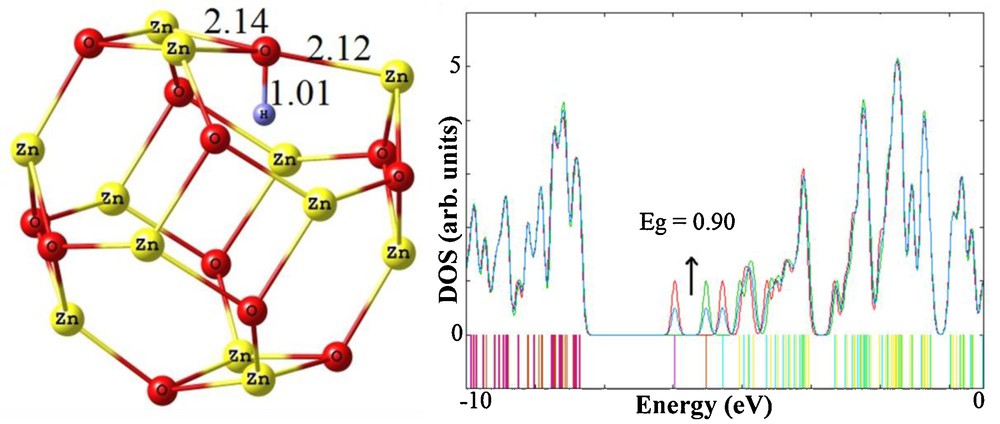
Geometrical parameters of endohedral adsorption of an H atop on Zn12O12 nanocluster and its density of states (DOS) plot. Distances are in angstrom (Å).
Fig. 6 shows that the DOS plot for endohedral adsorption is significantly changed near the Fermi level and a new impurity state is appeared at the energy level of −3.95 eV, compared to that of pristine cluster. The Eg of the cluster is dramatically reduced from 4.04 to 0.90 eV which is nearly similar to its change upon the exohedral adsorption. Therefore, like the case of exohedral adsorption, herein the semiconductive cluster has been transformed to an n-type one due to a charge transfer of 0.32 e from the hydrogen atom to the cluster. Finally, it can be concluded that although the exohedral and endohedral adsorptions of an H atom on the Z12O12 cluster are energetically different, they are somewhat electronically similar.
4 Conclusion
The chemical adsorption of one or two H atoms on a Zn12O12 cluster was studied using DFT calculations. The results showed that one H atom prefers to be adsorbed atop an O atom of the cluster surface rather than the Zn one, and also it was found that 1,2 additions are energetically more favorable than 1,3 additions in Zn12O12H2. The average binding energy of the two adsorbed H atoms was obviously found to be higher than that of one adsorbed atom. The Eg of the cluster is dramatically reduced from 4.04 to 0.81 eV upon one H adsorption, suggesting that it is transformed to n-type semiconductor ascribed to the large charge transfer from the hydrogen atom to the cluster. All cases of H adsorption will facilitate the electron emission from the cluster by shifting the Fermi level to higher energies and decreasing the work function.