

1 Introduction
About 20 years ago, Goodall et al. patented the use of polyoxometalates activated by alkyl aluminium derivatives as catalysts for the Ring Opening Metathesis Polymerization (ROMP) of dicyclopentadiene and norbornene [1,2]. Two publications by the same authors described also these systems which operated in a solution formed by the monomers [3,4]. Finally McCann et al. showed that the polymerization could also proceed via a heterogeneous way by using a single crystal of the tetrabutylammonium salt of the polyoxometalate activated by the alkyl aluminium species [5]. However, in these papers, nothing was done on the mechanism of formation of the active species and, up to now, there is no other report on these systems. This is probably related to the fact that it was assumed that the alkyl aluminium compound gave quite the same reaction than with tungsten or molybdenum chlorides, leading to the formation of metal alkyl species and then, via α-H abstraction, to the active carbene. However, when reacting them with organometallic compounds we observed that polyoxometalates have a reactivity quite different from that of metal chlorides or hydroxides [6–9]. As a consequence, we decided to further investigate the polymerization of strained olefins catalyzed by activated polyoxometalates. When looking at all the data provided by the literature, only the following conclusions could be made:
- • a variety of polyoxometalate structures are active catalysts;
- • both tungstic and molybdic compounds are active catalysts;
- • upon addition of the alkyl aluminium species there is reduction of the polyanion (the solution or the solid became dark).
The aim of this article is to:
- • extrapolate the catalytic system (catalyst and cocatalyst) to other species;
- • to obtain indications on the possible mechanism and on what happens when the catalyst reacts with the cocatalyst.
For this purpose we studied various forms of the polyoxometalate (in solution, as an insoluble inorganic salt or supported on an oxide) and other alkylating agents. We studied also the effect of the solvent on the polymerization. Indications on the mechanism were obtained by preparing activated catalysts by reaction with 13C-enriched methyl magnesium iodide and recording their 13C NMR spectra.
2 Experimental
2.1 Materials and procedures
All chemicals were purchased from Aldrich and used as received. The solvents were distilled before use and kept under molecular sieves. All experiments described here were made in Schlenk tubes under argon. However, the polymerization occurred also when working in air but the amount of alkylating agent was higher.
2.2 Synthesis of catalysts
The organic salts of polyoxometalates (tetrabutyl ammonium and tetraheptyl ammonium) were prepared by addition of the corresponding chloride to an aqueous solution of the polyacid. The resulting powder was dried under vacuum during 2 days at room temperature. In the case of tetraheptyl ammonium, the salt could be recrystallized in acetone. The cesium salts were prepared by addition of the stoechiometric amount of cesium carbonate to a solution of the desired heteropolyacid. The resulting suspension was evaporated to dryness and then evacuated under vacuum during two hours at 200 °C. Solids with various amounts of protons (Cs3-xHxPM12O40, x = 0.0, 0.5, 1.0) were prepared by this way. The silica-supported heteropolyacid (60 wt.%) was prepared by classical impregnation with water as solvent and treated under vacuum at 200 °C during 2 h.
2.3 Catalytic experiments
All catalytic experiments were performed as follows: two to five equivalents of the cocatalyst were added to one equivalent of polyoxometalate followed by 200 equivalents of norbornene in solution in 1,2-dichloroethane. The polymerization occurred immediately as shown by the appearance of a transparent solid. The reaction occurred without destruction of the active sites as further addition of 200 equivalents of norbornene resulted also in a polymerization. The solids were isolated and characterized by solid-state 13C CP-MAS NMR. In all cases the polymer yield was quantitative or nearly quantitative.
2.4 Mechanistic studies
13C-enriched methyl magnesium iodide was prepared by reaction of 13C-methyl iodide with magnesium. It was then reacted with Cs3PM12O40 (M = Mo, W) (ratio catalyst/cocatalyst varying between 0.3 and 2). The solid was then dried under vacuum and its 13C CP-MAS NMR and infrared spectra recorded. To ensure that this treatment did not destroy the active sites addition of norbornene in solution in 1,2-dichloro-ethane was made and resulted in an immediate polymerization. The NMR spectra were recorded on a Bruker DSX-300 spectrometer operating at 75.47 MHz with a classical probehead allowing spinning rates of 10 kHz and with cross-polarization (contact time 5 ms).
3 Results and discussion
3.1 Study of other catalytic systems
The literature data are limited to the use of alkylammonium salts of polyoxometalates, in solution for Goodall et al. and in the solid-state for McCann et al. and to the use of alkylaluminium compounds as cocatalysts. In a first series of experiments we reproduced the results of the literature by using the tetraheptylammonium salts of 12-molybdophosphate and 12-tungstophosphate as catalysts and triethylaluminium as cocatalyst. We performed the polymerization of norbornene in solution in toluene. These salts were chosen as they were soluble in toluene. As previously reported, the reaction of triethylaluminium with the polyoxometalate leads to a black mixture and addition of norbornene results in a polymerization reaction. Due to the high sensitivity of triethylaluminium to water, these experiments were done under argon. A 13C CP-MAS NMR study of the solid obtained after reaction showed that it was polynorbornene keeping its double bonds. No polymerization occurred when only one of the two components, polyoxometalate or triethylaluminium, was present in the solution. In order to simplify the experiments, we decided then to replace triethylaluminium by other alkylating reagents more usual for organic chemists, typically CH3MgX (X = Br or I) and CH3Li. The same experiments than above were then made with these cocatalysts. In all cases, the polymerization occurred rapidly at room temperature when using 1,2-dichloroethane as a solvent and no special care should be taken for doing the experiments which could be made with the Schlenk tube opened to air. Classically, two to five equivalents per polyoxometalate of the alkylating agent were used and the three components, norbornene, polyoxometalate and cocatalysts could be introduced in any order. However, in the case of a large excess of cocatalyst (more than ten equivalents) and of introduction of norbornene after the cocatalyst no polymerization occurred and the system turned dark brown, probably due to a too strong reduction of the polyoxometalate (the highly reduced polyoxometalates are brown). In conclusion, the alkyl aluminium cocatalyst can be replaced by other compounds without loss of activity.
In a second series of experiments, we studied the effect of the solvent on the polymerization, a parameter which was not taken into account in the previous works. When the solvent was dichloromethane, 1,2-dichloroethane or toluene, all systems (homogeneous and heterogeneous, see below) were active, but the same systems were inactive when the solvent was acetonitrile or diethylether. The choice of the solvent had therefore a great effect on the catalytic results. Clearly the reaction proceeds in non-coordinating solvents but does not proceed in the presence of coordinating solvents. The reason is probably that the solvent will coordinate to the active site preventing the coordination of the olefin.
In a third series of experiments we studied heterogeneous catalysts. For that purpose, there are mainly two possibilities: supporting the polyoxometalate on an oxide support like silica or alumina or to prepare an insoluble inorganic salt of the polyoxometalate. In all cases we observed also a polymerization of norbornene. In the case of silica-supported polyoxometalates, as methyl lithium or methyl magnesium halides react also with the remaining hydroxyl groups of the support (this method is used to quantify the surface hydroxyl groups of silica), it is necessary to add a higher amount of cocatalyst. In the case of inorganic salts, as the reaction will proceed at the surface of the solid, it should be interesting to have a salt with a high surface area. This can be done by using ammonium or alkaline cations (K+, Rb+, Cs+). Cesium salts of 12-molybdophosphate and 12-tungstophosphate were then prepared and used in the polymerization of norbornene. To ensure that the protons had no effect on the catalytic activity we prepared also mixed salts Cs3-xHxPM12O40 (M = Mo or W). For these experiments, the solvent was 1,2-dichloroethane and a large excess of the cocatalyst was used. In all cases the polymerization occurred rapidly and no effect of the number of protons could be detected, in agreement with the expected assumption that reaction of protons with the alkylating agent should only lead to the evolution of methane.
In conclusion, a variety of polyoxometalates can be used not only as homogeneous catalysts but also as heterogeneous catalysts. One key point is the choice of the solvent which must be non-coordinating.
3.2 Information on a possible mechanism
First of all, it should be pointed out that the amount of active species is probably very low. Indeed, even when one or two equivalents of norbornene were added to the catalyst/cocatalyst system a solid polymer was formed. As a consequence it was very difficult to characterize this species. Another point is that no metathesis activity was observed with this system when using acyclic olefins such as pent-2-ene even after 1 day. Finally, the polyoxometalate in its original form cannot be the active species as it cannot coordinate the olefin, the metal atoms being surrounded by six oxygen.
Taking into account the above constatations, we decided to study the first step of the reaction, when the cocatalyst is added to the polyoxometalate. For that purpose, we undertook a study by 13C CP-MAS NMR of the reaction product of Cs3PM12O40 (M = Mo, W) with 13CH3MgI. It was not possible to study the soluble salt of the polyoxometalate as the signals were masked by those of the organic cation. In addition, most of the polyoxometalate precipitated due to the formation of a magnesium salt. In order to avoid the formation of any side products, the reactions were made in conditions of strict exclusion of air and water. The use of a 13C labelled Grignard reagent allowed to observe in a reasonable time the 13C NMR signals of the species formed during the reaction and to see minor species. The spectra are shown on Figs. 1 and 2, for Cs3PW12O40 and Cs3PMo12O40 respectively.

13C CP-MAS NMR spectrum of the solid resulting from the reaction of Cs3PW12O40 with 13CH3MgI.
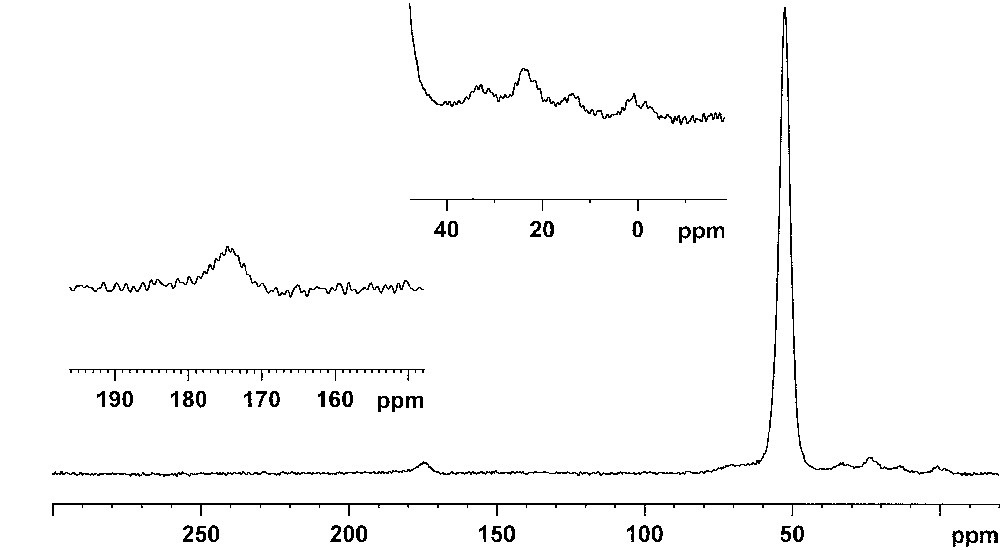
13C CP-MAS NMR spectrum of the solid resulting from the reaction of Cs3PMo12O40 with 13CH3MgI.
In both cases no signal attributable to a carbene species could be detected. The major species gives a signal near 52 ppm whatever the polyoxometalate and some small additional resonances can be shown (at ca. 65, 23, 13 and around 0 ppm for Cs3PW12O40 and 174, 70, 33, 23, 14 and ca. 0 ppm for Cs3PMo12O40). As observed above the solids were dark blue, showing a reduction of the polyoxometalate (this color is due to charge transfer bands between M(VI) and M(V) atoms in the polyanion). Luzgin et al. and Zhang et al. studied the reaction of methanol with H3PW12O40 by solid-state NMR and observed a signal at 59 ppm which was attributed to a methoxy species on the polyanion [10,11]. This methoxy species was formed according to:
We observed also quite the same species upon reaction of methane with the same polyacid, the evolved product being hydrogen instead of water [12]:
As in our case there are no protons such a reaction cannot proceed. However, the reduction of the polyoxometalate can be explained by assuming a reaction such as:
CH3− MgI + (PMVI12O40)3− → PMV2MVI10O39(OCH3)4− + Mg2+ + I−.
The slight shift from 59 to 52 ppm could be related to the fact that the polyoxometalate was reduced (note that the signals are relatively broad) (Δν1/2 = 600–700 Hz). In conclusion, it is clear that the expected alkylation reaction by the Grignard reagent results mainly in the formation on a methoxy group linked to the polyoxometalate. It should then be necessary to find another explanation for the creation of an active site. It cannot be excluded at this stage that the active site is formed by the partial decomposition of the polyoxometalate leading then to the formation of a multi-methylated metal giving the carbene by α-H abstraction. However this hypothesis seems unrealistic as:
- • a methylated species should be too reactive and so should decompose rapidly;
- • the driving force for the α-H abstraction is the steric hindrance.
There is no steric hindrance with methyl derivatives and this reaction is mainly observed with neopentyl derivatives. As a consequence, we can suppose a priori that the active species is formed from the polyoxometalate. When looking at the spectra on Figs. 1 and 2 there are the same minor peaks (quite the same in both cases) below 70 ppm. These peaks are probably due to impurities or some remaining solvent (the experiments were made in dichloroethane). More interestingly, the 13C NMR spectrum of Fig. 2 shows an additional signal near 175 ppm which can be attributed to a formyl species [13]. Such a species results from the oxidation of the methoxy group by the oxygen atoms of the polyoxometalate. We observed it also when heating the compound resulting from the activation of methane on H3PMo12O40 [12]. The scheme below displays a possible mechanism from the formation of this formyl group linked to the polyoxometalate:
For this purpose, we supposed that the methoxy group is located on a bridging oxygen atom of the polyoxometalate (such a species had been synthesized by Knoth some years ago [14]) but quite the same reaction could be made on a terminal oxygen atom. In a first step, one proton of the methyl group interacts with a terminal neighboring oxygen atom. In a second step, a concerted mechanism transfers the hydrogen atom and forms formaldehyde interacting with the two metal atoms. After elimination of formaldehyde, the lacunary system can be able to coordinate norbornene and then to initiate the polymerization.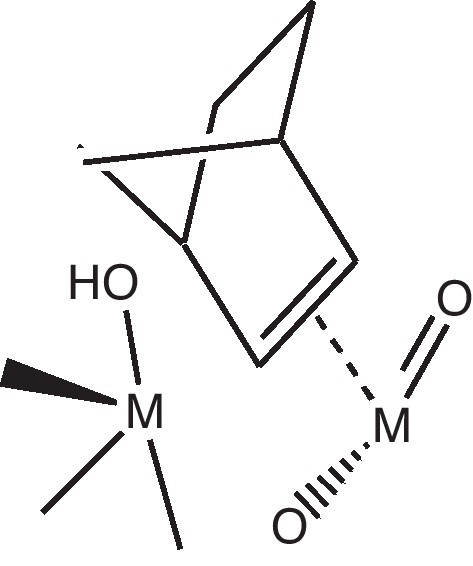
In the presence of a coordinating solvent, this coordination does not occur preventing the polymerization. At that step, the formation of the starting carbene can be only imagined by arising from the inverse of the Wittig reaction: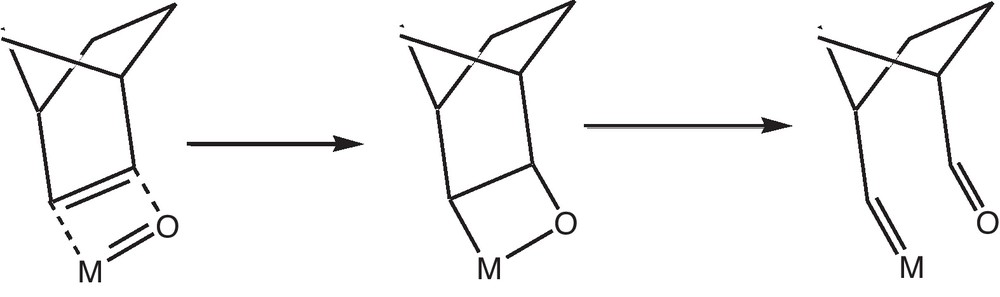
From a thermodynamic point of view, it is well known [15] that the reaction should be shifted to the left but, in this particular case, the driving force is probably the opening of the highly strained norbornene cycle, resulting in the formation of a small amount of carbene.
4 Conclusion
We have shown that the polyoxometalates, in various forms, can catalyze the ROMP of norbornene when activated by various alkylating agents. A study of the reaction of the catalyst with the cocatalyst has shown that there is an O-alkylation of the polyoxometalate rather than a M-alkylation. A model for the formation of the starting initiation carbene has been proposed from these data. It can be expected that, if the carbene species is part of a polyoxometalate entity, the nature of the starting polyoxometalate will have an influence not only on the activity but also on the structure of the polymer. Preliminary studies by 13C NMR of the polymers have shown differences depending on the catalyst. Further studies are in progress to correlate the structure to the polyoxometalate and attempts to isolate the initiation carbene are in progress.