

1 Introduction
2-oxo-1,2,3,4-tetrahydropyrimidines (THPMs) also known as Biginelli compounds [1] have shown a wide scope of important pharmacological and biological properties such as anti-hypertensive, antiviral, antitumor, anti-inflammatory and calcium channel blockers [2–7]. Oxidation of these compounds to their corresponding 2-oxo-1,2-dihydropyrimidines (DHPMs) facilitates the synthesis of new heterocyclic compounds. While few works were devoted to the oxidation of THPMs, the authors of [8–10], recently we reported the effective oxidation of these compounds bearing the acetyl, carboethoxy or carboxamide groups on the 5-positon of the heterocyclic ring under thermal and sono-thermal conditions and microwave or UV irradiations [11–16]. The results of our studies concerning thermal or photochemical oxidation of these compounds elucidate the effect of the nature, the steric hindrance and also their position either on the heterocyclic ring or on the aromatic ring attached to C4 atom on the rate of reaction.
Based on our proposed mechanism for the thermal oxidation of THPMs, the removal of hydrogen from heterocyclic ring occurs in the rate determining step [13,14]. This argument is supported by the influence of the steric and the electronic effects of the substituents on the rate of reaction. These observations prompted us to carry out the computational studies verifying these effects on the optimized structures of various 4-substituted 5-acetyl-, 5-carboethoxy- and 5-carboxamide-THPMs [17,18]. The computational results on these compounds have shown that independent of the type of the substituents located on the 4- or 5-positions, the pyrimidine core has a quasi-boat conformation (Fig. 1) in which the C4 substituent occupies the pseudo-axial position. The deviation of the C4 and N1 atoms of the heterocyclic ring from planarity (C2N3C5C6 plane) depends on the type of the substituent located on C4 and C5, and also on the position of the substituent on the phenyl ring located on C4, that also determining the dihedral angles of the aromatic ring and the carbonyl group with respect to the heterocyclic ring atoms. All these observations reflect the electronic and steric effects of the substituents.
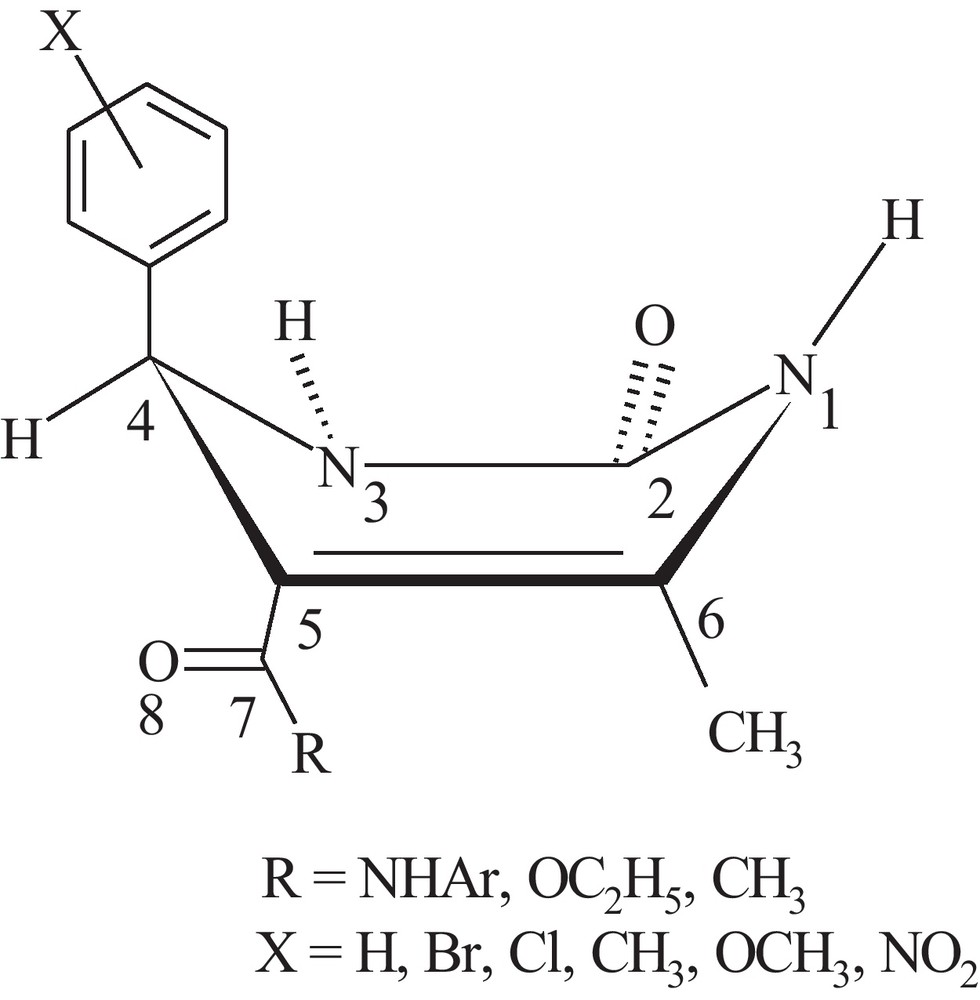
The pseudo-boat conformation of the heterocyclic ring in THPMs showing the pseudo-axial orientation of the C4 aryl group and out-of-plane positions of the C4 and N1 atoms.
The oxidative reaction of many organic compounds is described by an electron transfer mechanism, thus investigation of the electrochemical oxidation of these compounds is one confident way to elucidate the electronic and steric effects of various substituents on their oxidation potentials. Based on our literature survey, so far little works have been reported on the voltammetric studies of 2-oxo-1,2,3,4-tetrahydropyrimidines [19–21]. In our recent study, we investigated the electronic and steric effects of the substituents on the 4- and 5-positions of heterocyclic ring of various substituted 2-oxo-1,2,3,4-tetrahydropyrimidin-5-carboxamides using voltammetric techniques [22]. Analysis of voltammetric and computational studies in that study explained that several factors such as type and position of the substituent on the C4 and C5, deviation of the aryl group on C4 relative to the heterocyclic ring, orientation of the carbonyl group on C5 relative to the C5C6 bond of heterocyclic ring, and finally deviation of C4 and N1 atoms from the boat plane affect the facility of electrochemical oxidation and shift in the oxidation peak potential. Photo-oxidation of THPMs in CHCl3 solution is proposed to occur by an electron transfer from electronically excited THPMs to CHCl3 as electron acceptor molecule [12,15]. In continuation to the photochemical study of THPMs, we were interested to investigate the electron-donating ability of 1-, 4- and 5-substituted THPMs in the ground state towards photoexcited TiO2 nanoparticles in acetonitrile solution (Scheme 1) [23]. The results of this investigation explain that, besides the effect of the nature of solvent and the substituent on the 4- and 5-positions on the rate of reaction, the electronic and the steric effects of the substituent on the 1-position of the heterocyclic ring has a drastic effect. While, the presence of the methyl group on the 1-position of the heterocyclic ring (N1CH3) decreases the irradiation time needed for total conversion, the presence of the phenyl group (N1C6H5) increases this time as compared with that of the N1H derivatives. These observations lead us to investigate these effects by voltammetric methods also by the computational studies in the present work.
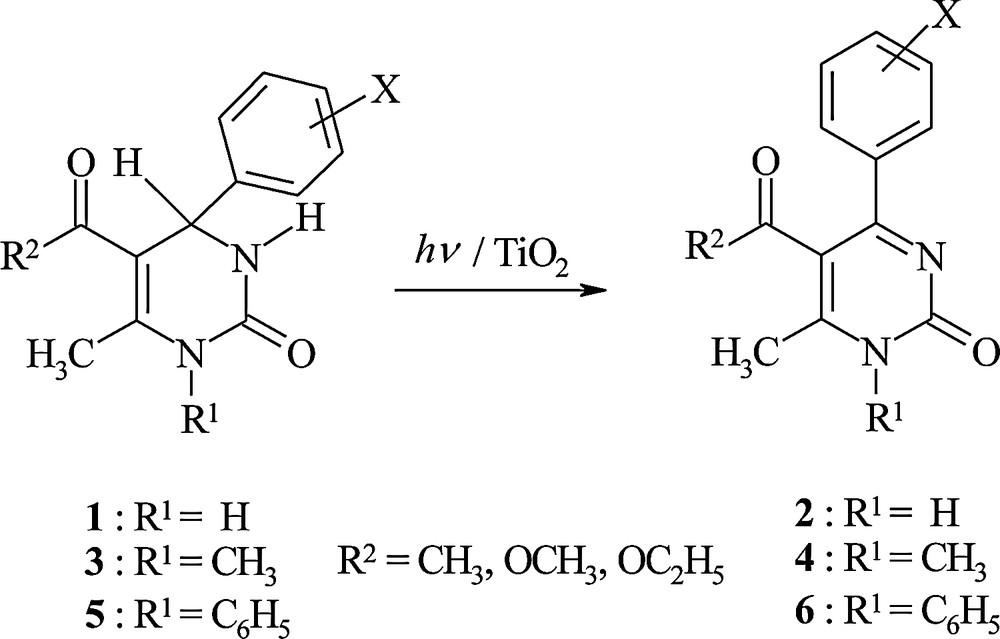
TiO2 sensitized oxidation of 2-oxo-1,2,3,4-tetrahydropyrimidines.
2 Results and discussion
The oxidation peak potential of the cyclic voltammograms obtained for various 4- and 5-substituted THPMs (1a-q), N1-methyl substituted THPMs (3a-q), N1-phenyl substituted THPMs (5a,g,k) are given in Table 1.
The oxidation peak potential (Ep), vs. ferrocene redox potential (0.583 V), obtained from cyclic voltammograms in acetonitrile for THPM (1a-q; R1 = H), N1-methyl substituted THPM (3a-q; R1 = CH3) and N1-phenyl substituted THPM (5a,g,k; R1 = Ph).
| 1 | X | R2 | E (V) | 3 | X | R2 | E (V) | 5 | X | R2 | E (V) |
| a | H | OC2H5 | 1.513 (1.439)a (1.265)b | a | H | OC2H5 | 1.410 (1.397)a (1.238)b | a | H | OC2H5 | 1.498 (1.455)a (1.335)b |
| b | 4-Br | OC2H5 | 1.554 | b | 4-Br | OC2H5 | 1.457 | ||||
| c | 4-F | OC2H5 | 1.573 | c | 4-F | OC2H5 | 1.426 | ||||
| d | 2-OCH3 | OC2H5 | 1.365 | d | 2-OCH3 | OC2H5 | 1.249 | ||||
| e | 3-OCH3 | OC2H5 | 1.472 | e | 3-OCH3 | OC2H5 | 1.371 | ||||
| f | 4-OCH3 | OC2H5 | 1.464 | f | 4-OCH3 | OC2H5 | 1.367 | ||||
| g | 4-NO2 | OC2H5 | 1.610 | g | 4-NO2 | OC2H5 | 1.502 | g | 4-NO2 | OC2H5 | 1.577 |
| h | H | OCH3 | 1.594 | h | H | OCH3 | 1.443 | ||||
| i | 4-Br | OCH3 | 1.608 | i | 4-Br | OCH3 | 1.482 | ||||
| j | 4-F | OCH3 | 1.583 | j | 4-F | OCH3 | 1.471 | ||||
| k | H | CH3 | 1.403 | k | H | CH3 | 1.266 | k | H | CH3 | 1.370 |
| l | 4-F | CH3 | 1.499 | l | 4-F | CH3 | 1.441 | ||||
| m | 2-OCH3 | CH3 | 1.352 | m | 2-OCH3 | CH3 | 1.226 | ||||
| n | 3-OCH3 | CH3 | 1.465 | n | 3-OCH3 | CH3 | 1.345 | ||||
| o | 4-OCH3 | CH3 | 1.456 | o | 4-OCH3 | CH3 | 1.330 | ||||
| p | 4-NO2 | CH3 | 1.600 | p | 4-NO2 | CH3 | 1.481 | ||||
| q | H | Ph | 1.488 | q | H | Ph | 1.373 |
a The measurements were carried out in propan-2-ol.
b The measurements were carried out in ethanol.
The results presented in Table 1 show clearly that the steric and electronic parameters of the substituents on the 1-, 4- and 5-positions of the heterocyclic ring influence the value of the oxidation peak potential (Ep). The following points contribute to these effects:
- • the presence of the methyl group on the 1-position of the heterocyclic ring (R1) in all THPMs decreases drastically the oxidation peak potentials compared with the hydrogen atom on this position, while the presence of the phenyl group causes little effect. These results are demonstrated in Fig. 2 presenting the cyclic voltammograms obtained for 1a, 3a and 5a as representative THPMs, in acetonitrile;
- • the presence of the carboethoxy group instead of the carbomethoxy group on the 5-position of the heterocyclic ring with the same substituent at 4-position decreases the oxidation potential (compare the values for compounds 1a-c with those of 1h-j);
- • in most cases, substitution of the ester groups with the acetyl group on the 5-position does not influence significantly the oxidation peak potential;
- • in contrast to the electron-withdrawing substituent on the phenyl group, the presence of the electron-releasing substituent enhances the electron density on the aromatic ring, which displays lowering of the oxidation peak potential. This is evidence that the sum of the negative inductive and the resonance effects of the nitro group on the 4-position of the aromatic ring compared with the balance of the negative inductive and the positive resonance effects of the methoxy group on the same position enhances the oxidation peak potential of the former case (compare these values for 1g, 1p, 3g, 3p with those of 1f, 1o, 3f and 3o, respectively);
- • the nature of solvent, polar protic vs. polar aprotic, affects the oxidation peak potential (compare the values reported for compounds 1a, 3a and 5a in acetonitrile, propan-2-ol and ethanol).

Cyclic voltammograms of compounds 1a, 3a and 5a and a blank sample in acetonitrile, demonstrating the effect of the substituent on the N1-position.
In order to clarify the above mentioned points on the obtained oxidation peak potentials, computational studies were carried out on THPMs, the species derived from them after first electron detachment THPṀ+, after proton removal TrHPṀ, and finally the oxidation products DHPMs. The geometrical parameters used in the analysis of the results introduced and presented in Table 2 where a sample set of the optimized values of these parameters are also reported. A full list of optimized geometrical parameters for all THPMs and their radicals, radical cations, and oxidized products are reported in Table S1 of the supplementary data. In addition to the gas phase calculations and due to the observed solvent effect on the oxidation peak potential, computational studies were also carried out in the bulk of solvent (acetonitrile) using the CPCM model and also in the presence of solvent molecule (acetonitrile, ethanol and propan-2-ol) for compounds 1a, 3a and 5a as representatives for N1-substituted THPMs.
Structural parameters of THPMs (1a, 3a and 5a) and the corresponding species obtained after electron detachment and proton removal.
| THPM | THPM·+ | ||||||||||||
| TrHPM | DHPM | ||||||||||||
| N1C2 | C2N3 | N3C4 | C4C5 | C5C6 | N1C6 | C4C11 | α | β | γ | μ (D) | hN | hC | |
| 1a | |||||||||||||
| THPM | 1.4015 | 1.3603 | 1.4687 | 1.5280 | 1.3648 | 1.3862 | 1.5346 | −174.0 | −42.7 | 81.5 | 3.9575 | 0.1267 | 0.3650 |
| THPM·+ | 1.4480 | 1.3502 | 1.4558 | 1.5006 | 1.4194 | 1.3399 | 1.5455 | −161.1 | −43.3 | 79.1 | 3.3783 | 0.0707 | 0.2822 |
| TrHPM· | 1.3848 | 1.3777 | 1.4112 | 1.4221 | 1.3863 | 1.3988 | 1.4552 | −53.1 | −31.9 | 150.1 | 3.1122 | 0.0369 | 0.0812 |
| DHPM | 1.4203 | 1.3752 | 1.3163 | 1.4493 | 1.3837 | 1.3555 | 1.4892 | −128.0 | −136.9 | 40.2 | 7.3147 | 0.0791 | 0.0436 |
| 3a | |||||||||||||
| THPM | 1.4104 | 1.3617 | 1.4591 | 1.5271 | 1.3673 | 1.4024 | 1.5371 | −166.0 | −31.6 | 92.0 | 4.1793 | 0.1708 | 0.4248 |
| THPM·+ | 1.4763 | 1.3467 | 1.4530 | 1.4941 | 1.4106 | 1.3511 | 1.5431 | −131.1 | −52.3 | 71.6 | 3.9311 | 0.0704 | 0.1790 |
| TrHPM· | 1.3947 | 1.3773 | 1.4052 | 1.4151 | 1.3895 | 1.4127 | 1.4546 | −59.2 | −30.8 | 149.9 | 2.7323 | 0.0377 | 0.0795 |
| DHPM | 1.4382 | 1.3701 | 1.3141 | 1.4412 | 1.3898 | 1.3644 | 1.4903 | −47.1 | −37.8 | 145.9 | 4.6002 | 0.1434 | 0.0723 |
| 5a | |||||||||||||
| THPM | 1.4160 | 1.3636 | 1.4600 | 1.5256 | 1.3644 | 1.4098 | 1.5369 | −165.7 | −31.4 | 92.3 | 4.0463 | 0.1456 | 0.4329 |
| THPM·+ | 1.4825 | 1.3468 | 1.4539 | 1.4959 | 1.4004 | 1.3674 | 1.5422 | 43.4 | −50.2 | 73.8 | 3.5349 | 0.1201 | 0.1327 |
| TrHPM· | 1.3999 | 1.3787 | 1.4052 | 1.4164 | 1.3878 | 1.4179 | 1.4545 | −57.9 | −30.5 | 149.9 | 2.4369 | 0.0576 | 0.1014 |
| DHPM | 1.4449 | 1.3716 | 1.3135 | 1.4428 | 1.3876 | 1.3695 | 1.4904 | −48.8 | −37.4 | 146.2 | 4.3195 | 0.1999 | 0.0712 |
By analysis of the optimized geometries and electronic characteristics of these species and the measured electrochemical oxidation potentials, it can be deduced that:
- • smaller deviation of the N1 atom from ring planarity allows better delocalization of the nitrogen lone pair towards the heterocyclic ring, especially established with the C5C6 double bond (as being enamine moiety, an electron rich double bond) or towards C2O9 double bond (as being amide group). Therefore, in contrast to the latter case, a decrease in the electrochemical oxidation potential is expected for the former case due to enhanced electron density on the heterocyclic ring;
- • smaller distance from coplanar orientation (0 or 180˚) of the carbonyl group (on the C5 atom) and the C5C6 double bond (defined as α) allows stronger conjugation, consequently decreases the electron density on the heterocyclic ring, which correlates with an increase in the electrochemical oxidation potential;
- • smaller deviation of the C4 atom from ring planarity increases possibility of having trihydropyrimidinyl radicals (TrHPṀ) with more allylic character (over radicals with benzylic character);
- • smaller inter-ring angle between the aryl group and the heterocyclic ring corresponds to more benzylic character for the trihydropyrimidinyl radical (TrHPṀ);
- • the positive inductive effect of the methyl group in contrast to the negative inductive effect of the phenyl group at 1-position is expected to enhance the electron density on the N-1 atom, which leads to a decrease in the electrochemical oxidation potential;
- • the electron detachment process in this study, namely, the electron transfer (ET) from a neutral molecule (THPM) to any acceptor species (A) which yields radical cation species (THPṀ+) and e–or (A–) and the back electron transfer (BET), should be considered like heterolytic bond cleavage in RX molecule to R+ and X– in the rate determining step of a SN1 reaction (Scheme 2).
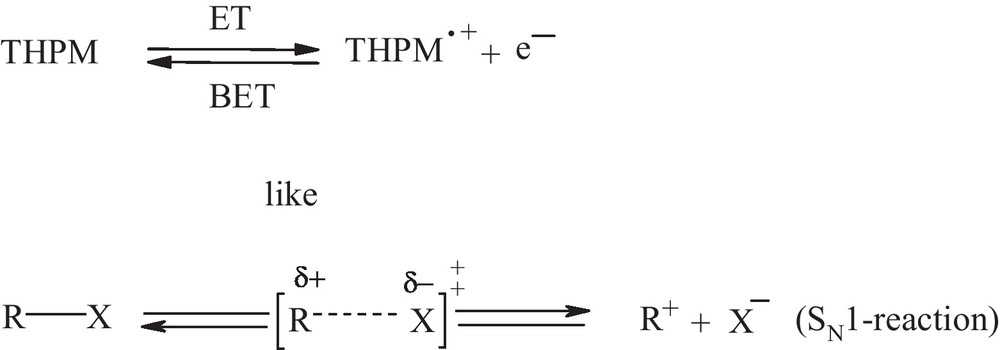
Comparison of THPM electron detachment with SN1 reaction.
It is known that, polar protic solvents, like alcohols (ROH) accelerates the bond cleavage step in SN1 reaction by two factors: firstly, Columbic attraction with the partial charge formed in the transition state which decreases simultaneously the activation energy of this step, and secondly, stabilizing the formed ions by solvation and preventing their recombination. Consequently, a decrease in the oxidation peak potential should be expected by changing the solvent from acetonitrile as a polar aprotic solvent to ethanol or propan-2-ol as polar portic solvent.
We have indeed expected that THPMs be better solvated in ethanol and propan-2-ol than in acetonitrile, and thus a higher oxidation peak potential would be expected when the measurements were carried out in alcohols. Surprisingly, the data reported in Table 1 show that electrochemical oxidation of THPMs is easier in alcohol than in acetonitrile. This can be attributed to the fact, as explained for the SN1 reaction, that the radical cation species (THPṀ+) formed after electron removal from THPM is solvated more strongly in alcohol due to coulombic attraction, as compared to the neutral THPMs. Therefore, back electron transfer (BET) is less favorable in alcohols than in acetonitrile. These results elucidate also that due to smaller size of ethanol than propan-2-ol, solvation of THPM and THPṀ+ is more favored in ethanol, which is reflected in lower oxidation potential in ethanol. This contribution of solvent is illustrated in Scheme 3.

Rational for the electron removal in protic and aprotic solvents.
Computational results obtained for the explicit interaction of 1a and its radical cation 1ȧ+ (as representative) with ethanol molecule (both with fully relaxed structures) are reported in Table 3.
Optimized structures of 1a (a), interaction of 1a with ethanol (b), 1ȧ+ (c) and interaction of 1ȧ+ with ethanol (d).
| a | b | c | d | ||||||||||||
| EtOH in THPM–ethanol | |||||||||||||||
| N1C2 | N1C6 | C5C6 | N1H | C2O9 | hN | hC | α | β | θ1 | θ2 | N1H···O | CO···HOEt | COH | CO | OH |
| 1a | |||||||||||||||
| 1.4015 | 1.3862 | 1.3648 | 1.0102 | 1.2267 | 0.1267 | 0.3650 | −174.0 | −42.7 | −4.3 | 0.9 | – | – | – | – | – |
| 1a–EtOH | |||||||||||||||
| 1.3903 | 1.3855 | 1.3653 | 1.0213 | 1.2387 | 0.1020 | 0.3403 | −174.0 | −49.1 | −3.2 | −0.2 | 1.9750 | 1.8902 | 110.4 | 1.4292 | 0.9799 |
| 1ȧ+ | |||||||||||||||
| 1.4480 | 1.3399 | 1.4194 | 1.0175 | 1.2267 | 0.0710 | 0.2825 | −161.1 | −43.3 | 0.6 | −0.1 | – | – | – | – | – |
| 1ȧ+–EtOH | |||||||||||||||
| 1.4420 | 1.3338 | 1.4239 | 1.0534 | 1.2145 | 0.0516 | 0.2388 | −153.4 | −48.8 | −0.5 | 0.5 | 1.6949 | – | 108.4 | 1.4593 | 0.9679 |
The results obtained for the interaction of other compounds (3a and 5a), and their corresponding radical cations (3ȧ+ and 5ȧ+) with ethanol and propan-2-ol are reported in Tables S2–S6 of the supplementary data. Analysis of the results shows that the interaction of 1a with the ethanol has caused:
- • decrease of hC and hN (i.e. less deviations from ring planarity), which are especially evident in the decrease in the dihedral angles θ1 and θ2 (θ1 = HN1C2O9, θ2 = HN1C6C10);
- • shortening of the N1C2, N1C6 bond lengths and simultaneous increasing of the N1H, C2O9 and C5O6 bond lengths, which are definitely due to better cross-conjugation of N1 lone pair with both unsaturated neighboring double bonds;
- • enlarging of COH angle, shortening the CO bond and increasing the OH bond in ethanol molecule, which is naturally expected for any hydrogen bond system.
The electron detachment from 1a occurs initially at the N1 center. The optimized structure of this radical cation species (1ȧ+) shows following changes compared with its neutral mother molecule 1a:
- • stabilization of the radical cation on the N1 center is achieved by better conjugation of the unpaired electron with C5O6 double bond as being a heteroallyl radical, which is evident from the drastic decrease in the N1C6 and simultaneous increase in the C5O6 and N1H bond lengths;
- • further decreasing of hC and hN (i.e. lesser deviations from ring planarity);
- • decreasing of the dihedral angle α (describing orientation of the CO group attached to C5 with respect to the neighboring double bond C5C6), and further decreasing of θ1 and θ2.
The stronger interaction (Columbic and hydrogen bonding) of ethanol with 1ȧ+ results in:
- • decrease in the N1C2, N1C6, C2O9 and increase in the C5C6 and N1H bond lengths;
- • decrease in hC, hN and α;
- • stronger hydrogen bond (i.e. shortened N1H······O and ethanol OH, and increased ethanol CO bond lengths);
- • interestingly, the missing of hydrogen bond interaction between the C2O9 oxygen atom and the OH hydrogen atom of ethanol which is due to the presence of the positive charge on the neighboring inducing N1 atom;
- • the calculated energy contents of 1a, 3a and 5a, and corresponding radical cation species in the gas phase and in the bulk of ethanol, illustrated in Fig. 3, indicates clearly the solvation effect of ethanol which stabilizes the radical cation species 1ȧ+, 3ȧ+ and 5ȧ+ more effectively than their neutral parent molecules 1a, 3a and 5a. The obtained comparative differential energy contents of these molecules indicate clearly different steric and electronic effects of the substituents located on the N1 atom, and on the delocalization of the N1 lone pair in the neutral molecules and unpaired electron in the radical cation species depending on the deviation of N1 atom (hN).
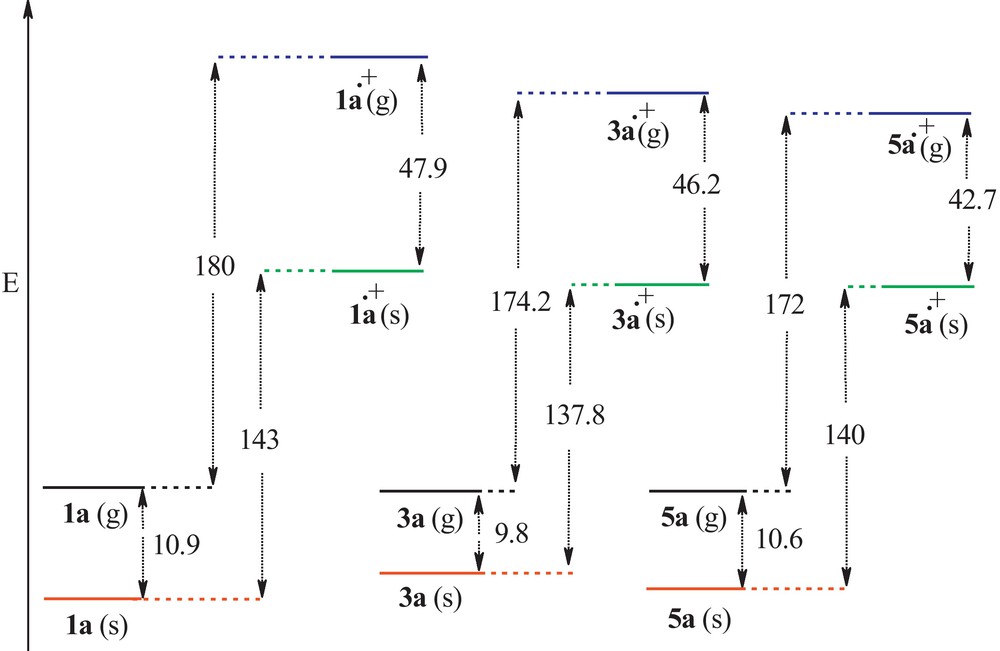
The calculated energy difference (kcal/mole) between 1a, 3a and 5a, and corresponding radical cation species in the gas phase (g) and in the bulk of ethanol (s).
The comparative behaviors of the presence of the methyl and the phenyl groups on the N1 atom of the heterocyclic ring vs. hydrogen atom are illustrated in Fig. 4 in terms of the correlation between differential charge on the N1 atom, (ΔqN1) and differential deviation of the N1 atom from ring planarity (ΔhN1) and the electrochemical oxidation potentials.

Correlation between electrochemical oxidation potentials E (eV) and (a) differential charge on the N1 atom [ΔqN1: q (THPM·+)–q (THPM)], and (b) differential deviation of N1 atom from ring planarity [ΔhN1: hN1 (THPM·+)–hN1 (THPM)].
Fig. 4a shows clearly that decrease in the electrochemical oxidation potential due to the methyl substitution is closely correlated to the increase in the differential charge (from the parent molecule to its radical cation species) on the N1 atom. This is while; increase in the oxidation potential due to the presence of the electron-withdrawing phenyl group, as compared to that of the methyl group, is correlated to the decrease in the differential charge on the N1 atom. It is expected that the greater decrease in the N1 negative charge (denoting the loss of electron density) observed for the methyl substituted THPM 3a as compared to that of the unsubstituted 1a in the first electron removal step, results in lower oxidation potential which is compatible with the experimental results. Fig. 4b shows the correlation of the electrochemical oxidation potential with the differential deviation of N1 atom from ring planarity, denoting the effect of the type of the substituent on the N1 atom compared to the hydrogen atom. A higher differential deviation correlates with easier electron removal from the parent molecule and stabilization of the formed radical cation on the N1 atom via delocalization. This diagram indicates clearly that the perpendicular orientation of the phenyl group on the N1 atom with respect to the N1 lone pair orbital and higher deviation of N1 from ring planarity in 5a due to the gauche interaction with 6-CH3 group, do not allow the phenyl ring to play its resonance role. Therefore, the phenyl group on N1 induces a negative inductive effect only. Thus, electrochemical oxidation potential of 5a does not differ significantly from that of 1a. In contrast, the presence of the electron-donating methyl group on the N1 atom and the reduced repulsion of this group with 6-CH3, leads to more lowering of hN1 (deviation of N1 from ring planarity) value, and consequently, to easier electrochemical oxidation. Numerical values of the relevant atomic charges are presented in Table S7 of the supplementary data.
We have expected that by substitution of the carboethoxy group with the acetyl group on the 5-position of the heterocyclic ring, due to higher electron-withdrawing nature of the acetyl group, to see an increase in the electrochemical oxidation potential, opposite to what was observed experimentally. This observation can be justified by comparison of the α dihedral angle formed between 5-CO and the C5C6 double bonds (–174.0˚,–166.0˚,–165.7˚,–175.0˚,–160.0˚ and –164.8˚ for 1a, 3a, 5a, 1k, 3k, and 5k, respectively) and hN1 values (0.1267 Å, 0.1708 Å, 0.1456 Å, 0.1535 Å, 0.1587 Å, and 0.1750 Å for 1a, 3a, 5a, 1k, 3k, and 5k, respectively). These values indicate that the larger deviation of hN1 and/or larger deviation of α values allow more electron density on the N1 atom, which contribute to the easier electron removal.
Effects of the electronic nature (electron-donating/withdrawing) of the additional substituent, vs. hydrogen atom, on the phenyl ring at the C4-position, on the electrochemical oxidation potentials are illustrated in Fig. 5.
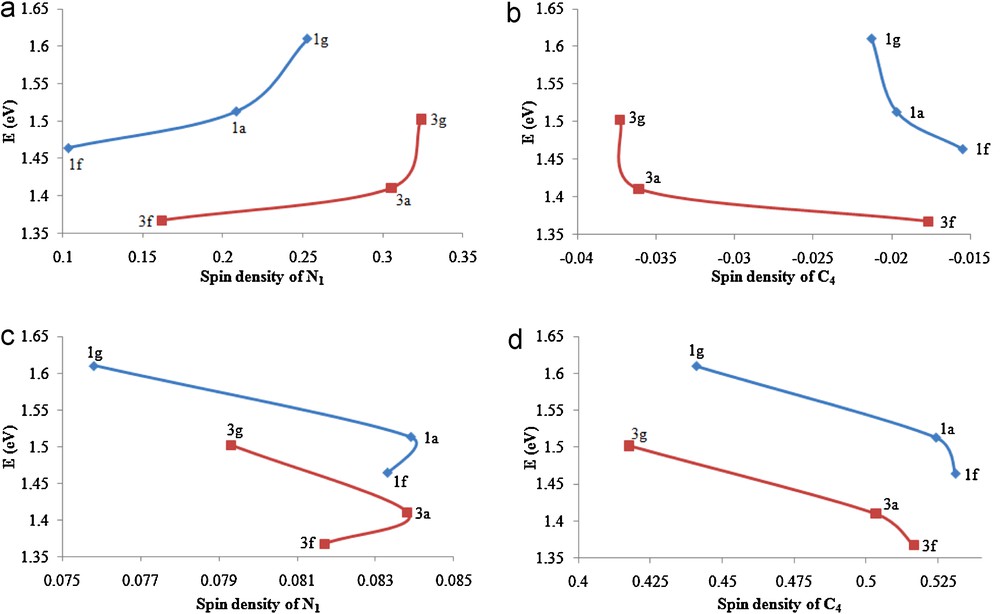
Correlation between electrochemical oxidation potentials E (eV) vs. the spin density on the N1 and C4 atoms in the radical cation species (a and b), and the spin density on the same atoms in the radical species (c and d).
It can be seen from Fig. 5 that C4 and N1 do not carry significant spin density in the radical cation and radical species, respectively. Correlations between E and the spin densities on the N1 and C4 atoms in the radical cations of the 1a, 1f, 1g, 3a, 3f and 3g series of THPMs, illustrated in Fig. 5a–d, show that the spin N1 density orders of 1f < 1a < 1g and 3f < 3a < 3g, corresponding to the reverse order, namely 1f > 1a > 1g and 3f > 3a > 3g, for the spin density on the C4 atom in the corresponding radical species (Fig. 5d) indicates that the presence of the electron-releasing 4-OCH3 group on the C4-phenyl ring (in 1f) facilitates electron removal from neutral molecule, whereas the presence of electron-withdrawing 4-NO2 group (1g) retards this process. This can be attributed to the fact that the balance of the negative inductive and the positive resonance effect of the 4-OCH3 group shift the electron density of the aromatic ring toward C11 attached to C4.
Further electron pushing through the sigma bonds stabilizes the formation of the positive charge on N1 atom during the electron removal step from 1f THPM, whereas 4-NO2 group in 1g THPM has an opposite effect as illustrated in Scheme 4.
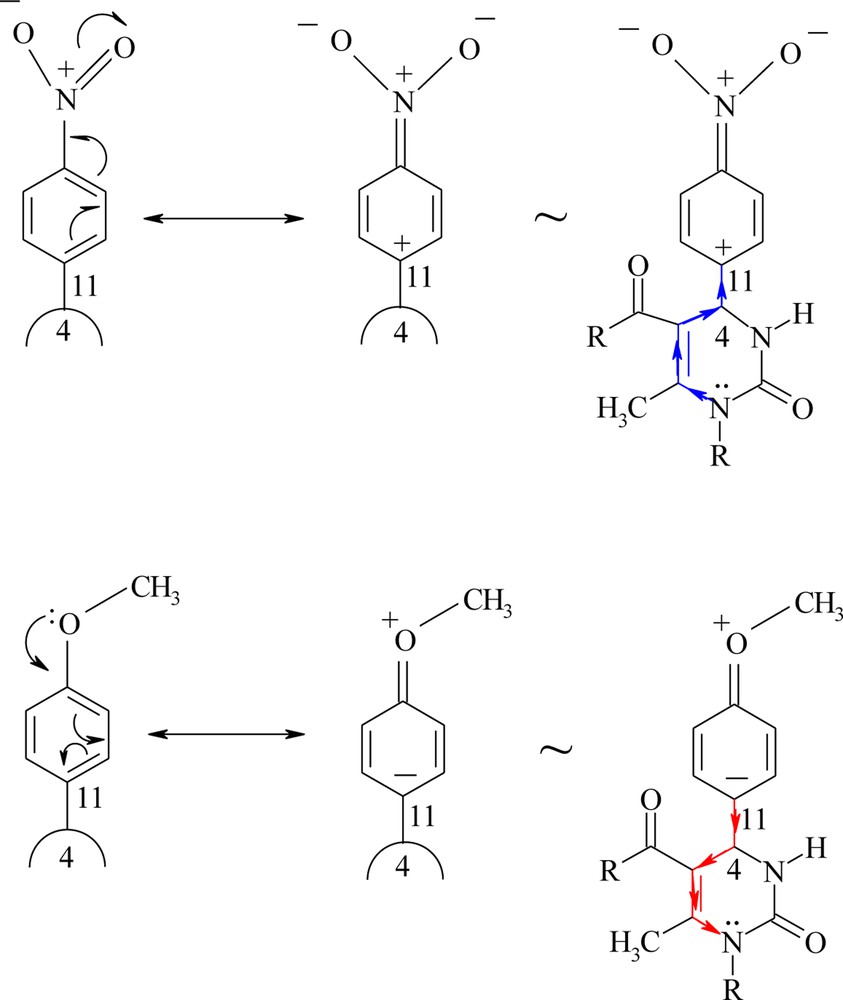
The inductive effect of the substituent on the electronic nature of heterocyclic ring.
This suggestion is supported by the lower spin density on C4, because the positive charge and the unpaired electron is delocalized dominantly over the N1 and C5 atoms than on the C4 atom, as shown in Fig. 4b. It should be noted that the electron-donating effect of the methoxy group is also supported by the obtained dihedral angles of the methoxy group on the 2-, 3- and 4-positions of the phenyl ring with respect to the aromatic ring atoms (between 176.1˚ and 179.8˚) and also the CaromaticOCH3 bond angle (between 118.5˚ and 118.9˚). This is shown in Fig. S1 in the supplementary data.
The interesting point is that after proton detachment from THPṀ+ and formation of TrHPṀ, the radical center formed on the C4 atom has simultaneously allylic and benzylic characters. The higher weights of the benzylic radical over the allylic radiacl of the g compounds compared with those of the f compounds (Table S8 of the supplementary data) are possibly due to:
- • the more effective SOMO-LUMO interaction over the SOMO-HOMO interaction facilitated by the presence of the 4-NO2 group;
- • closer orientation of the aryl ring to the coplanarity with the heterocyclic ring in the NO2-substituted g THPMs (e.g. 31.9˚ to 33.5˚ in 1f vs. 46.8˚ to 27.0˚ in 1g).
3 Conclusions
The effect of the substituent on the electrochemical oxidation of various in 1-, 4- and 5-positions substituted 2-oxo-1,2,3,4-tetrahydropyrimidines (THPMs) in acetonitrile is studied using cyclic voltammetry. The results clearly indicate that several factors affect the electrochemical behavior of these compounds. The important factor is the positive inductive effect of the methyl group vs. the negative inductive effect of the phenyl group in 1-position of the heterocyclic ring, the electronic effect of the additional substituent on the phenyl ring at C-4 position of the heterocyclic ring, and also the type of the substituent on the 5-position. The nature of solvent, protic vs. aprotic, has a drastic effect on the electrochemical oxidation potentials.
Computational study at DFT-B3LYP/6-31++G** level of theory is carried to calculate different structural and bonding characteristics, and to investigate their correlation with the measured electrochemical oxidation potentials. These computational results approve the proposed mechanism in which a protic solvent stabilizes the intermediate radical cation resulting in easier electrochemical oxidation of THPMs.
4 Experimental
4.1 Cyclic voltammetry measurements
Various substituted 2-oxo-1,2,3,4-tetrahydropyrimidines (1a-q, 3a-q and 5a,k,g) were synthesized according to the reported procedure [24]. Solvent used in this study, acetonitrile, ethanol and propan-2-ol were purified and dried by standard method. Solutions with concentrations of 1 mM of THPM and 100 mM of tetrabutylammonium perchlorate in acetonitrile, ethanol and propan-2-ol were prepared. The cyclic voltammetry (CV) measurements were performed using SAMA 500 Potentio/Galvanostat. All electrochemical experiments were carried out in a conventional three electrode system at room temperature. A silver electrode, a large area Pt plate (99.99%), and a glass carbon electrode (GCE) were used as reference, counter and working electrodes, respectively.
4.2 Computational methods
Geometries of THPMs and DHPMs were optimized using the density functional theory (DFT) B3LYP method with 6-31++G(d,p) basis set. All computations are carried out using G98 program package [25]. Compounds 1a, 3a and 5a (R1 = H, CH3 and Ph, respectively) all with X = H, are considered as representative THPMs to investigate comparatively the effect of the substitution on the aryl group in the C4-position and the substituent on the N1 atom on the optimized structures of compounds. Charge density distributions for all THPMs, and the radical cation (THPṀ+) and radical (TrHPṀ) species derived from them have also been studied using natural bond orbital (NBO) analysis [26–29]. To elucidate the effect of solvent on the structure and bonding characteristics, and the consequent effects on the electrochemical behavior, two sets of computations are carried out on these compounds; one in the bulk of solvent using the SCRF = CPCM model, and one in the presence of a solvent molecule (acetonitrile, ethanol or propan-2-ol) coordinating the primary oxidation site (i.e. the N1 atom of the heterocyclic ring). Note that, in the geometry optimization of the explicit THPM–ethanol interaction, another initial geometry in which the ethanol molecule faces the N3 atom is also adopted. However, the optimization resulted in exactly the same geometry as obtained when ethanol initially faced the N1 atom. This is shown in Fig. S2 in the supplementary data.
Acknowledgments
We thank the Center of Excellence (Chemistry), Research Council and Office of Graduate Studies of the University of Isfahan for their financial support. HRM is indebted to thank Dr. A. Rahimi, the former research vice chancellor for providing the SAMA 500 Potentio/Galvanostat.