

1 Introduction
Development of greener synthetic methodologies along with fast access towards the desired compounds is one of the fundamental objectives to speed up current organic synthesis [1]. In this regard, although the use of MWs [2], ultrasound waves [3], alternate reaction media [4], etc. are a few of the well accepted tools to expedite reaction rates, higher chemical efficiencies with remarkable rate acceleration can also be expected with the choice of an appropriate catalyst and absence of the reaction medium. The examples reported demonstrate that solvent-free reactions are generally faster, giving higher reactivity and involve easier work-up procedures [5,6].
α-Hydroxy phosphonates as well as α-hydroxy phosphonic acids have attracted much attention of organic chemists in recent years primarily because of their important biological activities such as antibacterial, antiviral, anticancer, rennin inhibitory, HIV-protease, enzyme inhibitor properties, etc. [7]. They also serve as useful precursors in the synthesis of other biologically important phosphonates like α-amino [8], α-keto [9], α-diketo [10], α-halo [11] as well as α-acetoxy phosphonates [12]. During the ongoing studies in our laboratory on the development of greener synthetic methodologies [13] in the recent past, we had reported an accidental result on the formation of α-hydroxy phosphonates during a three component condensation between an aldehyde, diethyl phosphite and diethyl amine [13a]. This initial observation led us to study the hydrophosphylation reaction in detail.
Hydrophosphylation of aldehydes (Pudovik reaction) is typically a base-catalyzed reaction [14]. Thus, the traditional synthesis of α-hydroxy phosphonates involves the reaction between an aldehyde and di/trialkyl phosphite in presence of a basic catalyst such as ethyl magnesium bromide [15a], quinine [15b], KF-alumina [15c], CsF [15d], MgO [15e], DBU [15f], LDA [15g], quaternary ammonium hydroxide [15h], etc. On the other hand, a few Brønsted acid catalysts such as HCl [16a], Amberlyst-15 [16b], guanidine-HCl [16c], oxalic acid [16d], potassium dihydrogenphosphate [16e], camphor sulfonic acid [16f], etc. have also been explored in recent years for the synthesis of α-hydroxy phosphonates. Each of these methods has its own merits. Interestingly, most of the base-catalyzed hydrophosphylation reactions are operable at ambient temperature while acid-catalyzed protocols require either thermal or ultrasound activation. Furthermore, a few of these protocols are plagued with the essentiality of more than a stoichiometric amount of catalyst or longer reaction time. With the rapid progress both in chemistry as well as biology of α-hydroxy phosphonates we surmised that the introduction of a simple, economic, high yielding and highly rapid protocol for the synthesis of α-hydroxy phosphonates at ambient temperature under solvent-free condition i.e. meeting closely to demands of an “ideal synthesis” [17] is desired.
2 Results and discussion
Potassium phosphate is an easy to handle, non-toxic and relatively inexpensive basic catalyst available commercially. It has been used earlier by us in Knoevenagel condensation [18a], Henry reaction [18b], thia-Michael addition reaction [18c], Claisen-Schmidt condensation [18d] as well as in few multicomponent reactions [18e,f]. In continuation of these studies, herein we report the use of potassium phosphate as catalyst in hydrophosphonylation of aldehydes (Scheme 1a). It is worthy to note that, apart from its remarkable basicity, choice of potassium phosphate as a catalyst was primarily concerned with its coordinating ability [18c,e,f] as well as the plausible mechanism of hydrophosphylation (Scheme 1b).
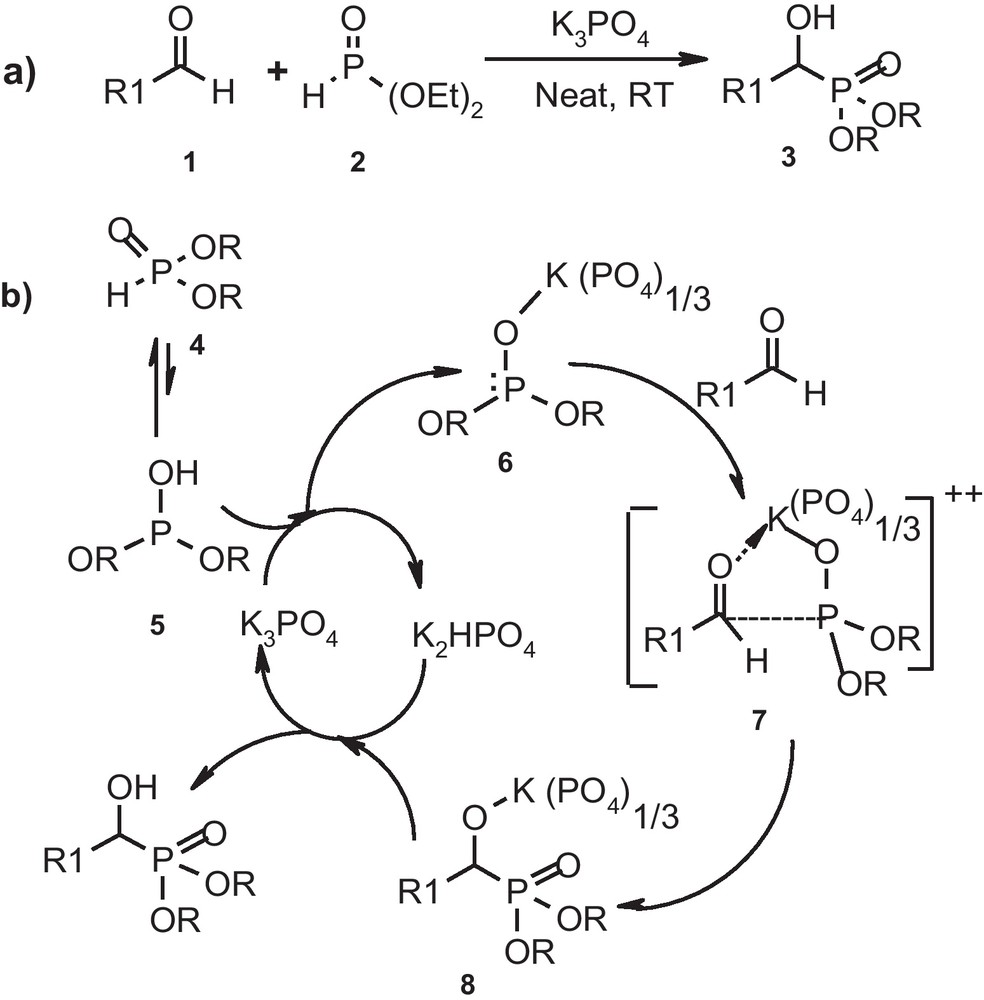
Plausible mechanism for expeditious formation of α-hydroxy phosphonate.
It is well known that dialkyl phosphite exists in equilibrium between its phosphite, 4 and phosphonate, 5, forms and under neutral condition, the equilibrium predominantly lies towards phosphonate form with sluggish reactivity [19a,b]. Thus, in achieving hydrophosphylation, facilitation of phosphonate-phosphite tautomerism is essential and a simple technique in accelerating this tautomerism is to effect deprotonation of phosphonate using a base [19]. The resultant dialkyl phosphite anion would then attack nucleophilically over carbonyl carbon in aldehyde to furnish corresponding α-hydroxy phosphonate. This explanation clearly justifies the choice of potassium phosphate as base in the synthesis of α-hydroxy phosphonates. However, it does not throw light upon the speed of reaction. In this context, taking clues from the mechanism proposed by us for Michael addition of thiols to enones, [18c] as well as from the mechanism proposed by de Naronha et al. [20] as well as Merino et al. [21] for hydrophosphylation of aldehydes we believed that by employing potassium phosphate as catalyst dialkyl phosphite anion would result through ligand exchange process. At the same time, potassium ion being sufficiently oxophilic (due to large size of counter ion viz. PO4−3), it would form a strong coordinate bond with oxygen atom in aldehyde. Thus, under solvent-free condition, there would exist a close contact between an aldehyde and phosphite nucleophile as shown in T.S. 7 and this would consequently result in increase in the rate of formation of α-hydroxy phosphonate (Scheme 1b).
Based upon this plausible mechanism, a model reaction was carried out under solvent-free condition between benzaldehyde and diethyl phosphite (1 eqv.; each) using potassium phosphate (20 mol %) as a catalyst. An exothermic reaction took place and upon completion of reaction, (< 5 min, TLC) desired α-hydroxy phosphonate, 3a, was obtained in almost quantitative yield in very high purity (NMR). Optimization of the reaction conditions with respect to catalyst loading alone revealed that, 5 mol % of potassium phosphate was sufficient enough to furnish the targeted α-hydroxy phosphonate, 3a, in same yield as well as purity with a slight compromise in time of the reaction (< 10 min against 5 min). Encouraged with this initial success, we then planned to expand scope of the developed protocol. Accordingly, diethyl phosphite component in the above reaction was replaced with dimethyl as well as di-iso-propyl phosphite. In both the cases, the corresponding α-hydroxy phosphonate was obtained in a very short time and in excellent yield. Interestingly, with the choice of trimethyl phosphite wherein the possibly of formation of phosphite nucleophile through ligand exchange process is not possible, the reaction required very long time (3 h).
Under the established reaction conditions for 3a in hand, generality as well as scope of the protocol was then explored by performing the reactions between a variety of aromatic, hetero-aromatic as well as conjugated aldehydes with diethyl, dimethyl as well as di-iso-propyl phosphite. It was observed that the hydrophosphylation of aldehydes containing electron-withdrawing as well as electron-donating groups was equally fast (Table 1). However, the reaction with aliphatic aldehyde was sluggish and did not go to completion even upon stirring the reaction mixture overnight (TLC) while ketones failed to furnish corresponding α-hydroxy phosphonates in acceptable yields. It is worth noting that all the isolated products were pure and they did not require any further purification. Since this mild and green protocol proved to be of wide generality to furnish α-hydroxy phosphonates in excellent yields as well as purity, we believed that, the protocol would be of general use if scaled up. Accordingly, scalability studies were performed using benzaldehyde and diethyl phosphite as substrates and we are happy to disclose that, without modification of reaction conditions, protocol was scalable on 10 g scale.
Potassium phosphate catalyzed synthesis of α-hydroxy phosphonates.
| No. | Product (3) | Ra = | Time (min) | Yield (%) | Reference |
| a | 3a, R = C2H5 | 6 | 98 | [16a] | |
| 3b, R = CH3 | 5 | 94 | [16b] | ||
| 3c, R = HC(CH3)2 | 5 | 95 | – | ||
| b | 3d, R = CH3 | 4 | 93 | [16b] | |
| 3e, R = C2H5 | 4 | 95 | [16a] | ||
| 3f, R = HC(CH3)2 | 6 | 96 | – | ||
| c | 3g, R = CH3 | 5 | 93 | [16d] | |
| 3h, R = C2H5 | 5 | 95 | [16a] | ||
| 3i, R = HC(CH3)2 | 6 | 95 | – | ||
| d | 3j, R = C2H5 | 4 | 96 | [13a] | |
| 3k, R = HC(CH3)2 | 5 | 95 | – | ||
| e | 3l, R = C2H5 | 5 | 93 | [13a] | |
| 3m, R = HC(CH3)2 | 5 | 94 | – | ||
| f | 3n, R = C2H5 | 4 | 96 | [16a] | |
| 3o, R = HC(CH3)2 | 5 | 95 | – | ||
| g | 3p, R = CH3 | 5 | 94 | [16b] | |
| 3q, R = HC(CH3)2 | 6 | 93 | – | ||
| h | 3r, R = C2H5 | 8 | 92 | [16a] | |
| 3s, R = CH3 | 7 | 90 | [16b] | ||
| i | 3t, R = CH3 | 6 | 96 | – | |
| 3u, R = HC(CH3)2 | 5 | 95 | – | ||
| j | 3v, R = C2H5 | 4 | 96 | – | |
| 3w, R = HC(CH3)2 | 5 | 95 | – | ||
| k | 3x, R = C2H5 | 5 | 94 | – | |
| 3y, R = HC(CH3)2 | 6 | 96 | – | ||
| l | 3z, R = C2H5 | 5 | 93 | – | |
| 3za, R = HC(CH3)2 | 7 | 94 | – | ||
| m | 3zb, X = O, R = C2H5 | 6 | 92 | [13a] | |
| 3zc, X = S, R = CH(CH3)2 | 5 | 94 | – | ||
| n | 3zd, R = CH3 | 24b | 20 | [16a] | |
| 3ze, R = C2H5 | 24b | 25 | [16b] | ||
| o | 3zf, R = CH3 | 24b | 30 | [16b] | |
| 3zg, R = C2H5 | 24b | 20 | [16a] |
a Reaction conditions: aldehyde and dialkyl phosphite (2 mmol, each), K3PO4 (5 mol %), rt.
b Time in hrs.
In view of green chemistry, we then turned our attention towards reusability of the catalyst. Upon completion of the reaction, methylene chloride was added to the reaction mixture and it was centrifuged using hand centrifuge machine. Organic extract was separated and the catalyst was washed with methylene chloride (3 × 5 mL). After removal of solvent, recovered catalyst was dried in oven for 2 h and reused for the subsequent runs. It was noticed that the catalyst could be reused for at least three cycles. However, in three consecutive runs, a gradual decrease in the yield was noticed (96, 90, and 80).
Finally, the efficacy of potassium phosphate was compared with that of other catalysts reported earlier in the synthesis of α-hydroxy phosphonate, 3a, as a model compound. The data summarized in Table 2 clearly highlights the superiority of potassium phosphate over to other basic as well as acidic catalysts, in terms of yield, cost, time, as well as ease of product isolation. It also reveals that amongst various base catalysts, potassium phosphate alone exhibits remarkable rate accelerating effect.
Comparison of the efficiency of K3PO4 vs. other catalysts in synthesis of 3a.
| No | Catalyst (g/mol %) | Temp (°C) | Time (h) | Yield (%)Ref |
| 1 | MgO (0.2 g) | rt | 2 | 90[15e] |
| 2 | CsF | rt | 0.5 | 98[15d] |
| 3 | KF-Alumina (0.6 g) | rt | 2 | 96[15c] |
| 4 | HCl (100)a | 0 | 24 | 74[16a] |
| 5 | Amberlyst (0.2 g)a | 50 | 1.5 | 95[16b] |
| 6 | Guanidine.HCl (5)a | 50 | 2 | 95[16c] |
| 7 | Oxalic acid (10)a | 80 | 1.5 | 98[16d] |
| 8 | K2HPO4 (5)a | rt,))) | 5b | 86[16e] |
| 9 | MoO2Cl2 (5) | rt | 24 | 89[18] |
| 10 | Et2NH(100) | rt | 5b,d | 98[20] |
| 11 | CSAc | rt | 0.5 | 91[16f] |
| 12 | K3PO4 (5) | rt | 6b | 98 |
a Using trimethyl phosphite.
b Time in min.
c Camphor sulfonic acid -))).
d Using 4-methyl benzaldehyde.
3 Conclusion
In conclusion, we have developed an expeditious, solvent-free and extremely simple protocol for hydrophosphylation of aldehydes using potassium phosphate as catalyst. To the best of our knowledge, literature to date contains no precedent for such a simple, scalable, economical and environmentally conscious protocol for the synthesis of α-hydroxy phosphonates.
4 Typical experimental procedure
To a stirred mixture of an aromatic aldehyde (2 mmol) and dialkyl phosphite (2 mmol) was added potassium phosphate (5 mol %) and stirring continued. Upon completion of the reaction (TLC), methylene chloride (10 mL) was added. After stirring for five minutes, the catalyst was separated by centrifugation and organic extract was syringed out. Removal of solvent from the organic extract furnished respective α-hydroxy phosphonate.
In case of aliphatic aldehydes (1n and 1o), after stirring the reaction mixture overnight, it was worked-up as described above and the residue obtained after removal of solvent from the organic extract was chromatographed over silica gel. Elution with hexane-ethyl acetate (8: 2, v/v) furnished pure α-hydroxy phosphonate.
Spectral data of representative α-hydroxy phosphonates is summarized below.
Di-iso-propyl-1-hydroxy-1-(4-iso-propylphenyl) methyl phosphonate, 3m: M.p. = 118–119 °C, IR (KBr): ν = 3270, 2978, 1513, 1238, 993 cm−1, 1H NMR (300 MHz, CDCl3): δ = 1.09 (d, J = 6 Hz, 3H), 1.23 (brs, 15H), 2.64 (br s, –OH), 2.89 (m, 1H), 4.55–4.68 (m, 2H), 4.88 (d, 1JHP = 10.8 Hz, 1H) 7.18 (d, J = 8.1 Hz, 2H, ArH) 7.40 (d, J = 8.3 Hz, 2H, ArH), 13C NMR (75 MHz, CDCl3): δ = 23.44 (d, 3JCP = 5.0 Hz), 23.81 (d, 3JCP = 5.25 Hz), 23.95 (2 × CH3), 24.03 (d, 3JCP = 3.75 Hz), 24.15 (d, 3JCP = 3.00 Hz), 33.81, 70.92 (d, 1JCP = 160.5 Hz, CPOH), 71.58 (d, 2JCP = 7.5 Hz), 71.92 (d, 2JCP = 7.5 Hz) 126.10, 127.31, 134.26, 148.53; HRMS: Mass calculated for C16H27O4P: 315.1726 (M + H) and 337.1544 (M + Na); Obs. mass 315.1547 (M + H) and 337.1077 (M + Na).
Di-iso-propyl-1-hydroxy-1-(3,4-methylenedioxyphenyl) methyl phosphonate, 3u: M.p. = 114–116 °C, IR (KBr): ν = 3263, 2982, 1505, 1225, 993, 523 cm−1, 1H NMR (300 MHz, CDCl3): δ = 1.18 (d, J = 6 Hz, 3H), 1.28 (d, J = 6 Hz, 9H), 3.74 (br s, –OH), 4.59–4.71 (m, 2H), 4.85 (d, 1JHP = 10.2 Hz, 1H), 5.95 (s, 2H), 6.72 (d, J = 8.1 Hz, 1H, ArH), 6.93 (d, J = 7.8 Hz, 1H, ArH), 7.03 (s, 1H, ArH), 13C NMR (75 MHz, CDCl3): δ = 23.63 (d, 3JCP = 5.25 Hz), 23.88 (d, 3JCP = 4.5 Hz), 24.03 (d, 3JCP = 3.75 Hz), 24.16 (d, 3JCP = 3.50 Hz), 70.83 (d, 1JCP = 154.5 Hz, PCOH), 71.71 (d, 2JCP = 7.5 Hz), 72.03 (d, 2JCP = 7.5 Hz), 101.02 (CH2), 107.14, 120.94, 130.54, 147.33, 147.52; HRMS: Mass calculated for C14H21O6P: 317.2946 (M + H) and 339. 2764 (M + Na); Obs. mass 317.0837 (M + H) and 339.1026 (M + Na).
Di-iso-propyl-1-hydroxy-1-(4-allyloxyphenyl) methyl phosphonate, 3y: M.p. = 74–76 °C, IR (KBr): ν = 3219, 2979, 1610, 1511, 1386, 1210, 1050, 995, 838, 568 cm−1, 1H NMR (300 MHz, CDCl3): δ = 1.13 (d, J = 6.3 Hz, 3H) 1.26 (d, J = 6.3 Hz, 9H), 3.28 (br s, –OH), 4.52 (d, J = 2.7 Hz, 2H) 4.54–4.68 (m, 2H,), 4.87 (d, 1JHP = 10.2 Hz, 1H), 5.27 (dd, J = 10.5 Hz & 1.5 Hz, 1H), 5.40 (dd, J = 17.1 & 1.5 Hz, 1H), 5.98–6.11 (m, 1H), 6.89 (d, J = 8.7 Hz, 2H, ArH) 7.40 (d, J = 8.7 Hz, 2H), 13C NMR (75 MHz, CDCl3): δ = 23.58 (d, 3JCP = 5.25 Hz), 23.87 (d, 3JCP = 4.5 Hz), 24.06 (d, 3JCP = 3.0 Hz), 24.16 (d, 3JCP = 3.0 Hz), 68.78 (OCH2), 70.55 (d, 1JCP = 144.75 Hz, PCOH), 71.68 (d, 2JCP = 8.0 Hz), 71.74 (d, 2JCP = 7.5 Hz), 114.43, 117.62, 128.54, 128.62, 128.98, 133.18, 158.41; HRMS: Mass calculated for C16H25O5P: 329.3484 (M + H) and 351.3302 (M + Na); Obs. Mass 329.3671 (M + H) and 351. 3886 (M + Na).
Di-iso-propyl-1-hydroxy-2-(thiophenyl) methyl phosphonate, 3zc: M.p. = 64–66 °C, IR (KBr): ν = 3245, 2984, 1386, 1231, 986, 704, 555 cm−1,1H NMR (300 MHz, CDCl3): δ = 1.19 (d, J = 6.3 Hz, 3H), 1.31 (d, J = 6.3 Hz, 9H,), 3.59 (br s, –OH), 4.67–4.74 (m, 2H), 5.16 (d, 1JHP = 10.8 Hz, 1H), 6.99 (t, J = 4.1 Hz, 1H), 7.18 (t, J = 3.8 Hz, 1H), 7.28 (1H, dd, J = 4.0 Hz and 1.2 Hz), 13C NMR (75 MHz, CDCl3): δ = 23.58 (d, 3JCP = 5.25 Hz), 23.86 (d, 3JCP = 4.5 Hz), 24.03 (d, 3JCP = 3.75 Hz), 24.18 (d, 3JCP = 3.00 Hz), 67.24 (d, 1JCP = 166.5 Hz, PCOH), 72.05 (d, 2JCP = 3.75 Hz), 72.44 (2JCP = 3.75 Hz), 125.49, 126.09, 126.66, 139.22, HRMS: Mass calculated for C11H19O4PS: 279.3128 (M + H); Obs. Mass: 279.0441 (M + H).
Acknowledgements
The authors (M.A.K. and U.V.D.) are thankful to U.G.C., New Delhi for financial assistance [F. No. 34-362/2008 SR]. We are also thankful to D.S.T. and U.G.C., New Delhi for providing NMR and elemental analyzer facilities to the Department of Chemistry, Shivaji University, Kolhapur under FIST and SAP programme, respectively.