

1 Introduction
Nanoparticles have attracted great attention in modern science [1]. While bulk materials or atomic structures show specific physical properties, the physical responses of nanoparticles are size-dependent [2]. These are manifestations of quantum size effects, [3–5] which can be observed, for instance, in electronic [6,7] and optical properties [8], or in superparamagnetism [9].
Despite some recent progress related to the self-assembly of nanoparticles, problems still remain. Conventional nanoparticles generally have an intrinsic size distribution, which leads to imperfect lattices and can causes severe disorder on the atomic level. In contrast to molecular crystals, lattices of nanoparticles have little to no translation order on an atomic scale. Furthermore, compositional homogeneity, i.e., homogeneous size distribution of nanoparticles, can be difficult to achieve and the interpretation of physical properties can therefore be hindered. Of course, such materials escape X-ray structure determination within atomic resolution, which is a fundamental requirement for a detailed understanding of the chemical and physical properties of nanoparticles.
Recently, we developed an approach to overcome this problem, wherein we combine well-defined building blocks via self-assembly to new binary compounds, which are called intercluster compounds (IC) [10]. ICs exist in combination with fullerides and cationic complexes [11], polyoxometalates (POM) with clusters [12–15]. These ICs were analyzed via single-crystal diffraction, which showed that all the precursor building blocks mostly retained their original constituents [16,17].
Many ICs that configuration are composed from two different building blocks (binary ICs) can be reduced to basic structure types of ionic solids. As examples, the structures of [Au9(PPh3)8][PW12O40] [15] and [Ag8(2,20-bipy)6(C≡CtBu)4][BF4]4 [14] exhibit NaCl type-packing. ICs can also be used to understand the fundamental principles of structure-directing effects, and long- and short-range bonding interactions [13,18].
We intended to extend this concept and to explore whether similar structural relationships are valid for quasi-ternary ICs and basic structure types of ternary compounds based on atomic constituents.
A few quasi-ternary compound systems consisting of a polyoxometalate and two different cationic components exist [19,20], but none of these structures were intentionally prepared, and the arrangements of their basic structure type were not investigated. These kinds of ternary systems could offer a completely new approach for the synthesis of ICs, and could also be used for the production of the multimetallic compounds used as single-source precursors for metal oxides [21].
For the study of a ternary IC, we have chosen three constituents: a triply charged anion, phosphomolybdic acid (the POM in this instance), a dicationic organic molecule, o-xylylenebis(triphenylphosphonium), and a singly charged cationic cobaltocenium complex.
We first investigated the quasi-binary systems, where the cobaltocenium complex and the organic ligand were each assembled separately with the POM. The packing behaviors of these binary systems were studied to determine the presence of basic structure types. Thereafter, the quasi-ternary IC was synthesized via self-assembly and we investigated its structural behavior.
The synthesis, characterization, and study of the structure-directing effects of the quasi-ternary IC and, as references, both related quasi-binary compounds, are presented here.
We chose to use the term quasi-binary and quasi-ternary, since we are disregarding the counter ions or solvent molecules.
2 Results and discussion
In order to have a better understanding of the quasi-ternary compound, we will first present both quasi-binary compounds, one comprising POM and cobaltocenium, and the other composed of POM and the organic molecule (o-xylylenebis(triphenylphosphonium)).
2.1 Crystal structures
2.1.1 [PMo12O40][CoC10H10]3 * 2(C2H6OS), 1
When phosphomolybdic acid and bis(cyclopentadienyl)cobalt(III) hexafluorophosphate (also known as cobaltocenium hexafluorophosphate) are mixed in DMSO, yellow crystals can be obtained after crystallization.
The asymmetric unit of 1 consists of one [Mo12O40P]3− polyanion, three cobaltocenium cations, and two DMSO solvent molecules. Compound 1 crystallizes in the triclinic space group P-1. The ratio of the Keggin ion to the cobaltocenium cations is 1:3 and is different from the observed ratio in the very similar ferrocenium system [22]. Fig. 1 shows the packing of 1 in the solid state along the x-direction, with each cobalt ion in a different color for enhanced visualization.
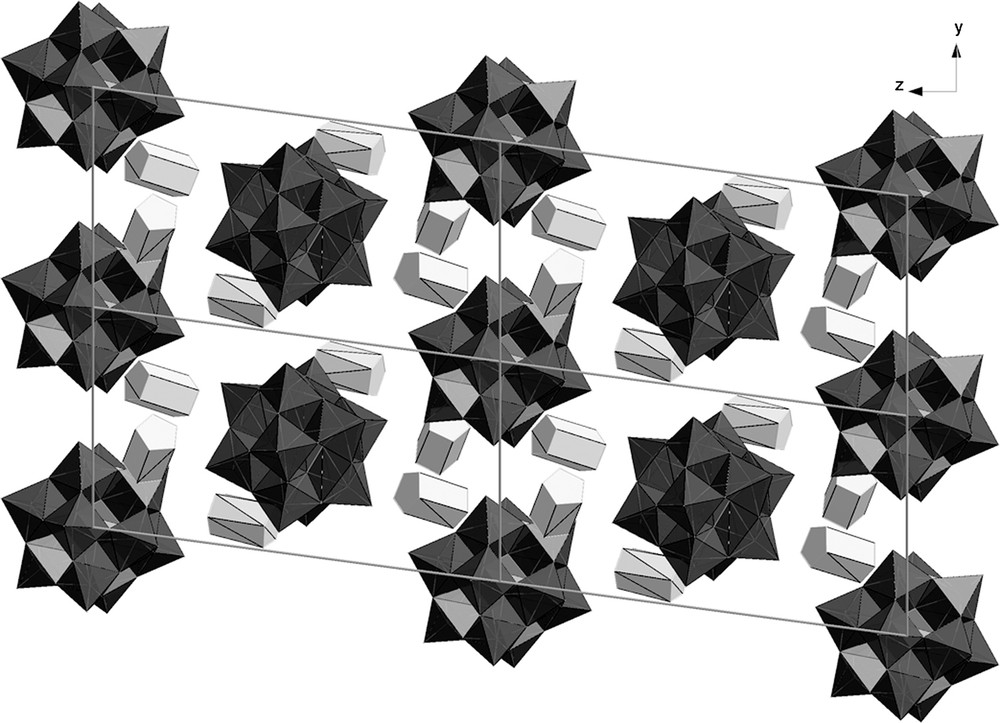
Packing in the x-direction black polyhedra = POM, grey polyhedra = cobaltocenium (hydrogen atoms and DMSO solvent molecules are omitted for clarity).
Each anion is surrounded by three cobaltocenium cations, with a distance between the framework oxygen atoms of the anion and the cyclopentadienyl rings of the cobaltocenium moieties of 3.074 to 3.213 Å. This indicates that the interactions between the cobaltocenium cations and the polyanions in 1 are much stronger than the previously observed interactions in ferrocenium compounds [22]. The closest Co…Co distance among the neighboring cobaltocenium cations is 7.191 Å. Interactions also exist between the polyanions along b (O24…O24: 3.296 Å) and along a (O6…O6: 3.392 Å), at the closest distance of the terminal oxygen atoms to the neighboring Keggin anions, which might be considered as 1D Keggin anion chains. The DMSO solvent molecules fill the voids formed by the POM and interact via hydrogen bonds between the methyl groups and the polyanions. Further H-bonding can also be observed between the hydrogen atoms of the cyclopentadienyl rings and the oxygen atoms of the polyoxometalate framework. These H-bonds range from about 2.37 Å to 2.60 Å. Table 1 shows a selection of the most relevant contacts.
Hydrogen-bonding geometry of 1 (Å,°).
| DH...A | DH | H...A | D...A | DH...A |
| C5H5…O24i | 0.9300 | 2.5900 | 3.37 (2) | 142.00 |
| C6H6…O42 | 0.9300 | 2.5200 | 3.415 (19) | 162.00 |
| C7H7…O4 | 0.9300 | 2.4800 | 3.329 (16) | 152.00 |
| C100H10A…O26 | 0.9600 | 2.6000 | 3.46 (3) | 150.00 |
| C102H10…O28ii | 0.9600 | 2.5300 | 3.32 (4) | 140.00 |
| C13H13…O29 | 0.9300 | 2.5300 | 3.13 (2) | 123.00 |
| C17H17…O48i | 0.9300 | 2.5300 | 3.289 (18) | 139.00 |
| C23H23…O5iii | 0.9300 | 2.4700 | 3.39 (3) | 170.00 |
| C30H30…O8 | 0.9300 | 2.3700 | 3.22 (3) | 151.00 |
The packing scheme can be derived from the Li3Bi structure type, in which the POMs form a face-centered likes arrangement. Co2 and the POMs form a NaCl structure, and Co1/Co3 with POM form a CaF2 lattice. In total, the Li3Bi structure is obtained, where Co1 and Co3 are situated in the tetrahedral voids, while Co2 is located in the octahedral voids (Supporting informations).
2.1.2 [PMo12O40][C44H38P2]2[Br] * 2(C3H7NO), 2
The crystal structure of 2 consists of the [Mo12O40P]3− anion, two o-xylylenebis(triphenylphosphonium) cations, one bromide anion, and two DMF solvent molecules. The quasi-binary compound 2 crystallizes in the monoclinic space group C2/c. The asymmetric unit is formed by half of the anion, one o-xylylenebis(triphenylphosphonium) cation, one half occupied bromide anion, and one DMF molecule. Four of the six phenyl “pods” of the o-xylylenebis(triphenylphosphonium) cation point toward the polyanion, giving the impression that the anion is capped with an organic moiety (Fig. 2). The two remaining phenyl pods, which are located on the same side as the xylylene moiety, and the xylylene moiety itself, point to the neighboring polyanion and also form short contacts to the free bromide ion and the solvent molecule. The relevant non-bonded CH…O interactions [23] are listed in Table 2.

Visualization of different H-bonds from the organic moiety to the polyanion (green, blue) and short contact to the bromide ion (pink).
Hydrogen-bonding and short contact geometry of 2 (Å,°).
| DH...A | DH | H...A | D...A | DH...A |
| C3H3...O31 | 0.9300 | 2.5200 | 3.35 (2) | 148.00 |
| C100H10 C...O20 | 0.9600 | 2.3800 | 3.05 (5) | 126.00 |
| C14H14...O36iv | 0.9300 | 2.5600 | 3.327 (19) | 140.00 |
| C16H16...O100iv | 0.9300 | 2.5900 | 3.32 (4) | 136.00 |
| C19H19A...O6 | 0.9700 | 2.5500 | 3.465 (15) | 158.00 |
| C19H19B...O15vi | 0.9700 | 2.5200 | 3.379 (13) | 147.00 |
| C23H23...O100iii | 0.9300 | 2.5800 | 3.51 (3) | 174.00 |
| C26H26A...Br1 | 0.9700 | 2.8500 | 3.818 (11) | 172.00 |
| C26H26B...O15vi | 0.9700 | 2.5200 | 3.356 (12) | 144.00 |
| C32H32...O3vi | 0.9300 | 2.4400 | 3.32 (2) | 156.00 |
| C35H35...O10vii | 0.9300 | 2.5900 | 3.507 (17) | 168.00 |
| C40H40...Br1 | 0.9300 | 2.8800 | 3.804 (13) | 170.00 |
| C42H42...O16i | 0.9300 | 2.3200 | 3.107 (17) | 143.00 |
The shortest intermolecular distance involves a carbon atom of one of the phenyl rings of o-xylylenebis(triphenylphosphonium) and one oxygen atom of the polyoxomolybdate anion [C42..O16 3.107(17) Å]. The resulting packing arrangement shows rows in the y-direction consisting of alternating polyanions, organic moieties, and free bromine anions, which are aligned between the organic moieties (Fig. 3).
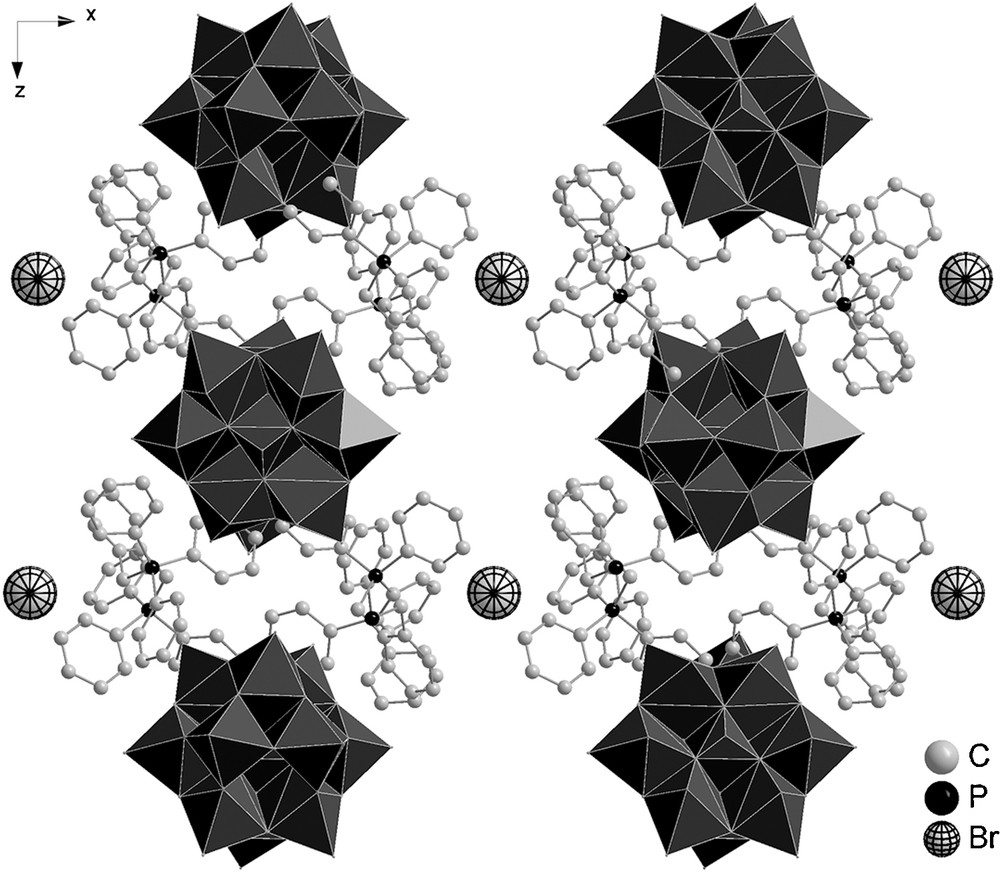
Packing of 2 in the y-direction (hydrogen atoms are omitted for clarity).
Four π–π interactions [24] can be observed between the phenyl rings of the o-xylylenebis(triphenylphosphonium). One π–π-stacking shows a T-shaped conformation with a distance of about 5.19 Å, measured between the centroids of the phenyl rings C1C6 and C33C39. The two other stacking interactions have typical parallel displacements, with a distance of 4.844 Å (measured between the centroids) between phenyl rings C1C6 and C41C46, and one further stacking between C7C12 and C27C32 (4.279 Å, measured between both centroids). An intermolecular CH…π bond can be found between phenylring C1C6 and methyl group C100H10D of the DMF solvent molecule in a distance of 2.81 Å measured between the H-atom and centroid.
In the reduced packing scheme, the polyanions are surrounded by organic moieties, which form strongly distorted octahedral layers, as observed in CdCl2 and CdI2, respectively (Supporting informations). These layers are shifted against each other in such a way that a clear assignment is not possible. Bromine intercalation is observed between the layers of the polyoxomolybdate and organic molecules.
2.1.3 [PMo12O40][C44H38P2][CoC10H10] * 3(C3H7NO), 3
In a one-pot reaction, phosphomolybdic acid, bis(cyclopentadienyl)cobalt(III) hexafluorophosphate, and o-xylylenebis(triphenylphosphonium) bromide were mixed in DMF. Compound 3 crystallizes in the triclinic space group P-1. Crystallization via gas diffusion affords green crystals of 3. The asymmetric unit of this compound is built from one [PMo12O40]3− anion, one dicationic o-xylylenebis(triphenylphosphonium), one monocationic cobaltocenium, and three DMF solvent molecules (Fig. 4).
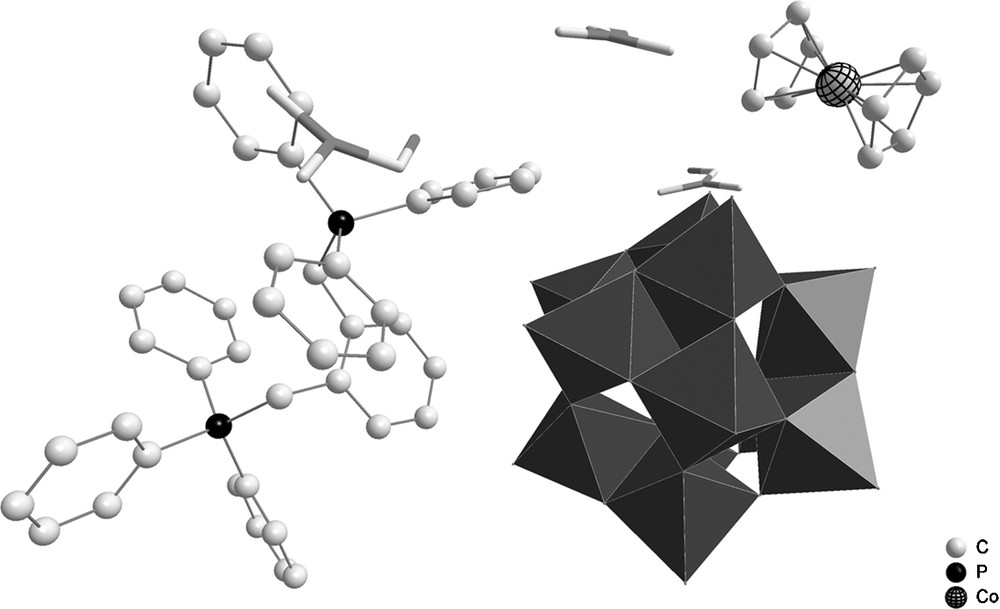
Asymmetric unit of 3; H-atoms are omitted for clarity.
The carbon atoms of the cyclopentadienyl ring of the cobaltocenium group are close to the oxygen atoms of the anion framework, from ca. 3.215 to 3.517 Å. This distance is approximately 0.2 Å longer than that in 1, therefore showing a weaker interaction. The closest Co…Co distance between neighboring cobalt complexes is 6.916 Å, also 0.2 Å shorter than that in 1.
The o-xylylenebis(triphenylphosphonium) cation has nearly the same arrangement around the POM as in 2. Here also, the organic ligands cap the polyanions with four of the phenyl rings; the other phenyl pods hydrogen bond to the free DMF solvent molecule or to neighboring polyanions. Further H-bonding is summarized in Table 3. Different π–π interactions [24] can be described between the phenyl rings: two T-shaped conformations, between phenyl ring C7C12 to phenyl ring C27C32 of a neighboring molecule (5.125 Å, measured between the centroids), and from phenyl ring C33C38 to phenyl ring C1C6 of a neighboring molecule (5.386 Å, measured between the centroids); and one parallel displaced π–π-stacking between phenyl ring C33C38 and phenyl ring C7C12 (4.688 Å, measured between the centroids).
Hydrogen-bonding geometry of 3 (Å,°).
| DH...A | DH | H...A | D...A | DH...A |
| C2H2…O200i | 0.9300 | 2.3800 | 3.305 (9) | 177.00 |
| C3H3…O30ii | 0.9300 | 2.5500 | 3.262 (8) | 134.00 |
| C16H16…O13iii | 0.9300 | 2.4800 | 3.260 (8) | 141.00 |
| C19H19B… O200i | 0.9700 | 2.4400 | 3.371 (8) | 162.00 |
| C22H22…O14 | 0.9300 | 2.4700 | 3.201 (6) | 135.00 |
| C26H26B… O400iv | 0.9700 | 2.3000 | 3.251 (14) | 168.00 |
| C303H30F…O33v | 0.9600 | 2.5800 | 3.419 (16) | 145.00 |
| C34H34…O400iv | 0.9300 | 2.3700 | 3.297 (14) | 172.00 |
| C38H38…O300vi | 0.9300 | 2.5200 | 3.340 (11) | 14 7.00 |
| C402H40A…O11 | 0.9600 | 2.3800 | 3.293 (15) | 159.00 |
| C402H40B…O14v | 0.9600 | 2.4800 | 3.303 (16) | 144.00 |
| C41H41…O17i | 0.9300 | 2.4800 | 3.239 (7) | 138.00 |
| C53H53…O10v | 0.9300 | 2.5900 | 3.289 (8) | 132.00 |
| C53H53…O32v | 0.9300 | 2.5800 | 3.405 (8) | 148.00 |
| C53H53…O35v | 0.9300 | 2.5000 | 3.198 (8) | 132.00 |
| C55H55…O3 | 0.9300 | 2.6000 | 3.517 (9) | 169.00 |
| C58H58…O10vi | 0.9300 | 2.5200 | 3.333 (8) | 146.00 |
The packing of the building blocks shows a layer formation in y-direction, with a ABCCBA system (where A = o-xylylenebis(triphenylphosphonium), B = POM, C = cobaltocenium) can be observed (Fig. 5).
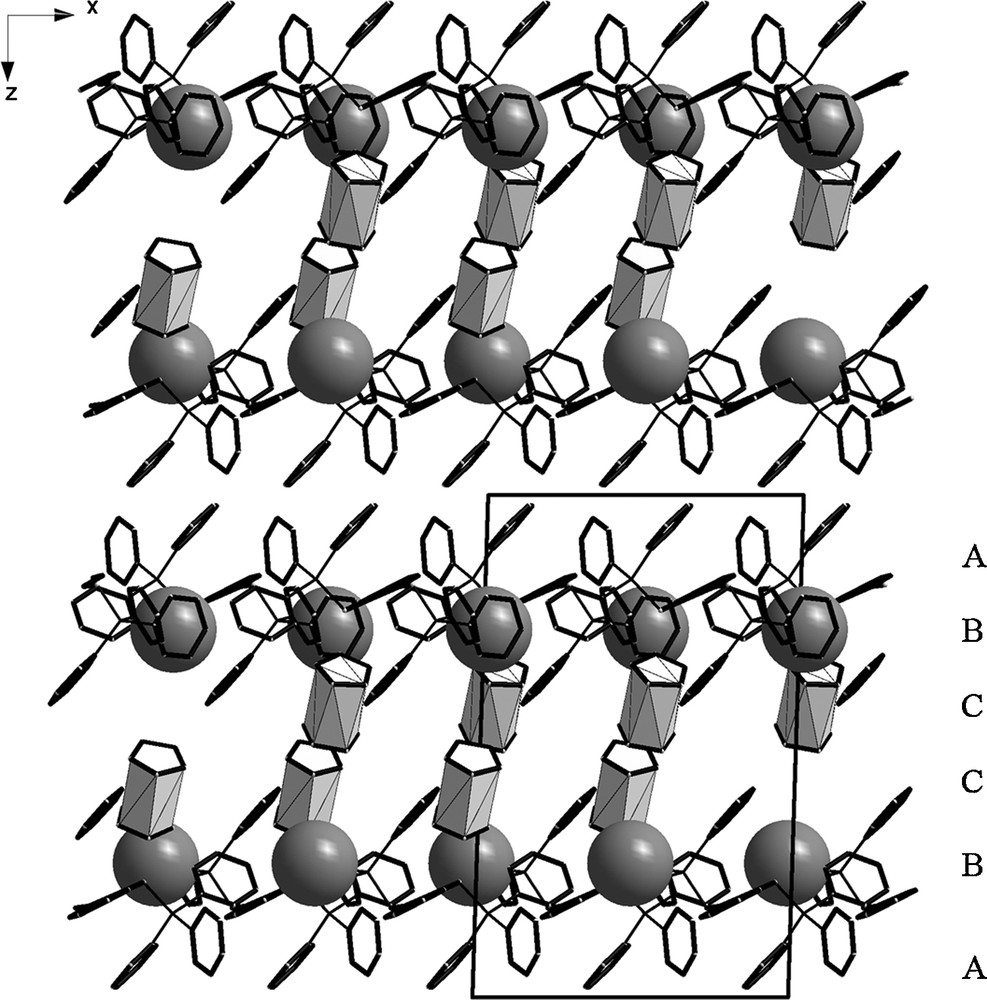
Compound 3 in y-direction. With simplified scheme of the packing (white polyhedra = cobaltocenium, grey balls = P1 of POM, black sticks = organic moieties).
The thickness of the layer is of about 20 Å (almost the full length of the c-axes of the unit cell) and the layer-motive is repeated in z-direction.
The phenyl units C13C18 points in the interface between two sandwich-layers, where also the three DMF solvent molecules are intercalated. The distance between these phenyl rings is about 3.52 Å (between C15 and C15 of the phenyl group in the neighbor row) and therefore at the upper limit of π–π stacking range.
For the reduction of the structure we used atom P1 as marking point for the polyoxometalates, atom C25 for the ligand molecules and Co1 for the cobaltoceniums. The POMs are surrounded by four ligand molecules, while equally also the ligand molecules are surrounded by four POMs. This leads in a first view to a strongly distorted NaCl structure (Fig. 6a). Due to this strong distortion of the NaCl-cube it looks like each second cube is empty, but watching from a different direction it is visible that in each tetrahedral void one cobaltocenium is located (Fig. 6b), which leads to an overall strongly distorted anti-PbFCl structure type [25].

a and b: two representations of the reduced structure of compound 3 (white = Co1, grey = P1 of POM, black = C25).
2.2 Vibrational spectroscopy and thermogravimetry
The vibrations of the polyoxometalate are visible in all spectra and correlate with known frequencies [26–28]. The IR frequency band observed at 1060 cm−1 (signature IR1, for all three compounds in Fig. 7) corresponds to asymmetric stretching of the PO and MoOt bonds of the polyoxometalate [26], while the broad band at about 960 cm−1 (IR2) corresponds to the symmetric stretching of these bonds. The shoulder at about 880 cm−1 (IR3) is assigned to asymmetric stretching of the MoOMo bonds. In the Raman spectrum, the bands at 986 cm−1 and 964 cm−1 (R1, for all three compounds) are attributed to the symmetric and asymmetric vibrations of MoOd [27,28]. A further band near 245 cm−1 (R2, all three compounds) can be attributed to MoOa. The typical CpCo frequency at 316 cm−1 is visible for compounds 1 and 3 (R3). Also observed in the spectra of both 1 and 3 are the different vibrations in the IR spectra for the cyclopentadienyl rings: CHδ (ring) at about 1414 cm−1 (IR4) and MCring at 460 cm−1 (IR5). For 2, the characteristic bands in the Raman spectra are weak only: CC (aliphatic chain) at about 666 cm−1 (R4), CH at 1030 cm−1 (R5), and a very weak band at 1582 cm−1 for CP. In the IR, the typical bands are: 1188 cm−1 for PO (IR7), a CC deformation vibration at 1440 cm−1 (IR8), and the typical vibrations at 2936 cm−1 and 3115 cm−1 for CC and CC, respectively.
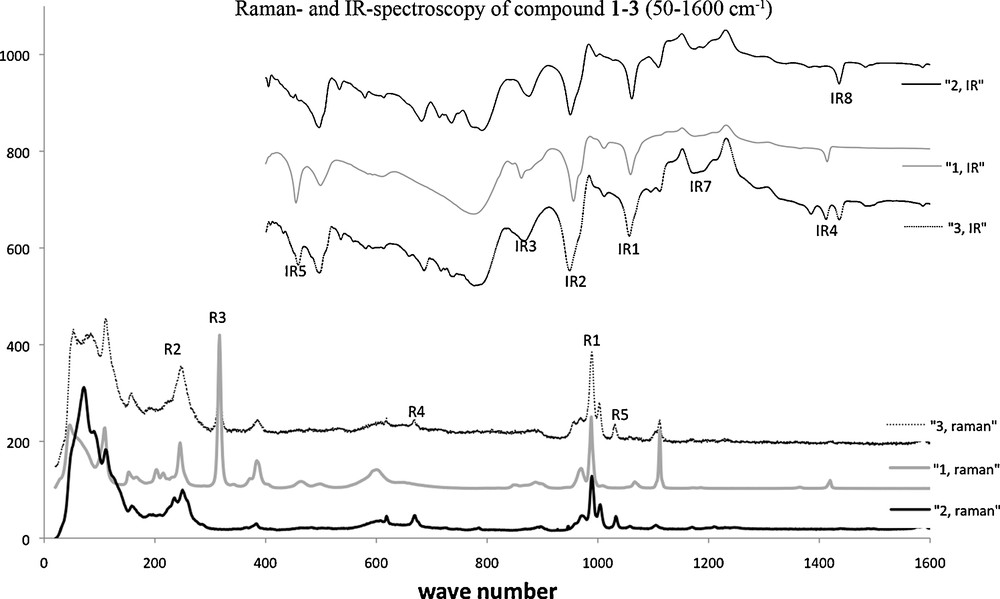
IR and Raman spectra for compounds 1, 2, and 3.
Thermogravimetric analysis was carried out in a flow of argon (Supporting informations). The polyanions start to decompose near 450 °C for all three compounds. Compound 1 shows the highest stability, with decomposition of the cobalt complex first at about 420 °C, while compounds 2 and 3 show the onset of decomposition for the organic ligand at 300 °C. Decomposition of the solvents (DMF, DMSO) can be observed before 150 °C.
3 Conclusion
In summary, it has been shown that building a multicomponent assembly using quasi-ternary ICs is an alternative route for creating well-defined and well-characterized nanostructured materials. The quasi-binary IC systems as well as the quasi-ternary IC itself were studied with regard to intermolecular interactions, such as hydrogen-bonding, and π–, and van der Waals-interactions. It was shown that the reduction to an ionic solid structure type was possible for the binary systems and for the ternary systems. Therefore, it is possible to extend the concepts of structural relationship also on quasi-ternary systems. Selective introduction of different building blocks (complexes, clusters, or organic compounds) will offer access to new compounds, which could show different materials properties.
4 Experimental
4.1 General
All starting materials were purchased commercially and used without further purification. The syntheses of compounds 1–3 can be performed with basic laboratory equipment in air and at room temperature.
4.2 [PMo12O40][CoC10H10]3 * 2(C2H6OS), 1 C34H42Co3Mo12O42PS2
Phosphomolybdic acid hydrate (1 g, 0.54 mmol) was dissolved in acetone (15 mL) and precipitated with an excess of cobaltocenium hexafluorophosphate (0.8 g, 2.39 mmol) in acetonitrile (15 mL). The yellow precipitate was filtered, washed thoroughly with acetone and acetonitrile, and dried under vacuum. The precipitate was dissolved in a minimum volume of DMSO (about 8 mL) and layered with diethyl ether. After a few days, yellow crystals of 1 appeared. Alternatively, the crystals can be grown using the gas diffusion of diethyl ether into the reaction solution. For quantitative analysis by inductively coupled plasma optical emission spectroscopy (ICP-OES, carbon analyzer), the ratio of Mo/Co/P/C in [PMo12O40]* 3(CoC10H10)* 2(C2H6OS) was determined as 44.96 (expected 45.24)/6.77 (6.95)/1.26 (1.22)/15.57 (16.05).
Single-crystals at different orientations were used for Raman spectroscopy. Raman (cm−1): 58 (m), 108 (m), 149 (w), 199 (w), 246 (m), 316 (s), 380 (m), 449 (w), 484 (w), 590 (broad, m), 881 (w), 967 (m), 987 (s), 1061 (w), 1110 (s), 1416 (w). Infrared spectroscopy on bulk material (cm−1): 1415 (w), 1188 (m), 1112 (w), 1063 (s), 955 (s), 865 (m), 783 (m), 505 (m), 458 (s).
After prolonged standing of the crystallization solution (about 1 months), another modification of compound 1 was observed. The asymmetric unit of 1a contained two polyanions and six cobalt complexes; more details are available in the Supporting informations.
4.3 [PMo12O40][C44H38P2]2[Br] * 2(C3H7NO), 2 C94H90BrMo12N2O42P5
Phosphomolybdic acid hydrate (2 g, 1.1 mmol) was dissolved in acetone (20 mL) and precipitated with o-xylylenebis(triphenylphosphonium) bromide (1.5 g, 1.9 mmol) in acetonitrile (30 mL). The precipitate was filtered, washed thoroughly with acetone and acetonitrile, and dried under vacuum. The light yellow precipitate was dissolved in a minimum volume of DMF and layered with diethyl ether. After three days, plate-shaped yellow-greenish crystals of 2 appeared. The crystals easily lose solvent molecules and need to be measured using a cooling system. For quantitative analysis (ICP-OES, carbon analyzer), the bulk material was filtered and dried in vacuo to remove excess solvent. This afforded a ratio of Mo/P/C ([PMo12O40][C44H38P2]2[Br], without solvent molecules) of: 36.22 (expected 36.44)/4.34 (4.89)/33.62 (33.45). For completeness, we performed powder X-ray diffraction (PXRD) with Pawley refinement on the dried bulk material. The cell volume (11646 Å3) was slightly smaller than the volume of the original cell (12043 Å3). This confirmed the loss of solvent molecules and, therefore, also supported the inaccurate results of the elemental analysis (refined parameters and spectra are included in the Supporting informations). Single-crystals at different orientations were used for Raman spectroscopy. Raman (cm−1): 70 (s, broad), 145 (w), 246 (m), 380 (w), 590 (w, broad), 607 (w), 665 (w), 902 (w, broad), 967 (m), 987 (s), 1002 (m), 1031 (m), 1087 (w), 1578 (w). Infrared spectroscopy on bulk material (cm−1): 1439 (w), 1186 (m), 1114 (w), 1063 (s), 958 (s), 880 (m), 792 (m), 686 (w), 500 (m).
4.4 [PMo12O40][C44H38P2][CoC10H10] * 3(C3H7NO), 3 C63H69CoMo12N3O43P3
Cobaltocenium hexafluorophosphate (0.09 g, 0.27 mmol), xylylenebis-triphenylphospine dibromide (0.21 g, 0.27 mmol), and phosphomolybdic acid hydrate (0.5 g, 0.27 mmol) were dissolved in DMF (25 mL) and stirred for 2 h. Dark green crystals of 3 were grown after a few days, using gas diffusion with diethyl ether. The crystals were collected via filtration and washed with small amounts of DMF. The crystals were dried quickly and the analyses were performed as fast as possible to avoid fragmentation into the binary compounds, which occurred after drying and storage in air. For storage purposes, it was better to leave the crystals in their mother liquor. Quantitative analysis (ICP-OES, carbon analyzer) resulted in a Mo/Co/P/C (C63H69CoMo12N3O43P3) ratio of: 40.39 (expected 40.32)/1.97 (2.06)/3.46 (3.25)/26.18 (26.5). Single-crystals at different orientations were used for Raman spectroscopy. Raman (cm−1): 70 (s, broad), 108 (s), 145 (w), 246 (s), 316 (s), 380 (w), 590 (broad, w), 607 (w), 881 (w), 967 (m), 987 (s), 1031 (m), 1110 (s), 1578 (w). Infrared spectroscopy on bulk material (cm−1): 1441 (w), 1417 (w), 1390 (w), 1188 (s), 1115 (w), 1059 (s), 952 (s), 876 (m), 792 (s), 500 (m), 462 (m).
4.5 Single-crystal X-ray diffraction studies
All crystal structures were solved by direct methods (SHELX97) and subsequent difference Fourier maps, and refined on F2 (SHELXTL) [29]. The measurements were made on an AXS Bruker Smart APEX diffractometer with Mo Kα radiation (l = 0.7103 Å) in the w scan mode (compound 2). Empirical absorption correction (SADABS) [30] was applied on 2. Compounds 1 and 3 were measured on a Stoe IPDS 2 with Mo Kα radiation. Numerical absorption correction (X-SHAPE) [31] was performed. Further details of the measurements can be found in Table 4.
Crystallographic data for compounds 1, 2, and 3.
| 1 | 2 | 3 | |
| Formula | C34H42Co3Mo12O42PS2 | C94H90BrMo12N2O42P5 | C63H69CoMo12N3 O43P3 |
| Mr | 2545.84 | 3305.79 | 2859.33 |
| Cryst. size, mm3 | 0.15 × 0.10 × 0.06 | 0.25 × 0.20 × 0.10 | 0.30 × 0.25 × 0.18 |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group | P-1 | C2/c | P-1 |
| a, Å | 12.747(3) | 35.578(4) | 13.125(3) |
| b, Å | 13.196(3) | 14.052(13) | 17.885(4) |
| c, Å | 21.691(4) | 24.097(2) | 20.949(4) |
| α, deg | 82.11(3) | 90 | 93.11(3) |
| β, deg | 87.44(3) | 91.456(2) | 91.80(3) |
| γ, deg | 61.54(3) | 90 | 111.38(3) |
| V, Å3 | 3176.3(11) | 12043(2) | 4565.5(16) |
| Z | 2 | 4 | 2 |
| Dcalcd, g cm−3 | 2.691 | 1.847 | 2.080 |
| μ(Mo Kα), cm−1 | 3.229 | 1.684 | 1.904 |
| F(000), e | 2426 | 6520 | 2776 |
| Refl. measd./unique | 55207/15176 | 37208/8724 | 68800/22351 |
| Param. refined | 857 | 699 | 1120 |
| Final R indices [I > 2σ(I)] | 0.0676/0.1490 | 0.0780/0.1953 | 0.0458/0.1032 |
| GoF (F2) | 1.043 | 1.048 | 1.028 |
| Δρfin (max/min), e Å−3 | 2.468/−2.279 | 1.191/−2.135 | 1.730/−1.715 |
All the Keggin ions in compounds 1 and 2 show the same type of crystallographic disorder as found in many other crystal structures with the Keggin anion. The center atom is surrounded by a cube of eight oxygen atoms, with each oxygen site half occupied; the central atom and these oxygen atoms formed two groups of tetrahedrons [32]. The disorder remains also while using non-centrosymmetric space groups.
Compound 1: All atoms but H-atoms were refined with anisotropic displacement parameters. Hydrogen positions were calculated using the HFIX command.
Compound 2: All atoms but H-atoms were refined with anisotropic displacement parameters. Diffuse solvent molecules could not be found properly in the difference Fourier map. Their contributions were eliminated by the SQUEEZE subprogram [33].
Compound 3: All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen positions were calculated using the HFIX command. Three DMF molecules per unit cell could be refined with anisotropic displacement parameters. The rest were treated by the SQUEEZE procedure.
CCDC-884550 (1), 886143 (1a, Supporting informations), 884551 (2), and 884552 (3) contain the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgements
The authors thank the Swiss National Foundation for generous support. We also thank Dr. Jürgen Nuss and Dr. Liana Vella-Zarb, for help with reduction to structure type and with PXRD refinement.