

1 Introduction
The palladium/copper co-catalysed arylation and vinylation of alkynes from organic halides is probably the most widely employed methodology to yield enynes. A related version of the reaction introduced by R. Heck exclusively requires a palladium catalyst, and is generally known as “copper-free” alkynes arylation. The pioneering works by Stephens and Castro in 1963 [1], then, in 1975 by Cassar [2], Dieck and Heck [3], and Sonogashira, Tohda, and Hagihara [4], have initiated a great number of studies in the field of enyne formation [5–7]. The original reactions and their experimental conditions, as disclosed by these pioneering authors (Scheme 1), revealed both a straightforward applicability and a very large substrate range, which is probably at the origin of their success.
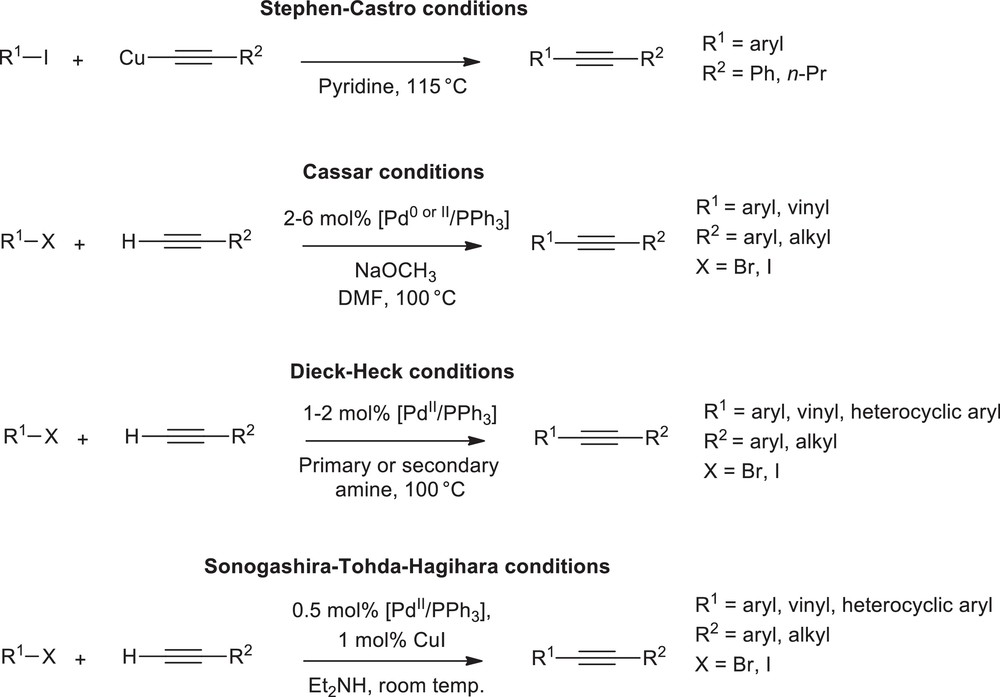
Pioneering conditions for direct arylation of terminal alkynes.
We initiated in 2004 a program aiming to develop several aspects of terminal alkynes arylation regarding different aspects of the reaction and related to (i) improving catalytic efficiency and (ii) products selectivity, (iii) facilitating general processing, and finally (iv) extending the scope of enyne molecules formed. The present account reviews all this work, as it has been presented by the corresponding author at GECOM–CONCOORD-Metabief as recipient of the 2012 European journal of Inorganic Chemistry Young Investigator Award.
2 Polyphosphine ligands for reactivity improvement using bromo- and chloroarenes
Terminal alkynes arylation reactions are usually carried out with catalytic amounts of palladium in the presence of auxiliary ligands and additives. A critical improvement in this field, directly connected to both sustainable chemistry and economic concerns, is the necessity to minimize the consumption of metallic resources, and especially palladium [8]. In the early 2000s, several examples of polyphosphines tetradentate ligands have shown excellent performances in C–C cross-coupling reactions in the presence of very low amounts of metal (less than 0.01 mol%) [9,10]. We consecutively anticipated that geometrically “more simple” and adaptable tridentate ferrocenyl triphosphines might lead as well to active systems in palladium-catalysed alkynes arylation. In an effort to improve the performances of the ferrocenyl phosphine ligands, we chose to electronically enrich one of the phosphorus atoms with the view of getting beneficial effects in the activation of reluctant bromo- and chloroarenes [11]. This strategy prompted us to design and synthesize the mixed ferrocenyl aryl/alkyl triphosphine 1,2-bis(diphenylphosphino)-1′-(di-isopropylphosphino)-4-tert-butylferrocene (Fig. 1) L1, [Fc(P)2t-Bu(Pi-Pr)]. The polyphosphines L1–L3 are conveniently obtained from the successive reaction of anhydrous FeCl2 with the appropriately substituted cyclopentadienyl lithium salts [12,13].
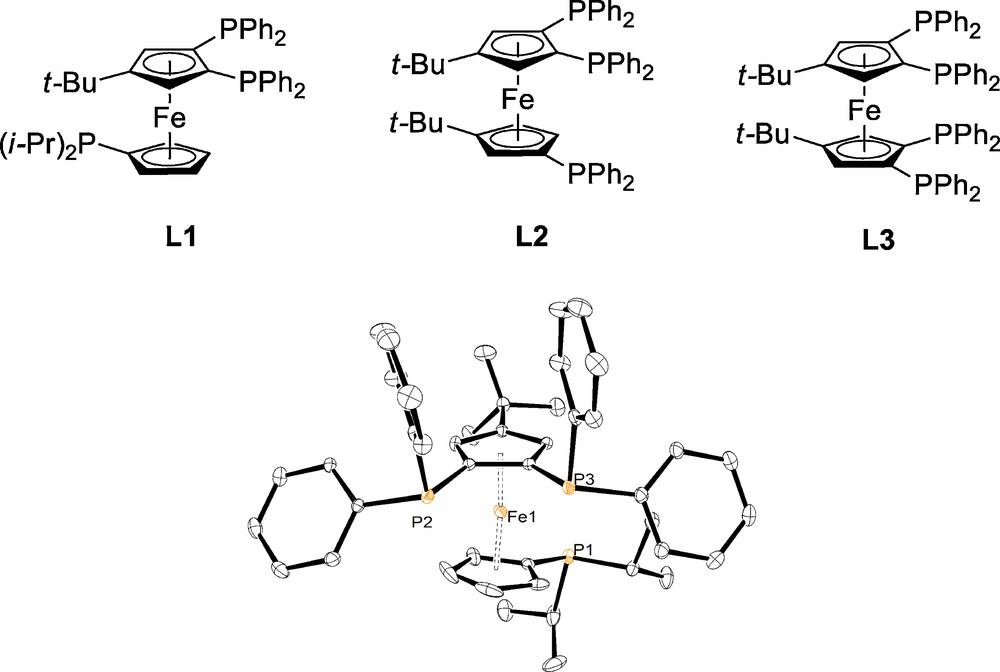
Triphosphine L1 and analogous polyphosphines L2 and L3 (top). X-ray molecular structure of ligand L1 (bottom).
The scope and limitations of a L1/palladium system for the coupling of a variety of aryl bromides with alkynes has been investigated (see Scheme 2 for selected examples) [14,15]. When the activating groups are present on the aryl bromide, high reaction rates were observed. For instance, the coupling of 4-fluorobromobenzene, 4-bromoacetophenone and 4-bromobenzonitrile with phenylacetylene, using only 0.001 mol% of palladium complex led to the coupling products in 20,000, 250,000 and 46,000 turn over numbers (TONs), respectively. The quantitative conversion of the strongly deactivated 4-bromoanisole requires a higher catalyst loading (0.4 mol%) and resulted in a satisfactory TON of 520. Reactions with activated aryl chlorides, such as 4-chlorobenzonitrile and 4-chloroacetophenone in the absence of the copper promoter led to the desired products in notable TONs of 540 and 205, respectively, and in very good yields using 0.4 mol% catalyst. Aliphatic alkynes, such as dec-1-yne or but-1-yn-4-ol were coupled to 4-bromoacetophenone with good TONs of 620 and 460, respectively. Expectedly, a very high TON of 95,000 was obtained for the reaction of iodobenzene with phenylacetylene.
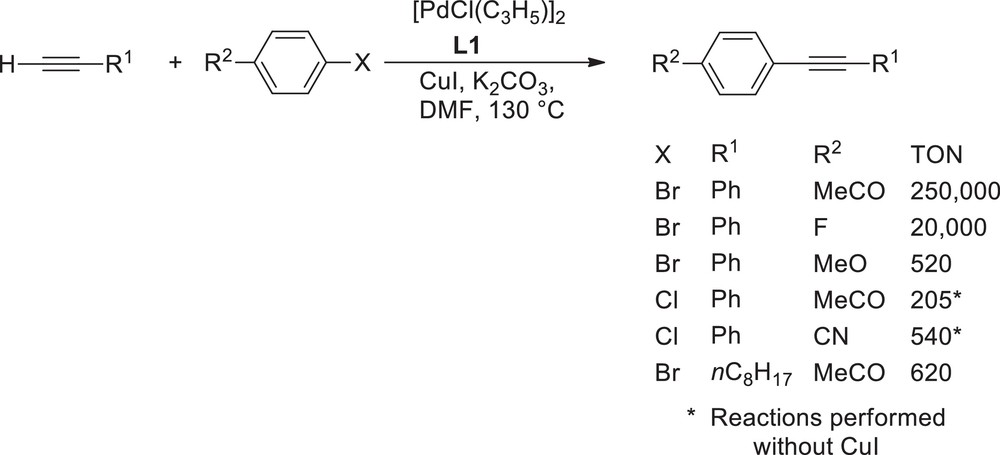
A triphosphine for high TONs coupling of aryl bromides and chlorides.
From this example and others [16], polydentate ligands clearly offered some of the highest TONs observed in Sonogashira–Heck–Cassar cross-coupling. Demanding substrates, such as deactivated aryl bromides and aryl chlorides were coupled with excellent TONs from polydentate auxiliaries. In order to obtain such high TONs, temperatures above 120 °C are required, but under these conditions, the use of robust polydentate ligands helps to stabilize the catalytic species. In addition, these ligands may also have a positive influence as the accelerator of the reductive elimination step of the catalytic cycle as demonstrated recently in C–O cross-coupling reactions [17]. Mechanistic studies, such as those ones may provide key information for the development of polydentate ligands, which can be resource-economic in metal catalysis.
3 Polyphosphines–copper adducts and the decreasing of the homocoupling of alkynes in Sonogashira reaction
3.1 Synthesis and characterization of tetraphosphine–copper adducts
The cross-coupling of terminal alkynes to other organic fragments, such as aryl, amide, or azide fragments catalysed by palladium and/or copper often suffers from oxidative homocoupling of the terminal alkynes as side-reactions of the cross-coupling catalytic cycle (Scheme 3, right). Acetylenic coupling, when intended, is a powerful tool in molecular construction [18]. However, it could be significantly detrimental as a side-reaction, especially since it can be promoted by both copper(I) salts [18,19], and palladium(II) complexes [20,21].
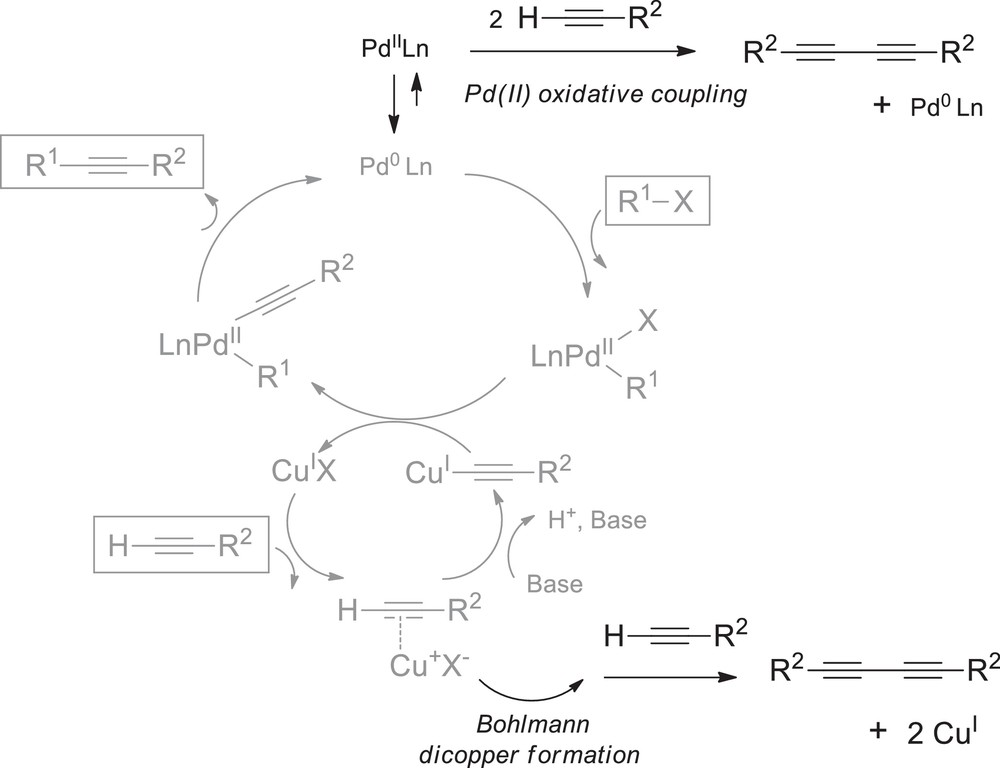
Sonogashira catalytic cycle and alkyne dimerization side-reactions.
As shown in Scheme 4 (top), alkyne dimerization promoted by palladium has been assumed to be achieved via redox reactions involving Cu(I)/Cu(II) and Pd(0)/Pd(II) couples [21]; the presence of an oxidant being necessary for regenerating Pd(II) from Pd(0) and Cu(II). Additionally, CuI, which is generally employed in excess comparatively to the palladium catalyst, is responsible for transmetalation. Tetracoordinated bridging copper dimers were also proposed by Bohlmann as species responsible for acetylenic coupling (Scheme 4, bottom), as reviewed by Diederich [18,22].
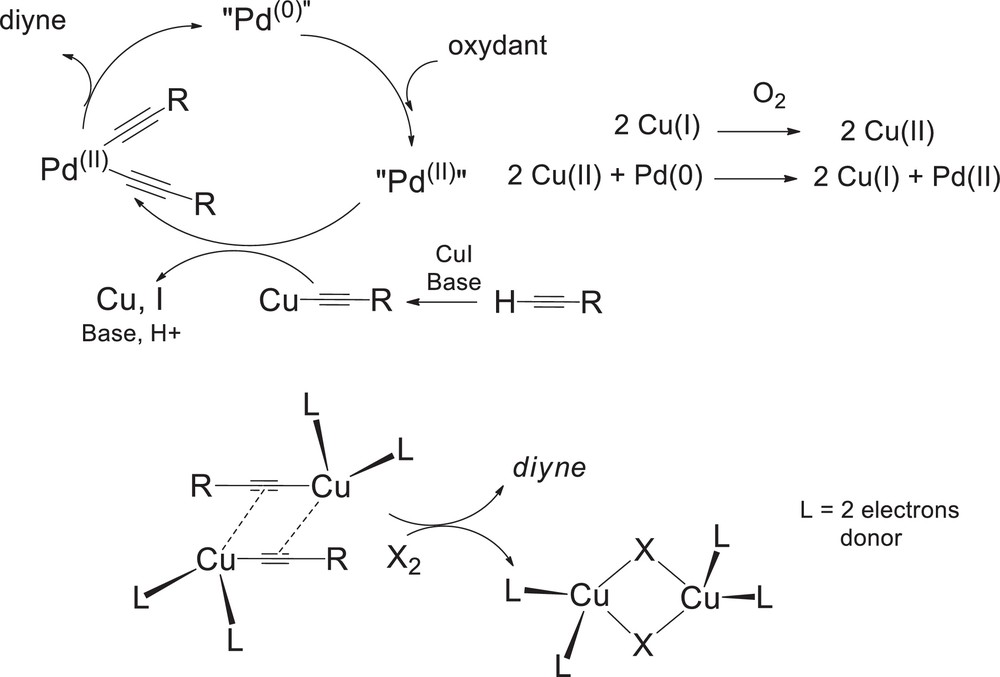
Palladium(II) oxidative coupling of alkynes–role of copper salts.
Thus, typically selective Sonogashira reactions require inert and oxygen-free conditions to restrain oxidative homocoupling of alkynes. On the other hand, more convenient handling conditions for instance, without the need of a glove-box or deoxygenating the reagents, especially the liquid participants, are preferred for large-scale industrial applications. We additionally realised that the mechanistic studies about Sonogashira cross-coupling had never address the interference of copper as coordinating transition metal via the formation of stable LnCuI adducts due to the presence in solution of an excess of Ln auxiliary ligands, aiming at stabilizing palladium. Such hypothetical “interference” appeared an interesting parameter to study, which could also bring helpful mechanistic information. We additionally envisioned that such copper adducts may potentially be an innovative way to inhibit diyne formation by stabilizing Cu(I) salts, and thus, avoiding Cu(II) formation (and consequently, Pd(II) alkyne dimerization). Finally, these may be also a way to hamper the copper nucleus dimerization responsible for Hay-Glaser copper-based acetylenic coupling.
Following these lines, the first examples of copper(I)-ferrocenyltetraphosphine complexes have been synthesized [23]. The molecular structures of complex (1) {P,P′,P′′-[1,1′,2,2′-tetrakis(diphenylphosphino)-4,4′-di-tert-butylferrocene]-iodo-copper(I)} (incorporating tetraphosphine L3, Scheme 5) and also complex {P,P′,P′′-[1,1′,2,3′-tetrakis(diphenylphosphino)-4,4′-di-tert-butylferrocene]-iodo-copper(I)} were solved by X-ray diffraction studies (exemplified Fig. 2, left). From the mixture of four equivalents of anhydrous CuI with L3 in deoxygenated acetonitrile, complex (1) was also isolated showing its predominant thermodynamic stability, but then, the excess of CuI co-crystallizes as white square-shaped crystals of a polymeric inorganic copper compound catena-[(3-iodo)-(acetonitrile-N)-copper(I)] identified by X-ray diffraction studies (Fig. 2, right) [2].

Copper-tetraphosphine adduct (1), and catenane (2) formed from the excess of copper iodide.
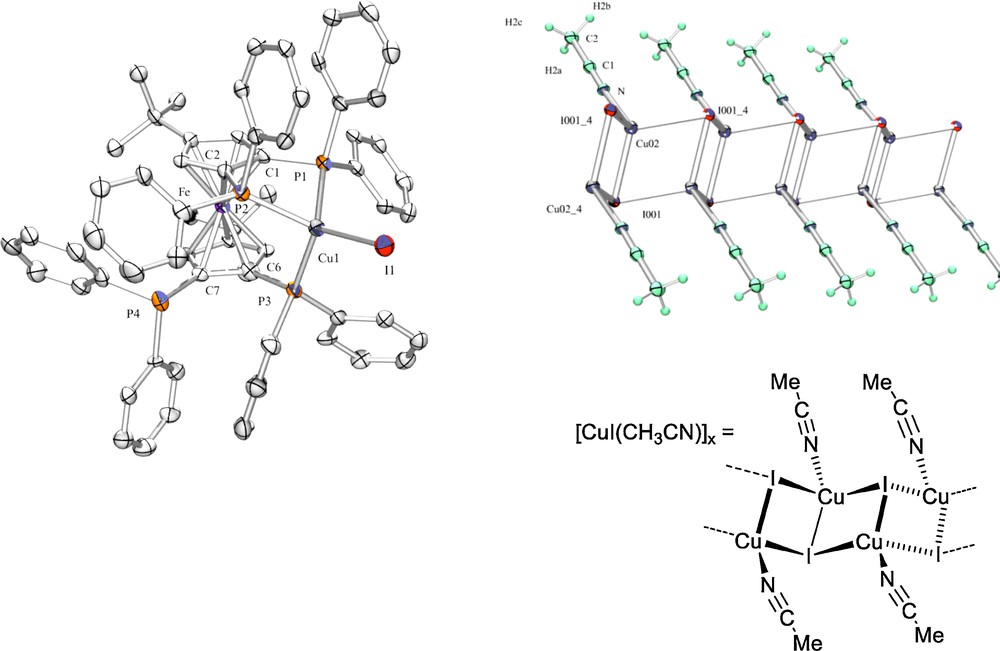
Molecular structure of copper-tetraphosphine adduct (1) and inorganic catenane (2).
The X-ray molecular structure obtained for complex (1) (Fig. 2, left) indicated that the copper(I) atom is bonded to three phosphorus atoms of the tetraphosphine in a distorted tetrahedral environment completed by an iodide [Cu–I 2.5848(14) Å]. The three metal-phosphorus bonds are substantially different [Cu–P(1) 2.310(3) Å, Cu–P(2) 2.355(2) Å, Cu–P(3) 2.273(2) Å] with a relative variation around 3.5% observed between the shortest and the longest one. In general, for the non-solvated complexes [(PPh3)3CuX] (X = halide), X-ray structural data have shown P–Cu–P and P–Cu–X angles close to the ideal tetrahedral angle of 109.5°. In (1), the major deviation is observed for the closed angle P(1)–Cu–P(2) = 85.9(1)°, originating from the 1,2-P chelating pattern, and P–Cu–I angles lie between 116 and 121°; several molecules of acetonitrile are present in the crystals. Nevertheless, to accommodate its fac-tricoordination, the ferrocenyltetraphosphine ligand undergoes severe constraints. In solution, we identified a surprising dynamic behaviour of (1), which has shown that the CuI fragment coordination is fluxional over three of the four P atoms.
31P NMR spectroscopy studies were conducted on (1) in deuterated dichloromethane. At ambient temperature, two signals at −23.4 and −24.6 ppm were detected (Fig. 3, top), the first one was a clear singlet, whereas the second one was broader and ill-defined. Low temperature NMR conducted down to −94 °C remarkably confirmed the X-ray structural studies. Upon cooling down the temperature, a fluxional behaviour of the copper complex is evidenced through coalescence (0 °C to −40 °C) and decoalescence (−60 °C) of the broader signal. Three new signals appeared at –75 °C. The occurrence of four different phosphorus from which three are in a dynamic exchange (centred at −15.1, −22.1 and −36.6 ppm) was ascertained at −94 °C. The appearance and chemical shift of the singlet was not modified by the variation of temperature. This dynamic phenomenon was found reversible increasing back the temperature.
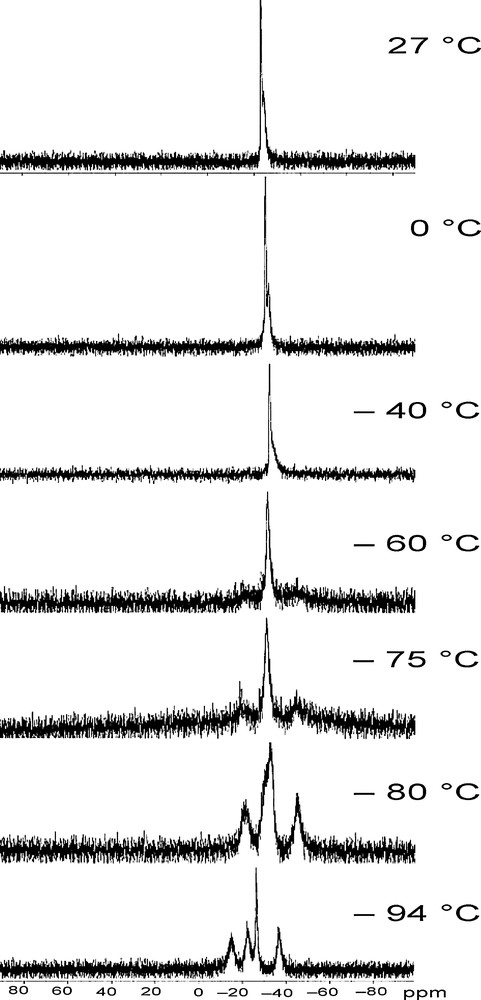
3 VT-31P{1H} NMR of crystals of (1) solubilized in CD2Cl2.
3.2 Synthesis and characterization of triphosphine–copper adducts
The preferential triligated coordination mode of CuI led us to explore whether simpler triphosphines, such as L1 and L2 (Fig. 1) were also good candidates for the production of stable copper adducts [24]. The copper complexes (3) and (4) were obtained in high yields (Scheme 6) and characterized by using VT-31P NMR, in which contrary to the tetraphosphine analogue (1), no fluxional behaviour was detected.

Synthesis of triphosphine–copper adducts (3) and (4) from L1 and L2.
In the solid state, XRD studies confirmed the expected tridentate coordination of the ligands L1 and L2 to one copper iodide (Fig. 4). The distorted tetrahedral environment for the copper centre in (3) and (4) is analogous to the one found for adduct (1), the main general difference being the uncoordinated phosphorus donor atom present in the tetraphosphine Cu adduct (1). Additionally, the molecular structure of (3) displays a plane of symmetry, which includes the CuI fragment, the Fe atom, and the quaternary C of t-Bu, in contrast to the structures of (1), (4) and uncoordinated triphosphine L1.
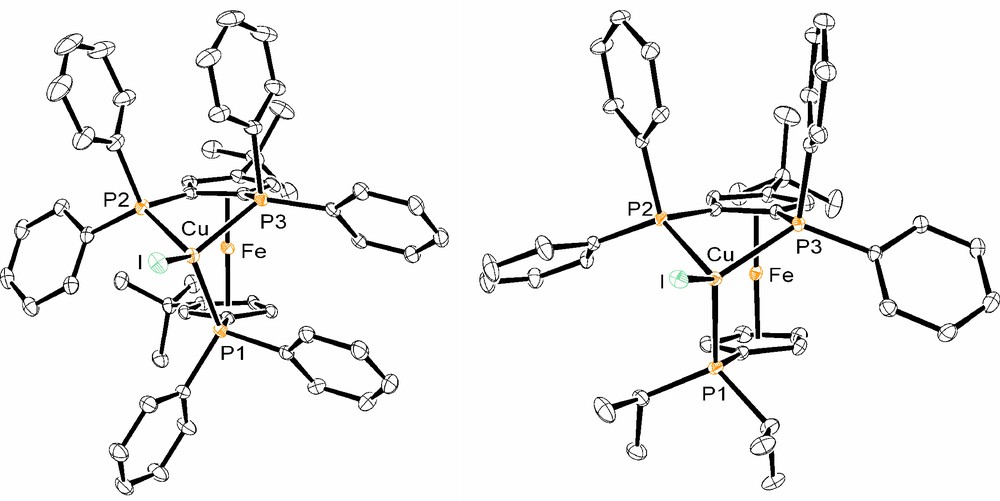
Molecular structure of copper-tetraphosphine adducts (4), left, and (3), right.
3.3 Performances of polyphosphine–copper adducts employed in the arylation of terminal alkynes
Experimental conditions and results in Sonogashira alkyne arylation for the catalytic system combining the palladium(II) precursor complex [PdCl(η3-C3H5)]2 with the tetraphosphine L3 are presented in Table 1 in the absence of any preformed copper adduct [23]. Electronically-neutral, activated and deactivated aryl bromides were coupled to phenylacetylene. The nature of the substituent on the organic halide plays an important role. Indeed, in the absence of an electron-withdrawing substituent on the aryl partner the concomitant phenylacetylene dimerization hampered a total alkyne arylation due to the consumption of the acetylenic reagent (see in Table 1 entries 2 and 3).
Sonogashira arylation with a [Pd/L3] catalytic systema.
| Entry | Aryl bromide | Yield (%) |
| 1 | 100 | |
| 2 | 32b | |
| 3 | 20b |
a Catalytic conditions (alkyne:aryl) 2:1, 2 equiv of K2CO3, 24 h in 10 mL DMF at 130 °C; GC average yields from two or three runs are given.
b Phenylacetylene dimerization to enynes, as described in ref. [21], is responsible for lower yields, in all runs unconverted bromobenzene (entry 2) or bromoanisole are observed (entry 3) in more than 30% yield.
These three, respectively, electronically-neutral activated and deactivated aryl bromides were then used to investigate the performances obtained in the arylation of phenylacetylene with various [(Pd)/(Cu)/(tetraphosphine)] systems. Table 2 summarizes the conditions and results obtained in this case. First, the origin of phenylacetylene dimerization and enynes formation that hampered the coupling of the more demanding substrate 4-bromoanisole in the catalytic reactions reported in Table 1 was checked. Experiments, with phenylacetylene in the presence of the sole copper source were conducted (CuI, or complex (1), or mixture of CuI/L3, see Table 2, entries 1 to 5). In these experiments, the alkyne and aryl halide reagents were recovered unchanged. It clearly appeared that palladium is necessary; otherwise no reaction occurs with the alkyne, neither in the presence nor in the absence of 4-bromoanisole. The pivotal role of palladium in alkyne dimerization was confirmed in the subsequent run (Table 2, entry 6), in which the coupling of bromoanisole to phenylacetylene in the presence of CuI and in the absence of tetraphosphine ligand was attempted. Under these conditions, only 5% of the coupled (aryl)alkyne was obtained, while diyne and enynes were formed in appreciable amounts. In the next runs (Table 2, entries 7 to 9), copper-free arylation conditions were used, showing that 4-bromoacetophenone and bromobenzene can visibly be coupled to phenylacetylene (100% and 89% yield, respectively) in the absence of copper. More importantly, it was shown that copper has a deleterious effect for bromobenzene coupling when compared to the preliminary results reported in Table 1 (entry 2). Conversely, the coupling of 4-bromoanisole to phenylacetylene was not very successful (22% of coupling product) in the absence of copper (Table 2, entry 9). Finally, we checked the usefulness of copper adduct (1) as a source of copper (Table 2, entries 10 to 12). The three electronically-neutral, activated and deactivated aryl bromides were very efficiently coupled to phenylacetylene, and in particular, the 4-bromoanisole substrate that had been under the previous conditions explored very reluctant to coupling (80 to 90% yield in the several runs conducted). An apparent reason is that the use of [Pd(allyl)Cl2/copper adduct (1)] system is not at all prompt to lead to diyne and enynes formation contrary to the other [Pd/(CuI)] systems we investigated. This was the first time that such kind of high selectivity was induced in Sonogashira reaction by initial ligand complexation to copper instead of palladium.
Investigation of alkyne arylation with [(Cu)/(Pd)/(Tetraphosphine)] systemsa.
| Entry | Palladium source | Copper source | Additional L3 | Aryl bromide | Yield in coupling product (%) |
| 1 | / | 5 mol% CuI | / | / | 0 |
| 2 | / | 5 mol% CuI | 1 mol% | / | 0 |
| 3 | / | 1 mol% Complex (1) | / | / | 0 |
| 4 | / | 5 mol% CuI | / | 4-bromoanisole | 0 |
| 5 | / | 1 mol% Complex (1) | / | 4-bromoanisole | 0 |
| 6 | 0.5 mol% [PdCl(allyl)]2 | 5 mol% CuI | / | 4-bromoanisole | 5b |
| 7 | 0.5 mol% [PdCl(allyl)]2 | / | 1 mol% | 4-bromoacetophenone | 100c |
| 8 | 0.5 mol% [PdCl(allyl)]2 | / | 1 mol% | 4-bromobenzene | 89d |
| 9 | 0.5 mol% [PdCl(allyl)]2 | / | 1 mol% | 4-bromoanisole | 22e |
| 10 | 0.5 mol% [PdCl(allyl)]2 | 1 mol% Complex (1) | / | 4-bromoacetophenone | 100f |
| 11 | 0.5 mol% [PdCl(allyl)]2 | 1 mol% Complex (1) | / | 4-bromobenzene | 98g |
| 12 | 0.5 mol% [PdCl(allyl)]2 | 1 mol% Complex (1) | / | 4-bromoanisole | 91h |
a Catalytic conditions (alkyne:aryl) 2:1, 2 equiv of K2CO3, 24 h in 10 mL of DMF at 130 °C, GC average yields from two or three runs are given.
b 95% unreacted ArBr, phenylacetylene dimerization diyne and enyne products are obtained.
c Phenylacetylene dimerization and unidentified heavier products are obtained from phenylacetylene excess.
d 5% of the unreacted ArBr and 5% of the heavier benzene products.
e 70% of unreacted ArBr and 7% of anisole.
f Traces of diyne from phenylacetylene dimerization.
g 2% of the unreacted ArBr.
h 9% of the unreacted ArBr.
The influence of the copper(I)-iodide triphosphine adducts (3) and (4) in Sonogashira reaction of deactivated aryl halide substrates was investigated in the light of the previous results obtained by employing (1) [24]. Table 3 summarizes the screening of catalytic performances in the coupling of the deactivated electron-rich 4-bromoanisole with phenylacetylene from various systems incorporating the copper iodide polyphosphine adducts. In the absence of palladium, the systems incorporating only either CuI or the adduct (1) were inefficient (Table 3, entries 1-2). As known from the literature, in the absence of ligand, the coupling of 4-bromoanisole is difficult (Table 3, entry 3). In our case, substantial amounts of alkyne dimerization were concurrently obtained. In the absence of a copper source, a low 20% yield of coupling was obtained from Pd/(1) with however, no diyne or enyne formation (Table 3, entry 4). The use of copper adducts (1) and (3) in the presence of [Pd(allyl)Cl]2 was more efficient in this coupling than a direct mixture of palladium with polyphosphines L1 or L3 and 5% CuI (Table 3, entries 7 and 9 compared to 5 and 6), resulting in a significant inhibition of diyne formation. The use of the copper adducts (1), (3) and (4) confirmed that the pre-stabilization of copper iodide with a multidentate ferrocenyl phosphine ligand promotes the palladium cross-coupling of demanding halides with phenylacetylene in a selective way (Table 3, entries 7–9).
Phenylacetylene coupling to 4-bromoanisolea.
| Entry | Copper source | Palladium/ligand catalytic system | Arylation product (%)b | Alkyne dimerization |
| 1 | 5 mol% CuI | / | 0 | No |
| 2 | 1 mol% (1) | / | 0 | No |
| 3 | 5 mol% CuI | 0.5% [Pd(allyl)Cl]2 | 25 | Moderate (40%c) |
| 4 | / | 0.5% [Pd(allyl)Cl]2/L3 | 20 | No |
| 5 | 5 mol% CuI | 0.5% [Pd(allyl)Cl]2/L3 | 10 to 30 | High (> 60%c) |
| 6 | 5 mol% CuI | 0.5% [Pd(allyl)Cl]2/L1 | 40 to 65 | Substantial (50%c) |
| 7 | 1 mol% (1) | 0.5% [Pd(allyl)Cl]2 | 80 to 90 | Low (< 10%c) |
| 8 | 1 mol% (4) | 0.5% [Pd(allyl)Cl]2 | 85 | Traces |
| 9 | 1 mol% (3) | 0.5% [Pd(allyl)Cl]2 | 99 | No |
| 10 | 0.4 mol% (3) | 0.2% [Pd(allyl)Cl]2 | 96 | Traces |
| 11 | 0.1 mol% (3) | 0.05% [Pd(allyl)Cl]2 | 83 | Traces |
a Reaction conditions: 4-bromoanisole (3.38 × 10–3 mol), phenylacetylene (2 equiv), K2CO3 (2 equiv), 120 °C, 20 h, 10 mL DMF under argon.
b GC yields average of two consistent runs or more.
c Based on phenylacetylene consumption, determined by GC.
The near quantitative conversion to (4-methoxyphenyl)phenylacetylene obtained from the system employing the adduct (3) in 1 mol% with 1 mol% Pd (Table 3, entry 9) led us to focus our screening on the minimization of the metal compounds employed in the catalytic reactions. A very high conversion of 96% 4-(methoxyphenyl)phenylacetylene was obtained in the presence of 0.4 mol% of copper and palladium (Table 3, entry 10). From 0.1 mol% of the metals, the conversion decreased to a still satisfactory 83% yield (Table 3, entry 11). While the decrease of the quantity of palladium is rather common in cross-coupling reactions, most of the Sonogashira reactions are reported with CuI amounts ranging between 5 to 10 mol%. Since the use of 5 mol% CuI is often correlated with the formation of substantial amounts of alkyne dimerization products, the use for coupling of demanding substrates of only 0.4 mol% of CuI (which is 10 to 25 times less than commonly reported conditions) is a valuable achievement.
The scope of this system was further examined under low metal content conditions. Table 4 shows the performances obtained for the coupling of phenylacetylene with a variety of halide substrates. By employing 0.4 mol% of (3) and the palladium source, the electron-rich, 4- and 3-bromoanisole were efficiently coupled in yields over 90% (96 and 92% respectively, Table 4, entries 1–2). Other deactivated electron-donating substituted aryl halides were tested, showing that the system tolerates either para-, meta- or ortho-substitution. However, 2-bromotoluene gave only 80% conversion (Table 4, entry 4) compared to the less hindered 3-bromotoluene for which 96% conversion was obtained (Table 4, entry 3). The sterically hindered ortho-substituted 2-bromonaphthalene was almost quantitatively converted into the desired product (Table 4, entry 5). We were also pleased to see that a highly efficient bis-arylation of terminal diynes was possible with 1,2-dibromobenzene (Table 4, entry 6).
Scope of [Pd/3] catalytic performances at low metal loadingsa.
| Entry | 3/Pd (mol) | Aryl halide | Coupling product | Yield (%)b |
| 1 | 0.4%/0.2% | 4-bromoanisole | 96 | |
| 2 | 0.4%/0.2% | 3-bromoanisole | 92 | |
| 3 | 0.4%/0.2% | 3-bromotoluene | 96 | |
| 4 | 0.4%/0.2% | 2-bromotoluene | 80 | |
| 5 | 0.4%/0.2% | 2-bromonaphtalene | 98 | |
| 6 | 0.4%/0.2% | 1,2-dibromobenzene | 99 | |
| 7 | 0.4%/0.2% | 4-chloroanisole | 0 | |
| 8 | 0.1%/0.05% | 4-chlorobenzonitrile | 27 | |
| 9 | 0.4%/0.2% | 4-chlorobenzonitrile | 46 | |
| 10 | 2.0%/1.0% | 4-chlorobenzonitrile | 66 | |
| 11 | 0.4%/0.2% | 4-chloroacetophenone | 36 |
a Reaction conditions: aryl halide (1 equiv), phenylacetylene (2 equiv), K2CO3 (2 equiv) and the catalytic system stirred at 120 °C over 20 h in 10 mL of DMF under argon.
b GC yields.
Under the successful conditions established for electronically or sterically demanding aryl bromides, the catalytic system was tested on aryl chlorides. The coupling of the deactivated 4-chloroanisole was ineffective (Table 4, entry 7). However, electron-poor, 4-chloroacetophenone and 4-chlorobenzonitrile were activated to yield moderate to good conversion to alkynylated aryls (Table 4, entries 8–11).
In general, when the conversions of expected product were low, we observed that the triphosphine system was producing more alkyne dimerization than the tetraphosphine system. We were, thus, interested to obtain further information on the interactions possibly existing in solution between polyphosphines and the metal centers copper and palladium. In particular, important questions arise concerning the adducts stability, and the possibility of ligand transfer from one metal to the other in the course of the tandem catalytic reaction (see Scheme 3 and 4).
3.4 Ligand exchange study between copper and palladium
We investigated by 31P and 1H NMR experiments the interaction of copper adducts (1), (3), and (4) with palladium allyl chloride in various solvent (CDCl3, CD2Cl2, DMF, toluene) at temperatures between −90 °C and 80 °C [24]. We were particularly interested in evaluating the lifetime of copper interaction with phosphorus in the presence of palladium. Upon mixing the triphosphine-Cu adduct (3) in CD2Cl2 with 0.5 equiv of the dimeric palladium allyl chloride at room temperature, a portion of the triphosphine ligand L1 was transferred to palladium, giving rise to a palladium complex, clearly characterized in the 31P NMR by two sharp singlets at 37.3 ppm (chelating bonded PPh2) and 2.1 ppm (uncoordinated phosphino group P(i-Pr)2) of respective intensity 2:1. According to the corresponding 1H NMR, an equilibrium around 50:50 is obtained between copper adduct (3) and the new palladium complex. This equilibrium was immediately obtained and was found to be unchanged after 30 min. Upon heating the mixture for 1 h at 50 °C, the equilibrium sharing out the ligand between copper and palladium is almost not modified; however, a novel palladium species is obtained presumably due to allyl decoordination. The same kind of experiments were conducted with the triphosphine-Cu adduct (4) in CD2Cl2. Analogous behavior was observed with this triphosphine, showing a partial transfer of ligand from copper to palladium. The 1H NMR spectrum indicated after 10 min, a ratio 80:20 in favor of the palladium complexes. Conversely, the mixture of the Cu-tetraphosphine adduct (1) with 0.5 equiv of the dimeric palladium chloride at room temperature in CDCl3 immediately reaches an equilibrium, which is conserved after 24 h at 4 °C The presence of copper adduct (1) in 70% was estimated from 1H NMR. Upon heating the mixture of adduct (1) and palladium allyl chloride in CDCl3/toluene (1:1) for 1 h at 80 °C, it appears that mostly the palladium-phosphine area is changed while copper-phosphine species are still detectable around −25.0 ppm. Thus, CuI adducts stabilized by tridentate or tetradentate ligands lead to very different species when simply mixed with a palladium precursor at room temperature. Clearly, the CuI phosphine adduct 1 formed with the tetraphosphine L3 is more resistant to ligand transfer and higher temperatures. Concerning the triphosphine adducts (3) and (4), the ligand transfer can be very fast; in general the equilibrium obtained between palladium and copper species at room temperature is conserved at 50 °C but slowly modified at 80 °C. It would have been too speculative to establish a rationale for the general catalytic behavior observed for the three species at this stage. However, better results concerning the inhibition of diyne formation have been obtained in runs involving palladium and adduct (1): this might be related to the greater lifetime of this copper-tetraphosphine adduct. Such interesting observation on copper iodide adduct need to be confirmed with other highly stabilizing chelating ligands.
The pre-stabilization of copper iodide with a multidentate ferrocenylphosphine ligand, which lead to a molecularly defined adduct, promotes low metal (palladium and copper) cross-coupling of deactivated aryl bromides and electron-poor chlorides with acetylenes. This system has some practical and economical advantages:
- • its efficient inhibition of alkyne oxidative homocoupling;
- • the easiness of weighing and storing the CuI adducts;
- • the outstandingly low amount of copper needed (0.4 to 0.1 mol%).
The coordination behavior of polydentate ligands towards copper and palladium, and in particular, the transfer from one metal to the other has been investigated. These studies have shown that copper-ligand interactions may exist in traditional tandem palladium/copper cross-coupling reactions, opening thus, the way to new perspectives in mechanistic understanding. This could be even more important when nitrogen or oxygen ligand more copperphilic than palladophilic are involved.
4 Alkyne arylation in ionic liquids, towards recyclable systems?
4.1 Alkyne arylation from bromoarenes
Ionic liquids as novel solvents have shown great promise because of their negligible vapour pressure, ease of handling and potential for recycling. As a reaction medium, their high compatibility with transition metals and limited miscibility with common solvents enable the easy separation of organic products and possibly retention of the catalyst at the resting-state in the ionic phase [25]. Apart from the pioneering work reported by Ryu et al. [26], and by Alper and Park [27], which have been the first groups to disclose catalytic systems for the coupling of aryl iodides in 1-n-butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6]), alkyne arylation in ionic liquids had remained underdeveloped. We subsequently provided the first method that allows the coupling of a large array of activated and deactivated bromoarenes [28,29]. Furthermore, the system of highest efficiency which has been identified was surprisingly the copper-free simplest and cheaper combination that employed [Pd(η3-C3H5)Cl]2 and the ubiquitous tertiary phosphine PPh3 at only a 1 mol% loading, with pyrrolidine in 1-n-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]). In addition to the quantitative conversion of 4-bromoacetophenone (Table 5, entry 1), good to excellent conversions were obtained in 2 to 4 h for the coupling of phenylacetylene with the electronically activated 4-bromobenzonitrile (80% in 4 h, Table 5, entry 2), 3,5-(trifluoromethyl)bromobenzene (92%, 4 h, Table 5, entry 3) and with the unactivated bromobenzene (82%, 2 h, Table 5, entry 4). However, the most remarkable results were obtained in longer time for the electronically and/or sterically deactivated substrates: in 20 h, 4-bromanisole (95% yield, Table 5, entry 5), 4-bromotoluene (86%, Table 5, entry 6), 2-bromotoluene (99%, Table 5, entry 7) were efficiently converted. While cross-coupling with aryl halides, in which one or both the ortho-positions are substituted, generally suffers from additional complications of steric origin, 2-bromonaphtalene and bromomesitylene were quantitatively converted into the corresponding enyne in 4 and 48 h, respectively (Table 5, entries 8–9). A double arylation was possible starting from a dibromoaryle (98% conversion in enynes, with 60% of monosubstitution and 40% of disubstitution, Table 5, entry 10). Finally, the coupling of aryl bromides with a demanding (long chain) aliphatic terminal alkyne, namely 1-decyne, was also explored with success: in only 4 h, 4-bromoacetophenone was converted in 96% yield (Table 5, entry 11), the electronically and/or sterically deactivated 4-bromotoluene and 2-bromotoluene yielded 76% and 65%, respectively, of the expected arylation product (Table 5, entries 13, 14). For 4-bromoanisole, 20 h of reaction gave 76% conversion into the desired enyne (Table 5, entry 12). Our attempts to activate aryl chlorides with the Pd/PPh3 system have remained unsuccessful, as even 4-chloroacetophenone was not converted.
Performances in copper-free alkyne arylation with aryl bromides in the presence of 1%mol of Pd/PPh3 in [BMIM][BF4]a.
| Entry | Aryl bromide | Alkyne | t [h] | Enyne yield [%] |
| 1 | 2 | > 99 | ||
| 2 | 4 | 80 | ||
| 3 | 4 | 92 | ||
| 4 | 2 | 82 | ||
| 5 | 20 | 95 | ||
| 6 | 20 | 86 | ||
| 7 | 20 | 99 | ||
| 8 | 4 | 98 | ||
| 9 | 48 | > 99 | ||
| 10 | 24 | 98 | ||
| 11 | 4 | 96 | ||
| 12 | 20 | 76 | ||
| 13 | 4 | 76 | ||
| 14 | 4 | 65 |
a Catalytic conditions: 1.0 equiv of aryl bromide, 1.2 equiv of phenylacetylene, 1.2 equiv of pyrrolidine, 3 mL [BMIM][BF4], 130 °C; 0.5 mol% [Pd(allyl)Cl]2, 3 mol% PPh3.
4.2 Novel cationic P-ligands for catalytic systems anchoring
The system [Pd/3 PPh3] (1 mol%) in [BMIM][BF4] has been found efficient for the coupling of a variety of activated and deactivated aryl bromides to phenylacetylene; however, the recycling of this catalytic system was limited by detrimental leaching of the monophosphine ligand PPh3 throughout the extraction process with organic solvents, and after three recycling the performances dropped [8]. We, thus, decided to explore the potential of the less lipophilic charged donor-stabilized phosphenium adducts, previously described in our group [30], in alkyne arylation reactions [31]. Indeed, we anticipated that the presence of a cationic charge on the coordinated ligand should facilitate the separation of metallic residues from the organic products. We employed the catalytic systems combining palladium and the catalysis auxiliaries (5) and (6) (Scheme 7) in the presence of pyrrolidine as an organic base.
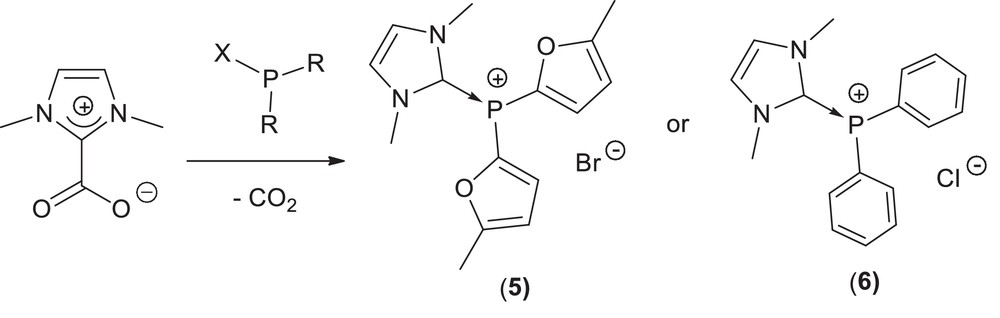
N-heterocyclic carbene phosphenium adducts 5 and 6.
Comparative coupling studies to phenylacetylene under copper-free conditions were conducted on four aryl bromides with different steric and electronic features: the activated electron-poor 4-bromoacetophenone, the deactivated electron-rich 4-bromoanisole, the ortho-substituted electronically deactivated 2-bromotoluene and the sterically hindered 2-bromonaphtalene. The palladium precursor combined with (5) was found to be a superior system, in all cases, than the analogous palladium complex containing (6), in terms of activity and reproducibility of the results [31].
Table 6 summarizes the scope and limitations of the [Pd/5/pyrrolidine] catalytic system for phenylacetylene arylation in [BMIM][BF4], together with the recycling runs conducted from the best conversions. Under the optimized conditions, and in the noticeable absence of CuI co-catalyst, the complete arylation of phenylacetylene were performed from bromobenzene, 3-bromotoluene and 4-bromoacetophenone (quantitative yields, Table 6, entries 1-3). Good to moderate coupling was also obtained from 3-bromoanisole (82% yield, Table 6, entry 8), 4-bromoanisole (58% yield, Table 6, entry 9) and 2-methyl bromobenzene (50% yield, Table 6, entry 4), indicating that either strongly unfavourable electronic or steric effects slightly hamper the performances of the system. However, the catalyst activity was clearly limited for the conversion of the trisubstituted 2-bromomesitylene (both electronically and sterically deactivated) even over a long reaction time (Table 6, entry 5, 26% yield of coupling after 48 h), although under similar conditions [Pd/3PPh3] gave a complete conversion.
Palladium arylation with various aryl bromide in [BMIM][BF4].
| Entry | Aryl bromide | Conditions T (°C)/t (h) | Run 1 yield (%) | 1st recycling yield (%) | 2nd recycling yield (%) |
| 1 | 125/40 | 99 (90) | 99 | 60 | |
| 2 | 125/40 | 99 (95) | 60 | 40 | |
| 3 | 90/40 | 99 (90) | 75 | ||
| 4 | 125/40 | 50 | |||
| 5 | 125/48 | 26 | |||
| 6 | 125/40 | 95 (85) | 70 | ||
| 7 | 125/40 | 95 (85) | 40 | ||
| 8 | 125/40 | 82 | 32 | ||
| 9 | 125/40 | 58 |
After ethereal extraction of the coupling product under argon, the resulting ionic liquid phase was reused without any treatment and refilled with the coupling reagents and the base (first recycling, Table 6 entries 1–3). While the recycling of bromobenzene gave excellent results (99% yield), functionalized aryl bromides, such as 3-bromotoluene and 4-bromoacetophenone showed a drop of activity (60 and 75% yield, respectively). A second recycling confirmed this tendency (see Table 6). 31P NMR experiments on the extracted phases did not point out any traces of ligand leak into the organic phase, in contrast to the experiments where triphenylphosphine was employed as the ligand. Much to our disappointment, although the catalyst remained insoluble in non-polar organic solvents and was thus successfully immobilized in the IL phase during recycling attempts, its activity in [BMIM][BF4] decreased from the first recycling. The deactivation was assumed to be due to a protonation reaction of the C→P bond of the ligand in the presence of the large quantity of the pyrrolidium salt formed upon the successive recycling. This observation suggests a certain incompatibility of such ligands with catalytic reactions performed in acidic media, or which generates easily transferred protons, especially when no treatments are done on the reused phase (water washing, etc.). Alternatively, a halide attack on the phosphenium with trapping of the released carbene is also possible [32]. However, in the platinum-catalysed hydrogenation of chloronitrobenzenes in [BMIM][BF4], the recycling potential of donor-stabilized phosphenium ligands was confirmed, since the catalytic system was successfully recycled seven times without noticeable metal leaching in the organic phase and any significant loss of activity [31].
5 Versatile coupling of heterocyclic bromides and chlorides
5.1 Alkyne heteroarylation using bromides and chlorides
In ionic liquid, excellent alkyne arylation performances had been obtained from the simple and cheap combination using [Pd/3PPh3] at only 1 mol% loading with 1.2 equiv of base and in the absence of copper salt. We thus decided to investigate the potential of this system for alkyne direct functionalization with heteroaromatics [33]. Preliminary results revealed that both temperature and reaction time are crucial parameters that differed from our previous studies of alkyne arylation [28,29]. Fig. 5 displays the products of copper-free arylation from a variety of six-membered heteroaryl bromides achieved with this catalytic system. 2-Bromopyridine was quantitatively coupled to phenylacetylene in 3 h at 100 °C. At higher temperature (130 °C) or beyond 3 h of the reaction time, the hydroamination of the formed 2-(phenylethynyl)pyridine by the excess of pyrrolidine is observed. 3-Bromopyridine and 3-bromoquinoline were also efficiently coupled to phenylacetylene or aliphatic alkynes. The coupling of N-diheteroaromatic pyrimidine bromides, in position -2 and -5, proceeds smoothly in 4 h. For a selective coupling of 2-bromopyrimidine, NEt3 is also required to avoid the predominant coupling of pyrrolidine to pyrimidine. Apparently, the halogenated position is not specifically problematic and the hindrance or electronic effect of the fused-rings of quinoline does not limit the coupling. Additionally, good selectivity and excellent yields were obtained when phenylacetylene was coupled with functionalized 3-bromo-5-methoxypyridine (91%), 3-bromo-6-methoxypyridine (90%) and with 3-bromo-6-methylpyridine (91%). An excellent yield was obtained under the same conditions for the sterically deactivated substrate, 3-bromo-4-methylpyridine. Since the efficient coupling of phenylacetylene with a variety of electron-rich pyridinyl bromides was also achieved, we examined the arylation reaction of some different terminal alkynes. A selection of electron-rich heterocycles was consequently reacted with short chain or long chain aliphatic acetylenics. The system was also successfully employed for alkyne arylation using a dihydro-2H-[1,5]benzodioxepin bromide as a much less studied substrate of industrial interest. Possibly due to the basic conditions or temperature, heterocyclic substrates, such as bromolactone, bromoindole, bromopyrrazole, bromouracil and bromoisoxazole were found to be problematic since either no coupling or the decomposition of the reagents was observed.
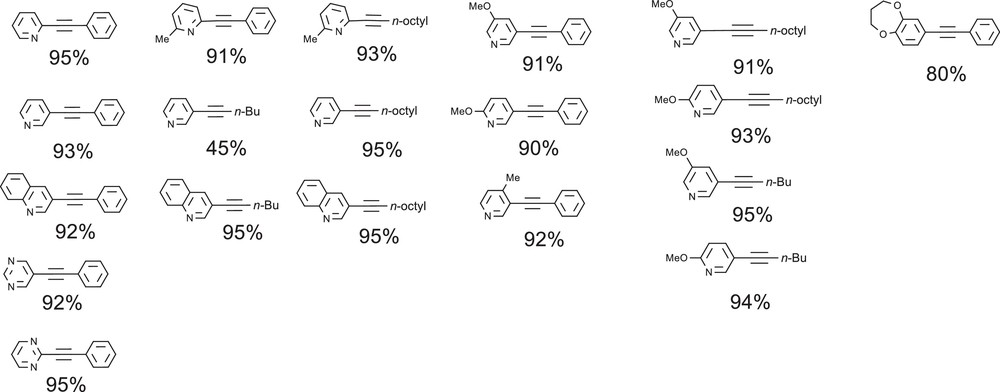
Products of alkyne arylation from six-membered heteroaryl bromides.
As shown in Fig. 6, clean and quantitative arylation reactions with the five-membered respectively O- and S-heteroaromatic, 3-bromofuran and 3-bromothiophene, were achieved with our system [33]. Notably, earlier reports underlined the failure of many systems, involving either PPh3 or more sophisticated ligands [34,35]. Higher temperature and longer time are mandatory to couple the 3-bromofuran. A first genuine limitation appeared from the S,N-diheteroaromatic 2-bromothiazole, in the presence of pyrrolidine, a coupling of this amine to thiazole was observed in majority. However, the selectivity towards (phenylethynyl)thiazole, which was then obtained in 72% yield, can be restored by using the tertiary base NEt3. The sterically hindered 2-methyl-3-bromothiophene was also successfully coupled to 1-octyne (94%). Longer reaction times were needed to couple the more volatile 1-hexyne with less satisfactory results (70%).

Products of alkyne arylation from five-membered heteroaryl bromides.
With regards to the relevant results we obtained in alkyne arylation from aryl bromides, we investigated the alkyne arylation reactions from more reluctant heteroaryl chlorides. Contrary to the case of bromides, heteroaryle substrates halogenated in position-3 were found unreactive under our conditions (3-chloropyridine, 3-chlorothiophene). Conversely, the coupling of phenylacetylene to mono- and diheteroaromatics chlorinated in a position adjacent to the heteroatom proceeds smoothly (Fig. 7) [33]. Thus, 2-chloropyridine and 2-chloropyrazine are quantitatively coupled. Interestingly, the selectivity of 2-chlorothiazole coupling is much improved compared to the bromide analogue (92% vs 72%) and 2-chlorobenzothiazole is an even more suitable substrate. N-diheteroaromatics pyrimidine and pyridazine were also very efficiently coupled, and we obtained in a remarkably high yield and clean reaction the comparatively electron-rich 3-(phenylethynyl)-6-methoxypyridazine.

Products of alkyne arylation of heteroaryl chlorides.
5.2 Extension to novel diynes
With regards to the relevant results obtained in the arylation of terminal alkynes in an ionic liquid, we investigated the double arylation of terminal diynes by using the same methodology (Scheme 8) [36]. One objective was to examine whether a first arylation of the terminal diyne would modify the course of a second arylation on this substrate. In this case, interesting unsymmetrical double arylation might be envisioned. Additionally, the resulting heteroarylated diynes may be interesting precursors for further intermolecular annulation towards indoles and carbazoles, for instance by catalysed hydroamination. This type of double cross-coupling reactions had been only rarely investigated, and exclusively on isolated examples, while the obtained products have shown great potential in medicinal chemistry or materials science [37,38].
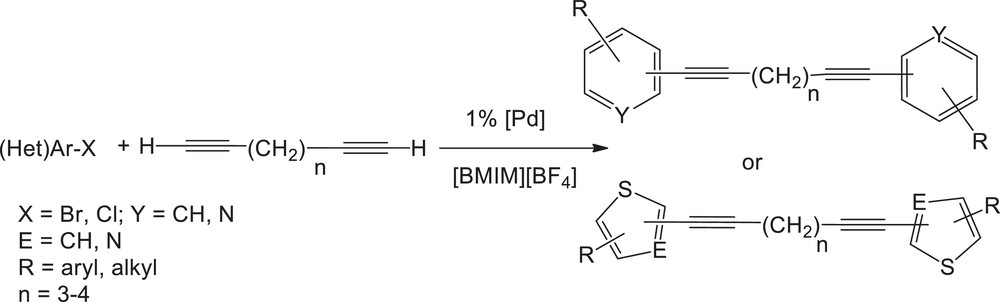
Palladium-catalysed double arylation of terminal diynes.
Table 7 summarizes the successful results obtained under conditions similar to the ones optimized for the simple arylation of monoalkynes (Table 6). 3-Bromopyridine was quantitatively coupled to 1,7-octadiyne to give 1,8-di-(pyridin-3-yl)octa-1,7-diyne (entry 1). 3-Bromoquinoline and 3-bromothiophene were also efficiently coupled to 1,7-octadiyne (entry 2 and 3). A limitation appeared for the coupling of the diheteroaromatic 2-bromothiazole since in the presence of pyrrolidine, a coupling of this base to bromothiazole was mainly observed again. Conversely, 1,8-di-(thiazol-2-yl)octa-1,7-diyne was obtained by using the tertiary base NEt3 (entry 4). 3-Bromoquinoline was also reacted with a slightly shorter chain diyne, 1,6-heptadiyne, under the same conditions (entry 5). Using heteroaryl chlorides substrates, good yields were obtained with 2-chloropyrazine and 2-chlorobenzothiazole coupled to 1,7-octadiyne (entries 6 and 7). Double arylation reaction by using functionalized aryl bromides was also examined: moderate to satisfactory conversions were obtained for the arylation of 1,7-octadiyne with 4-bromoacetophenone (entry 8) and bromobenzene (entry 9) at 130 °C for 40 h. 4-Bromanisole and 4-bromotoluene (entries 10,11) were also converted. In the latter cases, we detected a competitive oligomerization of the diynes present in excess, which diminishes the final yield in enynes. Our attempts to make unsymmetrical arylated diynes one-pot or stepwise remained unsuccessful, giving mixtures of the products. This suggests that contrary to our initial expectations, a first arylation of the diyne has little influence on the remaining terminal alkyne function.
Scope of the palladium-catalysed double arylation in [BMIM][BF4].
| Entry | Haloarene | Base/time (h)a | Product | Yield (%)d |
| 1 | pyrrolidine/5 | 98 | ||
| 2 | pyrrolidine/5 | 91 | ||
| 3 | pyrrolidine/5 | 93 | ||
| 4 | NEt3/5 | 54 | ||
| 5b | pyrrolidine/5 | 92 | ||
| 6 | NEt3/5 | 80 | ||
| 7 | NEt3/5 | 69 | ||
| 8c | pyrrolidine/40 | 30 | ||
| 9c | pyrrolidine/40 | 64 | ||
| 10c | pyrrolidine/40 | 51 | ||
| 11c | pyrrolidine/40 | 50 |
a Reaction conditions: 0.5 mol% [Pd(allyl)Cl]2, 3 mol% PPh3, 1.0 mmol aryl bromide (or chloride), 0.55 mmol of 1,7-octadiyne, 1.2 mmol of base, 3 mL [BMIM][BF4], 100 °C.
b 1,6-Heptadiyne.
c 130 °C.
d Isolated yields obtained after chromatography on silica column.
Thus, a double arylation of bifunctional terminal diynes can be efficiently performed by using the same simple catalytic system developed for arylation and heteroarylation. Both aryl and heteroaryl bromides are suitable substrates, as well as 2-chloroheteroarenes. Depending on the substrate, a primary or a tertiary amine has to be used as soluble base. The accessibility of the system featuring cheap PPh3 ligand, an organic base and the ionic [BMIM][BF4], makes these results of valuable interest for further development of Heck- and Sonogashira-type coupling reactions in ionic liquids. We importantly showed that besides facile iodoarenes, aryl and heteroaryl bromides and some chlorides are suitable substrates for copper-free palladium alkynes arylation in a highly polar ionic liquid.
Acknowledgements
Thanks are due to the undergraduate students which have been involved in this work (L. Gaillard, J. Boudon, E. Fayad). The authors thank the CNRS, the Université de Bourgogne, and “Conseil Régional de Bourgogne”, with the 3MIM program, and the ANR CP2D program for the project “CAMELOT”. Most of the polyphosphines described herein are commercially available since early 2012 from STREM INC, which are acknowledged for their trust. J.-C. H. sincerely thanks the editorial board and staff of European Journal of Inorganic Chemistry for the attribution of the EurJIC Y.I. award.