

1 Introduction
Energy is the main driving force for the advancement of civilization [1]. Currently, most of the world's energy originates from petroleum-based fossil fuels. However, petroleum is a finite energy source. Taking into account the consumption rate of petroleum, which increases each day, the world's reserves simultaneously decrease. In the short term, the situation is not sustainable, and various strategies must be implemented for the more efficient consumption of petroleum [1]. Furthermore, pollution and global warming minimize the effects of petroleum technology if stricter fuel quality specifications are not respected. In this context, diesel fuel can help solve the current economic and environmental problems if it has a high combustion efficiency and a high cetane number, which will result in a lower consumption of diesel fuel and preserve the existing petroleum reserves. The cetane number increases when the rings of naphthenic molecules are opened [2–4]. Naphthene ring opening, which has been extensively studied for methylcyclopentane (MCP) [5–8] in the petrochemical industry, forms products with cetane indexes greater than that of MCP. The ring-opening products were 2-methylpentane (2-MP), 3-methylpentane (3-MP) and n-hexane (n-H). Numerous industries have implemented atom-economical processes [9,10]; the petrochemical industry, in particular, has used this process in which the role of the catalyst is the cornerstone of this petrochemical challenge. Several types of catalysts have been investigated in this MCP reaction, including noble metallic-supported [11,12,5,13–15], and mesoporous oxide catalysts [16,17]. Among these, noble metallic-supported catalysts have a reputation for being the most selective for the ring opening of MCP. Based on previous studies [18–20], two mechanisms, in particular, have been distinguished: the cyclic mechanism, which involves five carbon atoms, and the bond-shift mechanism, which involves three carbon atoms. The cyclic, or “non-selective,” mechanism favors the formation of three isomers of MCP in statistical distribution (2-MP/3-MP/n-H = 2:1:2), whereas the bond-shift, or “selective,” mechanism favors the formation of 2-MP and 3-MP (2-MP/3-MP = 2:1) and not n-H. Over the past few decades, researchers have developed a significant interest in understanding the mechanism of the ring opening of MCPs because of their immense importance in catalysis. The ring-opening reaction of MCP on noble metals is generally accepted [19,20] to be a structurally sensitive reaction, especially when the catalysts are Pt nanoparticles supported on alumina. The non-selective mechanism operates on small Pt particles, and the selective mechanism operates on large Pt particles. Interestingly, a similar size effect was not observed on Ir particles [21–23]; however, another parameter, the support, should be investigated. The literature on supported metallic catalysts shows that the principal factors that contribute to the understanding of the ring-opening mechanism of MCP are the particle size and the support. The selective mechanism was favored when Ir was dispersed on a silica support, whereas the non-selective mechanism was observed when Ir was dispersed on an alumina support. Despite impressive studies and progress in the ring opening of MCP on Ir, Pt nanoparticles dispersed on non-reducible oxides such as SiO2 and Al2O3, few reports have been published that concern the reducible oxides. Among the reducible oxides used as supports for Ir and Pt nanoparticles, TiO2 [24–26] and MoO2 [7] supports have provided the most interesting results with respect to the strong metal–support interactions (SMSI) and the nature of active sites in the ring opening of MCP [7,24–26]. However, no studies on the ring opening of MCP on Ir, Pt, and Pt–Ir nanoparticles dispersed on a reducible WO2 support have been reported. The objective of this study is to understand the effect of a reducible support and the nature of active sites on these catalysts as a function of the reaction temperature to develop the design catalysts for the selective ring opening of MCP.
2 Experimental
2.1 Materials
WO2 (99.99%) was supplied by Strem Chemicals, and the H2PtCl6.6H2O (> 99.99%) and H2IrCl6.6H2O (> 99.99%) metal precursors were supplied by Sigma-Aldrich. All the reactants were used without previous purification. Deionized water was used in all experiments. The MCP was supplied by Aldrich (> 99%). Hydrogen (Linde), helium (Linde) and air (Linde) were used after purification by a gas-cleaning filtration system (Chrompack).
2.2 Catalyst preparation
The monometallic 0.5 wt.% Pt/WO2, 0.5 wt.% Ir/WO2 and bimetallic 0.25 wt.% Pt–0.25 wt.% Ir/WO2 catalysts were prepared by the incipient wetness impregnation and co-impregnation, respectively, of aqueous solutions that contained the requisite amounts of the respective metal precursor on a WO2 support. After the impregnation and co-impregnation procedures, the solids were dried overnight at 120 °C and then calcined under air at 350 °C for 4 h. The samples were designated as Pt/WO2 for 0.5 wt.% Pt/WO2; Ir/WO2 for 0.5 wt.% Ir/WO2; and Pt–Ir/WO2 for 0.25 wt.% Pt–0.25 wt.% Ir/WO2.
2.3 Physico-chemical characterizations
The specific surface areas of the samples were determined using the standard Brunauer–Emmett–Teller (BET) method by nitrogen sorption measurements performed at –196 °C on a TriStar apparatus. Prior to the N2 physisorption measurements, each sample was degassed at 200 °C under vacuum at 10−6 mmHg overnight. The PSD estimations were calculated from the Barett-Joyner-Halenda (BJH) equation. The morphologies of the samples were observed using a field-emission scanning electron microscopy (SEM). The images were obtained on a JEOL 6700 scanning electron microscope operated at 200 kV; the samples were dispersed as thin films on carbon grids. The crystal structures of the solid catalysts were identified using X-ray analysis. The powder X-ray diffraction patterns (XRD) of the samples were acquired at room temperature on a Bruker D8 powder diffractometer equipped with a Cu Kα radiation source (0.154 nm) operated at 40 kV and 100 mA. The diffractograms were recorded in the 2θ range of 10–80°. The XRD patterns were compared to those of known standards taken from the JCPDS database. The crystallite size and the lattice strain were determined from the half-widths of the diffraction lines using the Scherrer equation. The shape and the size distribution of the metallic particles before catalytic tests were estimated from transmission electron microscopy (TEM) images. These images were obtained on a TopCon 2100 FCs microscope operated at an accelerating voltage of 200 kV. The relative percentages of each metal in the solid catalysts were determined by the Service Central d’Analyse of CNRS-Solaize, France.
2.4 Catalytic tests
Catalytic measurements for the conversion of MCP were performed under atmospheric pressure using a pulse microreactor connected to a gas chromatograph equipped with a 50 m (CP-SIL-5CP) column and a flame ionization detector (FID). The mass of the catalysts used was 200 mg, and the volume of MCP injected was 10 μl. The gas flow used for treatments and catalytic tests was fixed at 40 cm3 min−1. Prior to use in the experiments, the catalysts were reduced in situ in pure hydrogen at 500 °C for 4 h.
3 Results and discussion
3.1 Preparation and physico-chemical characterization catalysts
The chemical analysis confirms that the metal (Pt, Ir) loading values in the monometallic and bimetallic solid catalysts were close to the theoretical values (Table 1).
Physico-chemical properties of the support and metallic catalysts.
| Catalyst | wt.% metal theoretical | wt.% metal depositeda | dp (Å)b | (Å)c | dmetal (Å)d |
| W18O49 | – | – | 45;75;110;170 | 260 | – |
| Pt/W18O49 | 0.5 | 0.51 | 45;75;110;170 | 260 | < 10 |
| Ir/W18O49 | 0.5 | 0.47 | 45;75;110;170 | 275 | < 10 |
| Pt–Ir/W18O49 | 0.25–0.25 | 0.25–0.23 | 45;75;110;170 | 290 | < 10 |
a Measured by ICP for the calcined samples.
b Pore diameter.
c Crystallite sizes of the WO2 nanorods calculated from the XRD analysis for the calcined samples.
d Metallic size from TEM analysis for the calcined samples.
The structures of the catalysts were revealed by XRD analysis. The X-ray diffractograms of the calcined support and metallic catalysts is shown in Fig. 1. Interestingly, the XRD spectra reveal that the calcined support corresponds to a suboxide, W18O49, with a monoclinic structure and crystalline character and not to WO2. This result can be explained by the contamination of WO2 due to oxidation during air exposure. The XRD patterns of the calcined support and catalysts are similar, and a perfect match was obtained with respect to W18O49 (JCPDS 00-005-0392), which confirmed their identity. The lattice parameters of the monoclinic structure are a = 18.28 Å, b = 3.775 Å, c = 13.98 Å and β = 115.20°. The typical crystallite sizes of the tungsten nanorods, calculated from the Scherrer equation [27] from the diffraction peak at 2θ = 23.61° for the calcined samples, are between 260–290 Å. For all the catalysts, no detectable diffraction peaks attributable to Pt and Ir metals were observed. This result suggests that both the Ir and Pt metals are highly dispersed with particle sizes smaller than the limit of detection of the apparatus.
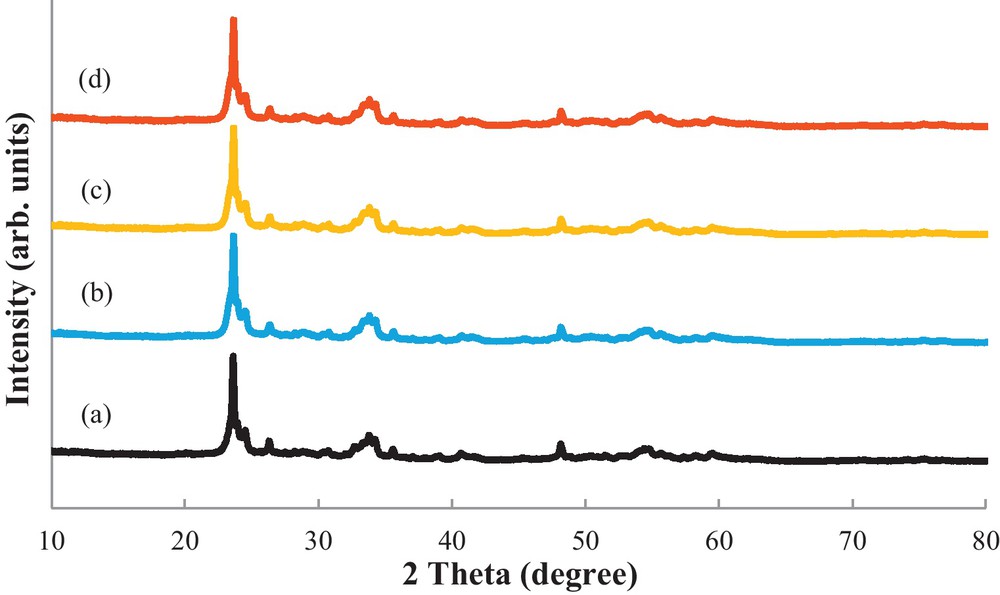
Wide angle XRD patterns of W18O49 (a), Pt/W18O49 (b), Ir/W18O49 (c) and Pt–Ir/W18O49 (d) catalysts calcined at 350 °C.
The high dispersion degree can also be confirmed through the TEM images of the metallic catalysts, which exhibit a clear lattice image of W18O49 with no distinguishable crystalline Pt or Ir particles visible on the support (Fig. 2). However, the shape of nanoparticle supported catalysts is difficult to characterise because of the small particle size, high dispersion and/or very low metal loading. Unfortunately, Ir and Pt particles were difficult to differentiate as observed already on reducible MoO2 support [7]. The shape of the particles was assumed to be conformed to icosahedral Mackay structures defined by (111) fcc planes [28,29], with a high density of atomic edges and corners.

TEM micrographs of W18O49 (A), Pt/W18O49 (B), Ir/W18O49 (C) and Pt–Ir/W18O49 (D) catalysts calcined at 350 °C.
Fig. 3 shows the experimental nitrogen adsorption–desorption isotherms for the support and metallic catalysts. Interestingly, the pattern for the catalysts appears to be a superimposition of the support pattern: all of the catalysts exhibit adsorption primarily at high p/p0 values, which implies that the pores have been filled. The isotherms are Type IIb, with a hysteresis loop that does not fall into any of the well-established categories. A compilation of the BET surface area and the average pore diameter of the support and metallic catalysts are presented in Table 1. Compared with the support, the metallic catalysts were not changed by impregnation of the metallic phase. The BET surface area of the support was 2 m2/g and was quasi-identical to that of the metallic catalysts. The BJH pore size distribution with respect to the adsorption isotherm branch is plotted in Fig. 4 in terms of the dV/dlog (D) pore volume versus the pore diameter. This result indicates that the W18O49 support has pore dimensions at the outskirts of the validity range of the BJH method, and because the curves appear to extend over the boundaries, i.e., 75, 110 and 170 Å, respectively, the corresponding average pore diameter values must be regarded with caution (Table 1). However, the majority of pores are in the mesoporous range with diameter maxima centered at approximately 45 Å.
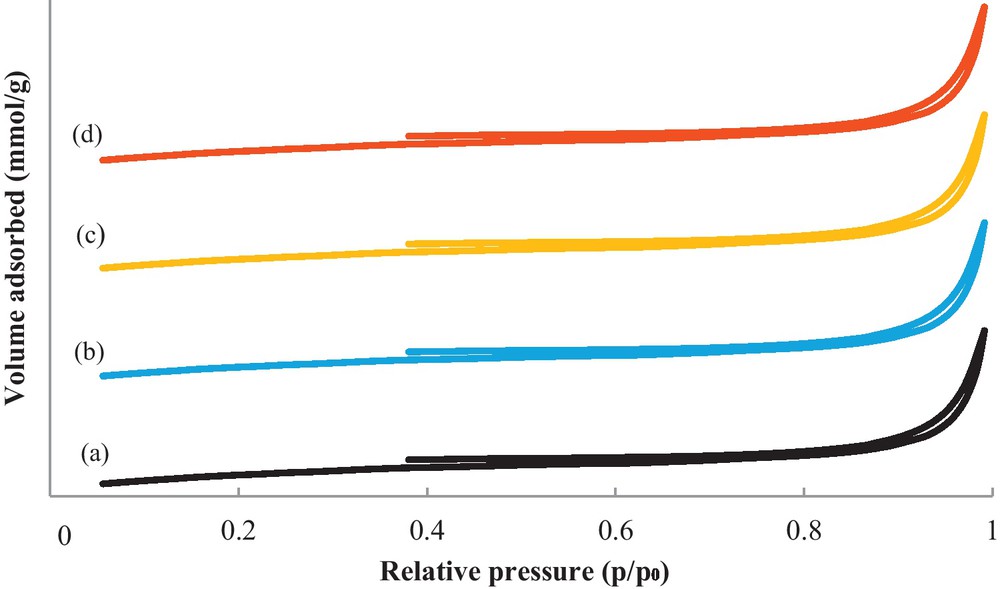
N2 sorption isotherms of W18O49 (a), Pt/W18O49 (b), Ir/W18O49 and Pt–Ir/W18O49 (d) catalysts calcined at 350 °C.
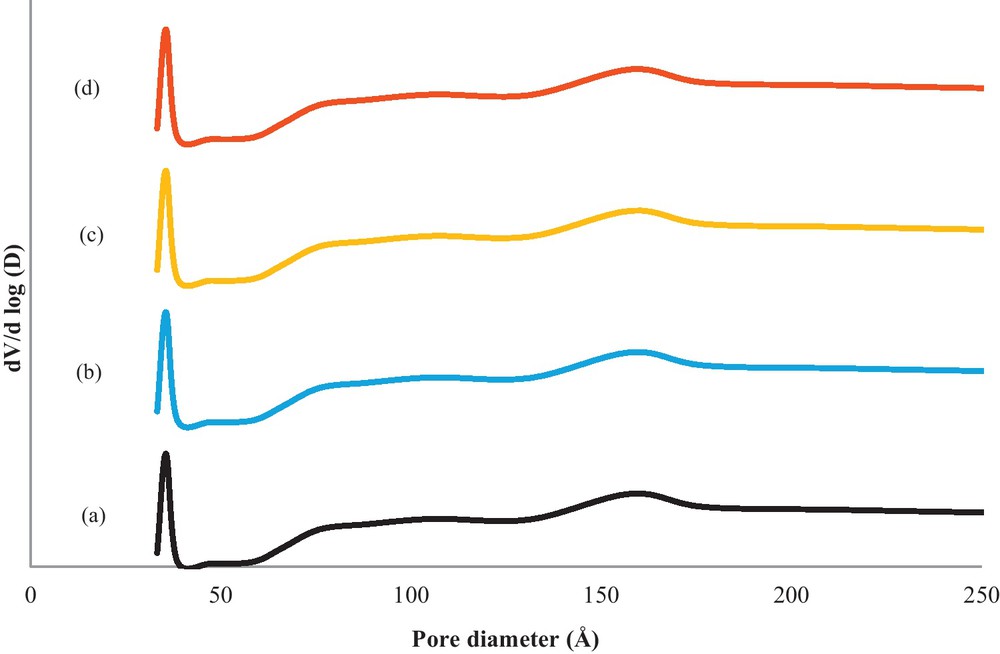
Pore size distribution of W18O49 (a), Pt/W18O49 (b), Ir/W18O49and Pt–Ir/W18O49 (d) catalysts calcined at 350 °C.
The morphology of the samples was observed using SEM analysis (Fig. 5). The overall morphology of the samples is composed of a large quantity of uniform nanorods with diameters typically in the range of 1000 Å and with lengths of up to several micrometers.

SEM images of W18O49 (A), Pt/W18O49 (B), Ir/W18O49 (C) and Pt–Ir/W18O49 (D) catalysts calcined at 350 °C.
These observations reveal that the nanorods have a uniform diameter along their entire length and a narrow diameter distribution.
3.2 Catalytic activity of WO2, Pt/WO2, Ir/WO2 and Pt–Ir/WO2 catalysts in the conversion of MCP
We attempted to determine if the dispersion of the monometallic and bimetallic particles on the acidic reducible support causes significant changes in catalytic activity with respect to the conversion of MCP. In particular, we investigated the changes in catalytic activity on the WO2 acidic reducible support, which, unlike the cases of Al2O3, SiO2, and TiO2 supports, has not been previously reported in the literature. The relative activities obtained for all the catalysts are presented as a function of the reaction temperature.
3.2.1 Reaction products observed in the conversion of MCP
The WO2 support and the metallic catalysts were tested in the conversion of MCP in the 180–500 °C temperature range. The different observed reaction products are presented in Table 2. With the exception of the Ir catalysts, which begin to exhibit catalytic activity at 340 °C, no reaction products were detected under our experimental conditions at temperatures less than 400 °C for the support and the metallic catalysts. No olefins were observed in the investigated temperature range.
Product distributions (% mol) as a function of temperature in the conversion of MCP over the support as well as over monometallic and bimetallic catalysts.
| Temperature (°C) | C1 | C2 | C3 | i-C4 | n-C4 | i-C5 | n-C5 | 2-MP | 3-MP | n-H | Ch | Bz |
| WO2 | ||||||||||||
| 400 | 2.3 | 2.8 | 1.1 | 1.2 | 0.4 | 0.5 | 0.9 | 0 | 0 | 0 | 0 | 2.0 |
| 440 | 11.5 | 4.9 | 8.5 | 0.4 | 1.8 | 0 | 0 | 0 | 0 | 0 | 0 | 4.3 |
| 470 | 54.1 | 18.2 | 12.0 | 1.4 | 2.8 | 0.8 | 0 | 0 | 0 | 0 | 0 | 3.9 |
| 500 | 79.7 | 11.7 | 4.7 | 0.3 | 0.6 | 0 | 0 | 0 | 0 | 0 | 0 | 2.5 |
| Pt/WO2 | ||||||||||||
| 400 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.1 | 0.4 |
| 440 | 4 | 0.6 | 2.4 | 0.6 | 0.9 | 1.1 | 1.6 | 0 | 0 | 0 | 2.7 | 3.4 |
| 470 | 5.9 | 3.8 | 4.0 | 1.8 | 2.1 | 1.5 | 0.8 | 1.1 | 0.1 | 1.1 | 0.8 | 6.1 |
| 500 | 16.8 | 11.0 | 9.9 | 2.3 | 2.6 | 1.4 | 1.6 | 3.5 | 1.1 | 1.4 | 0.3 | 5.2 |
| Ir/WO2 | ||||||||||||
| 340 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1.4 | 0.3 | 0.1 | 0.7 | 0 |
| 400 | 0.9 | 0.5 | 0.7 | 0.2 | 0.6 | 0.2 | 0.2 | 1.9 | 1.3 | 0.2 | 2.6 | 0.9 |
| 440 | 6.6 | 3.8 | 2.9 | 1.2 | 1.2 | 1.1 | 0.2 | 2.2 | 0.5 | 1.4 | 2.8 | 2.9 |
| 470 | 14.8 | 7.9 | 6.7 | 2.2 | 2.6 | 2.1 | 1.4 | 3.0 | 0.3 | 1.0 | 2.1 | 3.0 |
| 500 | 42.3 | 16.5 | 10.7 | 3.0 | 3.3 | 1.1 | 1.5 | 1.0 | 0.04 | 0.3 | 0 | 3.6 |
| Pt–Ir/WO2 | ||||||||||||
| 400 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5.2 | 1.2 |
| 440 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1.8 |
| 470 | 8.7 | 4.8 | 4.4 | 2.3 | 1.8 | 1.9 | 0.5 | 2.3 | 0.4 | 1.2 | 0 | 4.4 |
| 500 | 27.0 | 13.0 | 8.7 | 2.0 | 4.2 | 1.7 | 1.7 | 1.6 | 0.4 | 0.7 | 0 | 6.0 |
The reaction products observed on monometallic and bimetallic catalysts were those from cracking C1-C5, ring-opening products (2-methylpentane (2-MP), 3-methylpentane (3-MP), and n-hexane (n-H)) and ring-enlargement products (cyclohexane (Ch) and benzene (Bz)). In contrast, the reaction products on the support in the investigated temperature range were only those of cracking and ring-enlargement reactions (Table 2).
The catalytic activity of the catalysts was evaluated as a function of the total conversion (α), the cracking (SC), the ring-opening (SO) and the ring-enlargements (SE) selectivities. The level of MCP conversion and the selectivities achieved over the support and over both the monometallic and bimetallic catalysts is compared in Table 3.
Methylcyclopentane conversion as a function of temperature reaction and metal.
| Temperature (°C) | α (%) | SC (%) | SRO | SRE | 2-MP/3-MP | 3-MP/n-H |
| WO2 | ||||||
| 400 | 15.7 | 80.3 | 0 | 19.7 | – | – |
| 440 | 56.9 | 93.9 | 0 | 6.1 | – | – |
| 470 | 93.3 | 95.8 | 0 | 4.2 | – | – |
| 500 | 99.4 | 97.5 | 0 | 2.5 | – | – |
| Pt/WO2 | ||||||
| 400 | 0.9 | 0 | 0 | 100 | 0 | 0 |
| 440 | 17.3 | 64.5 | 0 | 35.5 | 0 | 0 |
| 470 | 29.0 | 68.5 | 7.6 | 23.9 | 11.0 | 0.1 |
| 500 | 57.2 | 79.9 | 10.4 | 9.7 | 3.2 | 0.8 |
| Ir/WO2 | ||||||
| 340 | 2.4 | 0 | 73.0 | 27.0 | 5.5 | 4.9 |
| 400 | 10.1 | 32.0 | 34.0 | 34.0 | 1.5 | 7.8 |
| 440 | 26.7 | 63.7 | 15.1 | 21.2 | 4.2 | 0.4 |
| 470 | 47.0 | 80.2 | 9.0 | 10.8 | 11.9 | 0.3 |
| 500 | 83.2 | 94.2 | 1.5 | 4.3 | 25.0 | 0.1 |
| Pt–Ir/WO2 | ||||||
| 400 | 6.4 | 0 | 0 | 100 | 0 | 0 |
| 440 | 6.8 | 0 | 0 | 100 | 0 | 0 |
| 470 | 32.7 | 74.6 | 11.9 | 13.5 | 5.8 | 0.3 |
| 500 | 66.7 | 87.2 | 4.0 | 8.8 | 4.2 | 0.6 |
3.2.2 Catalytic activity of WO2 support
The role of the support is a key factor in understanding the catalytic reactivity of metallic catalysts. Controlling the reactivity of the support under the same conditions used for the metallic catalysts is therefore important.
The support exhibited no activity at low temperatures but became active at 400 °C. At this temperature, the conversion value was 15.7%, and the conversion increased gradually with increasing temperature to 99.4% at 500 °C. The WO2 is only active in the cracking and ring-enlargement reactions in the studied temperature range, which indicates a dual character of the metallic and acidic sites. This behavior is different from that of the MoO2 support, which has the ability to open the ring of MCP [7], whereas the Al2O3 support [14] was inactive in the ring opening of MCP under the same conditions. Table 3 also shows the selectivity values of the cracking and enlargement reactions. The cracking reaction, with a selectivity between 80% (at 400 °C) and 97% (at 500 °C), is predominant. For all the temperatures studied, the formation of methane prevails, which reflects the metallic character of the catalysts. The methane content is 2.3% at 400 °C and increased gradually with increasing temperature to 79.7% at 500 °C. At 400 and 440 °C, all cracking products were formed. The only product that corresponds to a ring-enlargement reaction is benzene, which should be formed on dehydrogenation sites present on the support. As shown in Table 3, the ring-enlargement selectivity decreased with increasing reaction temperature.
3.2.3 Catalytic activity of Pt/WO2
The Pt/WO2 catalyst exhibited no activity below 400 °C; this behavior differs from that of Pt/Al2O3 [14] under the same conditions. On the non-reducible Al2O3 support, the Pt catalysts open the ring of MCP in three positions at approximately 180 °C [14]. At this temperature of 180 °C, the Pt/MoO2 catalyst converts 2.9% of MCP to ring-enlargement products only (SRE = 100%); the ring-enlargement products were Ch and Bz. Notably, under these conditions, the Pt catalyst favored the isomerization and the dehydrogenation reactions. The WO2 itself will contain both acidic and metallic sites, as discussed in section 3.2.2. Compared with the reducible WO2 support, which converts 15.7% of MCP under the same conditions, we observed that the addition of Pt metal did not improve catalytic activity and even diminished it (Table 3). However, the addition of Pt to WO2 changes the distribution of the products and their ratio compared to the products of the WO2 support alone. These observations collectively points toward a metal support interaction by adduct sites, where the acidic and metallic sites cooperate in a mutual interaction instead of behaving as separate sites. The adduct sites, which exhibit both acidic and metallic properties, acted as “collapsed bifunctional sites,” where the metal was considered “electron-deficient” [30]. The “electron deficiency” of noble metals was enhanced on WO2 because of its acidic properties. The enhanced “electron deficiency” of noble metals on acidic supports has been reported previously [31,32]. In our case, the adduct sites present on the supported catalysts took the form [(Mm)(Hx)x+], where m is the number of Pt or Ir atoms in the metallic aggregates, and x is the number of surface protons linked to Pt or Ir [7,33,34]. These sites are presented in Fig. 6. The agostic species is the reaction intermediate, as has been previously observed in other catalytic systems [7,35]. Our results agree with previous studies that invoked the existence of adduct sites and agostic-entity intermediates [7,36]. This mechanism is different from the classical bifunctional mechanism [37–39], where the reaction of MCP proceeds over the two separate catalytic sites. In the collapsed bifunctional route, the reaction occurs over only one adduct site, where the intermediates are not exchanged between sites. In this case, all of the reaction steps can be achieved during a single adsorption of reagent molecules. When the temperature was increased to 440 °C, the ring enlargement selectivity, with benzene as the prevailing product, decreased in favor of cracking selectivity.
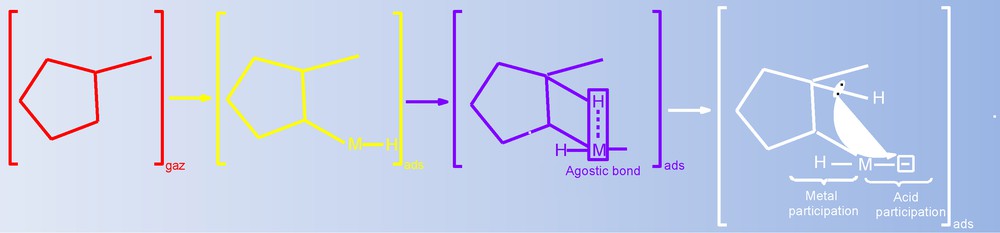
Methylcyclopentane adsorbed on a metal–proton adduct site.
Among the cracking products, the C1 product is dominant. For higher temperatures of 470 and 500 °C, ring-enlargement selectivity decreased in favor of cracking and ring-opening selectivities. The formation of ring-opening products suggests that the fraction of accessible Pt sites increased with temperature. At 500 °C, the catalyst favored the rupture of a single endocyclic CC bond in the secondary–tertiary positions of MCP, with the formation of 2-MP, 3-MP and n-H in a statistical distribution that follows a non-selective mechanism (2-MP/3-MP = 3.2 and 3-MP/n-H = 0.8). These results can be attributed to the relatively high rate of desorption of the formed ring-opening products into the gas phase relative to the desorption rate at lower temperatures. Whatever the temperature of the reaction, the cracking reaction dominates. The dominant cracking products for all temperatures are the C1 product. The cracking selectivity reaches 79.9% at 500 °C. This CC multiple-rupture property of the Pt/WO2 catalyst may be related to reactions that occur on acidic and metallic sites.
3.2.4 Catalytic activity of Ir/WO2
This catalyst was the most reactive toward the conversion of MCP. At 340 °C, it converts 2.4% of MCP into ring-opening (SRO = 73%) and ring-enlargement (SRE = 27%) products. Under the same conditions, the Pt/WO2 and Pt–Ir/WO2 catalysts and the WO2 support were inactive. However, the high activity of Ir relative to that of other metals is well established [7,14,23,40,41]. Based on these results, the addition of metal decreased the required temperature of reaction and improved the catalytic activity. This phenomenon was attributed to the hydrogenative and dehydrogenative properties of Ir as well as to hydrogen spillover. These results demonstrate the high propensity of Ir to promote the ring opening of MCP. The catalyst apparently exposed a large fraction of iridium atoms at the surface. Under these conditions, the high activity and selectivity is due to the metallic function of the catalyst. This metallic function is also responsible for the high hydrogen storage capacity of WO2, which can favor the reverse spillover of supplied hydrogen to the highly dispersed Ir nanoparticles. The MCP ring opening followed the selective mechanism and exhibited an enhanced rate of desorption of 2-MP and 3-MP from the Ir/WO2 catalyst surface into the gas phase. The ring-opening products were formed as primary products from the rupture of secondary C bonds and were formed in the order 2-MP (1.4%) > 3-MP (0.3%) > n-H (0.1%). At the reaction temperatures studied of 340 °C, the cracking reaction products were absent. This behavior, with high activity toward ring-opening products and negligible activity toward cracking, has been previously observed [42] and was attributed to the lower activation energy required for ring opening compared with that required for other reactions. Given the expectation of results comparable to those already reported [43], our results again confirmed that ring opening over Ir catalysts at low temperatures occurs mainly via the selective mechanism with a non-statistical distribution of 2-MP, 3-MP and n-H products (2-MP/3-MP = 5.5 and 3-MP/n-H = 4.9). Under these conditions, the rupture in the vicinity of the methyl group with the formation of n-H was strongly hindered. An increase in the reaction temperature to 400 °C increased the total conversion of MCP, which reached 10.1%, whereas the selectivity to ring opening (So) decreased to 34.0% in favor of cracking products (Sc = 32%). The selectivity to ring enlargement increases slightly to 34%. In this case, we observed that the rate of desorption of the products from the catalyst surface diminished with increasing temperature and that multiple CC ruptures occurred on Ir/WO2 at 400 °C. When the temperature was increased, the total conversion increased, and the maximum conversion (α = 83.2%) was achieved at 500 °C, where the SO decreased to 1.5% in favor of the cracking reaction (SC = 96.5%). In this range of temperatures, the cracking products formed were C1-C5. Among these products, the formation of methane (C1) as the dominant cracking product suggests that demethylation is favored on Ir/WO2.
3.2.5 Catalytic activity of Pt–Ir/WO2
The Pt–Ir/WO2 catalyst demonstrates catalytic activity at temperatures of 400 °C and above, which is identical to the performance of Pt/WO2 and WO2 and different from the performance of an alumina support when the reaction temperatures were significantly lower in the same conditions [14]. This last observation is attributed to the existence of a strong metal support interaction when a reducible acidic support is used for metal dispersion and part of the metallic phase is covered by the WO2. Moreover, at this temperature, the Pt–Ir/WO2 catalyst converted 6.4% of the MCP into the ring-enlargement products with 100% selectivity. The conversion rates are intermediate between those of monometallic catalysts. The primary ring-enlargement products formed on the Pt–Ir/WO2 catalyst were Ch (5.2%) and Bz (1.2%), which suggests the presence of adduct sites. The presence of adduct sites on the metallic acidic supported catalysts has also been reported for the MoO2 support [7]. Only ring-enlargement products were also observed at 440 °C. Under these conditions, the Pt–Ir bimetallic catalyst shows Pt-like character. An increase in the temperature to 470 or 500 °C resulted in a conversion rate intermediate between those observed for Ir and Pt under the same conditions, which apparently provides evidence of the mutual interaction of atoms of the individual two metals. At this high temperature, the cracking products were dominant, followed by modest activities for ring-enlargement and ring-opening reactions. Among the cracking products, the C1 is the major product formed by the multiple rupture of CC bonds of the MCP molecule, and the formation of Bz is favored over the formation of Ch. The formation of cracking products can be explained by the adsorption of the primary product formed onto the surface; this product may undergo repeated CC bond ruptures in the adsorbed state on acidic and metallic sites before desorption, as has been previously reported in the literature [44]. Ring-opening products (2-MP > 3-MP > n-H) were formed by the single rupture of endocyclic CC bonds in a statistical distribution that follows a non-selective mechanism. As previously mentioned, the Pt–Ir/WO2 catalyst exhibits bimetallic behavior with respect to the level of conversion; however, with respect to the nature of the ring opening of MCP via a non-selective mechanism, the Pt–Ir bimetallic catalyst shows monometallic Pt-like character. These results showed that the ring opening of MCP occurs on Pt active sites and that the bimetallic catalyst prepared by co-impregnation contained separate entities of the two metals, as previously shown [6,7,14,45]. Because the small particles with diameters of 1 nm are expected to have (111) fcc crystal planes and a Mackay icosahedral structure the presence of more Pt atoms than the Ir atoms at the surface of the particles cannot be inferred on platinum enrichment catalysts because of geometric thermodynamic arguments concerning topological segregation due to a low metal loading (0.25 wt.%). The presence of more Pt atoms can be explained instead by the spatial distribution of Pt and Ir atoms on the catalysts. More specifically, the edges and corners were populated with Pt atoms.
4 Conclusion
This study addressed the conversion of methylcyclopentane on Pt/WO2, Ir/WO2, Pt–Ir/WO2 in comparison with the WO2 in the temperature range of 180 to 500 °C. The results of this study show the following:
- • with the exception of Ir/WO2, which begins to exhibit catalytic activity at approximately 340 °C, all the catalysts convert the MCP starting at 400 °C. Although both the monometallic and bimetallic catalysts show relatively high activities, the catalysts also show a decrease in activity with respect to their support. Among the metallic catalysts, the Ir/WO2 shows a high activity in the conversion of MCP following the order Ir > Pt–Ir > Pt. For each catalyst, the activity increased with increasing reaction temperature;
- • the activity of the metallic-supported acidic support was lower than that of alumina- and titania-supported catalysts [14,45] under the same conditions. These unexpected results were associated with the strong metal–support interactions;
- • ring opening was the main reaction only on the Ir/WO2 catalysts at 340 °C, whereas at other temperatures and with other catalysts, the cracking reaction was the major reaction. The bimetallic Pt–Ir shows Pt-like behavior in the ring-opening reaction;
- • the Pt and Pt–Ir catalysts open the ring at the secondary–tertiary C bond via the non-selective mechanism, whereas Ir catalyst operates by the selective mechanism and opens the ring in secondary–secondary C bonds of the MCP ring;
- • the Pt/WO2 and Pt–Ir/WO2 show 100% selectivity toward the ring-enlargement reaction at 400 °C, which is attributed to the adduct sites. In this case, the Pt–Ir shows Pt-like character;
- • the low level of selectivity toward the ring-opening reaction is associated with the strong metal–support interactions that occur on the reducible WO2 support;
- • the results show that, under these conditions, the WO2-supported Pt, Ir and Pt–Ir catalysts are inappropriate for generating ring-opening products with atom economy. Compared with results in the literature [7,14,45], the ring-opening selectivity values showed conspicuous changes as a function of the nature of the support.
Acknowledgements
The authors thank the REALISE and the IDECAT excellence network for facilitating this study.