

1 Introduction
Benzimidazole structural motifs are found in numerous pharmaceutical agents with a diverse range of biological properties [1]. Benzimidazoles have commercial applications in various realms of therapy, including antiulcer, antihypertensive, antiviral, antifungal, antitumor, antihelminthic and antihistaminic agents in veterinary medicine [2–11]. The widespread interest in benzimidazole-containing structures has been promoted by extensive studies of their synthesis. While many strategic pathways are available for benzimidazole synthesis, there are two general methods for the synthesis of 2-substituted benzimidazoles. One is the coupling of phenylenediamines and carboxylic acids or their derivatives (see, for example, [11–15]). The other way includes the oxidative cyclo-dehydrogenation of Schiff bases, which are often generated from the condensation of phenylenediamines and aldehydes. Various oxidative and catalytic reagents such as nitrobenzene, 1,4-benzoquinone, sulfamic acid, air, oxone, iodine, DDQ, FeCl3·6H2O, Fe(NO3)3/H2O2, KHSO4, NaHSO3, MnO2, R(OTf)3 where R = In, Yb, Sc, (NH4)2Ce(NO3)6, microwave irradiation and ionic liquids have been involved [16–30].
One of the directions of our research is the silyl and germyl modification of biologically active compounds to improve their biological properties, such as increasing lipophilicity, achieving prolonged action, lowering toxicity and therapeutic dose in comparison with unsubstituted or organyl-substituted substances [31,32].
Based on the above mentioned facts, we decided to synthesize a series of 2-furyl-(thienyl)benzimidazoles with organosilicon and organogermanium substituent in the position 5 of heterocycle and to study their antitumor properties.
2 Experimental
2.1 Materials and methods
The 1H, 13C and 29Si NMR spectra were recorded on a Varian Mercury-400 instrument at 400, 100 and 80 MHz, respectively, in CDCl3 as a solvent, with (Me3Si)2O as a standard for 1H, TMS (external) as the standard for 29Si, and the signal on the residual proton of the solvent (δ 77.05 ppm) for 13C. The mass spectra under electron impact conditions were recorded on a GC-MS Agilent Technologies 7890 GC system with 5975 C EI/CI MSD (70 eV) on an HP-5 capillary column. High-resolution mass spectra were recorded on Q-TOF micro (micromass) electrospray ionizacion (ESL+). All solvents were dried on CaH2, metallic sodium and distilled prior to use. Thin-layer chromatography (TLC) was performed on a Merck silica gel 60 F254 with various eluents. Column chromatography was performed on silica gel (0.060–0.200 mm, pore diameter 6 nm, Acros), with different eluents. All starting aldehydes were prepared by the known methods [33–35].
2.2 Cytotoxicity in vitro
Monolayer tumour cell lines MG-22A (mouse hepatoma), HT-1080 (human fibrosarcoma) and NIH 3T3 (normal mouse fibroblasts) were cultivated for 72 h in standard Dulbecco's modified Eagle's medium (Sigma) without indicator and antibiotics [36]. After the ampoule was thawed not more than four passages were performed. The control cells and cells with tested substances in the range of 2–5 × 10–4 cell ml−1 concentration (depending on the nature of cell line) were placed into separate 96-well plates. Solutions containing test compounds were diluted and added to wells to give the final concentrations of 50, 25, 12.5 and 6.25 μg ml−1. The control cells were treated in the same manner only in the absence of test compounds. Plates were cultivated for 72 h. A quantity of survived cells was determined using crystal violet (CV), neutral red (NR) or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) coloration, which was assayed using a multiscan spectrophotometer (Tetretek Multiscan MCC/340). The quantity of living cells on the control plate was taken in calculations for 100% [36,37]. The concentration of NO was determined according to Fast et al. [37]. The mean lethal dose (LD50) was determined on 3T3 cells (alternative to LD50 in vivo test) according to the standard protocols [36].
2.3 Chemistry
2.3.1 General synthetic procedure for synthesis of benzimidazoles
The 2-furyl- or 2-thienylaldehyde with the same silicon or germanium substitution pattern of the desired final product (0.78 mmol) and o-phenylenediamine (0.78 mmol) was thoroughly mixed in DMF (4 ml), then 0.29 mmol NaHSO3 was added and heated at 80 °C for 2 to 3 h. The reactions were followed by thin-layer chromatography (TLC) and GC-MS. When reaction was finished, the solution was cooled to room temperature, and 35 ml of H2O drop wise was added with vigorous stirring. The oil was extracted into EtOAc, the organic phase was washed with H2O, brine and dried Na2SO4. Evaporation of solvent gave the crude product, which was diluted in Et2O and passed through Al2O3. After evaporation of ether, the corresponding benzimidazole was crystallized from hexane or ethanol, or purified by column chromatography. These experimental conditions led to good or moderate yields. The final products were identified by NMR (1H, 13C and 29Si), GC-MS, elemental analysis and HRMS spectra. In the present study, all new silicon (germanium) containing benzimidazoles were systematically characterized; the unsubstituted furyl- (1), and thienyl- (5) benzimidazoles have been previously reported [22,30].
2.3.1.1 2-(5-Trimethylsilyl-2-furyl)benzimidazole (2)
Compound 2 was prepared from 5-trimethylsilyl-2-furfural and o-phenylenediamine under heating for 3 h. Yield: 49% (after purification by Al2O3 and column chromatography, hexane as eluent). M.p. 228–230 °C. Anal. Calcd. for C14H16N2OSi: C, 65.59; H, 6.29; N, 10.93. Found: C, 65.78; H, 6.36; N, 11.04. 1H NMR δ ppm: 0.30 (s, 9H, SiMe3), 6.70 (s, 1H, NH), 6.75 (m, 1H, H4, J3.4 = 3.2 Hz), 7.21 (d, 1H, H3, J3.4 = 3.2 Hz), 7.26–7.28 (m, 2H, Ar), 7.60–7.65 (m, 2H, Ar). 13C NMR δ ppm: –1.89, 111.05, 115.12, 121.89, 122.93, 138.59, 144.39, 149.03, 163.11. 29Si NMR δ ppm: –10.05. GC-MS, m/z (%): 256 (M+, 72), 241 (M+ - Me, 100), 211 (M+ - 3Me, 32).
2.3.1.2 2-(5-Trimethylgermyl-2-furyl)benzimidazole (3)
Compound 3 was prepared from 5-trimethylgermyl-2-furfural and o-phenylenediamine under heating for 2.5 h. Yield: 47.9% (after crystallization from EtOH/activated carbon). M.p. 173–175 °C. Anal. Calcd. for C14H16N2GeO: C, 55.89; H, 5.36; N, 9.31. Found: C, 55.86; H, 5.34; N, 9.48. 1H NMR δ ppm: 0.37 (s, 9H, GeMe3), 6.50 (bs, 1H, NH), 6.62 (d, 1H, H4, J = 3.2 Hz), 7.20 (d, 1H, H3, J = 3.2 Hz), 7.23–7.25 (m, 2H, Ar), 7.60–7.62 (m, 2H, Ar). 13C NMR δ ppm: –2.00, 111.18, 115.05, 120.30, 122.93, 138.48, 144.37, 148.69, 164.34. GC-MS, m/z (%): 302 (M+, 65), 287 (M+ - Me, 100), 257 (M+ - 3Me, 47).
2.3.1.3 2-(5-Triethylgermyl-2-furyl)benzimidazole (4)
Compound 4 was prepared from 5-triethylgermyl-2-furfural and o-phenylenediamine under heating for 2 h. Yield: 46% (after crystallization from EtOH/activated carbon). M.p. 200–203 °C. Anal. Calcd. for C17H22N2GeO: C, 59.54; H, 6.47; N, 8.17. Found: C, 59.39; H, 6.35; N, 8.00. 1H NMR (DMSO), δ ppm: 1.01–1.12 (m, 15H, GeEt3), 6.84 (d, 1H, H4, J3.4 = 3.9 Hz), 7.16–7.30 (m, 1H, H3, J3.4 = 3.9 Hz, 2H, Ar), 7.50–7.52 (m, 1H, Ar), 7.62–7.64 (m, 1H, Ar), 12.73 (m, 1H, NH). 13C NMR δ ppm: 4.24, 8.72, 110.98, 115.04, 121.39, 122.70, 138.76, 144.68, 149.125, 162.50. GC-MS, m/z (%): 344 (M+, 16), 315 (M+ - Et, 42), 287 (16), 257 (M+ - 3Et, 31).
2.3.1.4 2-(5-Trimethylsilyl-2-thienyl)benzimidazole (6)
Compound 6 was prepared from 5-trimethylsilyl-2-thiophenecarbaldehyde and o-phenylenediamine under heating for 2.5 h. Yield: 44.5% (after purification by column chromatography, hexane: EtOAc 3:1, and crystallization EtOH/activated carbon). M.p. 187–189 °C. Anal. Calcd. for C14H17N2SSi: C, 61.49; H, 5.90; N, 10.24. Found: C, 61.33; H, 5.89; N, 10.18. 1H NMR (DMSO), δ ppm: 0.34 (s, 9H, SiMe3), 7.17–7.21 (m, 2H, Ar), 7.39 (d, 1H, H4, J3.4 = 3.6 Hz), 7.52–7.58 (m, 2H, Ar), 7.87 (d, 1H, H3, J3.4 = 3.6 Hz), 12.93 (bs, 1H, NH). 13C NMR δ ppm: –0.005, 115.27, 123.21, 128.24, 134.98, 137.91, 139.161, 144.57, 147.77. 29Si NMR δ ppm: –5.86. GC-MS, m/z (%): 272 (M+, 60), 257 (M+ - Me, 100), 227 (M+ - 3Me, 21).
2.3.1.5 2-(5-Trimethylgermyl-2-thienyl)benzimidazole (7)
Compound 7 was prepared from 5-trimethylgermyl-2-thiophenecarbaldehyde and o-phenylenediamine under heating for 2 h. Yield: 43.2% (after purification by column chromatography, hexane: EtOAc 5:1). M.p. 200–202 °C. Anal. Calcd. for C14H16N2GeS: C, 53.05; H, 5.09; N, 8.84. Found: C, 52.97; H, 5.20; N, 8.42. 1H NMR (DMSO), δ ppm: 0.48 (s, 9H, GeMe3), 7.15–7.22 (m, 2H, Ar), 7.31 (d, 1H, H4, J3,4 = 3.6 Hz), 7.48–7.60 (m, 2H, Ar), 7.87 (d, 1H, H3, J3,4 = 3.6 Hz), 12.87 (s, 1H, NH). 13C NMR δ ppm: –0.54, 122.94, 127.27, 133.48, 137.05, 145.69, 147.19. GC-MS, m/z (%): 318 (M+, 39), 303 (M+ - Me, 100), 273 (M+ - 3Me, 23).
2.3.2 General synthetic procedure for synthesis of N-substituted benzimidazoles
The targets compounds 8–22 were synthesized by heating (80 °C) of corresponding benzimidazole 2–7 (0.4 mmol) with propyl-, allyl- or propargylbromide (0.4 mmol) in two-phase benzene-potassium hydroxide (1.2 mmol) system and catalytic amounts of 18-crown-6 (18-C-6). The reactions were monitored by TLC. After 4 to 12 h, the reaction mixture was cooled and filtered. Evaporation of solvent gave the brown oil, which was diluted in Et2O and passed through Al2O3. After crystallization or purification by column chromatography and removal of the eluent, the new compound 8–22 was obtained. The final products were identified by NMR (1H, 13C and 29Si), GC-MS, elemental analysis and HRMS spectra.
2.3.2.1 2-(5-Trimethylsilyl-2-furyl)-N-propylbenzimidazole (8)
Compound 8 was separated by crystallization from hexane after 8 h heating at 80 °C of starting substances. Yield: 47.2%. M.p. 95–97 °C. Anal. Calcd. for C17H22N2OSi: C, 68.41; H, 7.43; N, 9.39. Found: C, 68.36; H, 7.33; N, 9.31. 1H NMR δ ppm: 0.34 (s, 9H, SiMe3), 0.99–1.03 (t, 3H, Me), 1.90–1.99 (m, 2H, CH2), 4.48 (t, 2H, CH2N, J = 7.4 Hz), 6.78 (d, 1H, H4, J3,4 = 3.4 Hz), 7.22 (d, 1H, H3, J3,4 = 3.4 Hz), 7.26–7.30 (m, 2H, Ar), 7.36–7.39 (m, 1H, Ar), 7.76–7.79 (m, 1H, Ar). 13C NMR, δ ppm: –1.69, 11.37, 23.67, 46.71, 109.54, 112.69, 119.68, 121.54, 122.46, 122.69, 135.69, 143.15, 144.36, 149.89, 162.53. 29Si NMR δ ppm: –10.00. GC-MS, m/z (%): 298 (M+, 100), 283 (M+ - Me, 100), 269 (M+ - Et, 24).
2.3.2.2 2-(5-Trimethylsilyl-2-furyl)-N-allylbenzimidazole (9)
Compound 9 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yield: 48.3%. M.p. 92–94 °C. Anal. Calcd. for C17H20N2OSi: C, 68.80; H, 6.80; N, 9.45. Found: C, 68.98; H, 7.00; N, 9.22. 1H NMR δ ppm: 0.32 (s, 9H, SiMe3), 5.06–5.12 (m, 3H, CH2), 5.20–5.22 (m, 1H, CH2), 6.03–6.12 (m, 1H, CH), 6.77 (d, 1H, H4, J3.4 = 3.6 Hz), 7.18 (d, 1H, H3, J3.4 = 3.6 Hz), 7.26–7.29 (m, 2H, Ar), 7.33–7.36 (m, 1H, Ar), 7.78–7.80 (m, 1H, Ar). 13C NMR δ ppm: –1.71, 47.31, 109.68, 113.03, 117.28, 119.64, 121.48, 122.85, 123.06, 132.37, 135.35, 144.28, 148.96, 163.03. 29Si NMR δ ppm: –9.89. GC-MS, m/z (%): 296 (M+, 49), 281 (M+ - Me, 27).
2.3.2.3 2-(5-Trimethylsilyl-2-furyl)-N-propargylbenzimidazole (10)
Compound 10 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yield: 45%. M.p. 118-120 °C. Anal. Calcd. for C17H18N2OSi: C, 69.35; H, 6.16; N, 9.51. Found: C, 69.25; H, 6.14; N, 9.33. 1H NMR δ ppm: 0.35 (s, 9H, SiMe3), 2.31–2.33 (m, 1H, CH,), 5.26–5.28 (m, 2H, CH2), 6.80 (d, 1H, H4, d, J3.4 = 3.2 Hz), 7.25 (d,1H, H3, J3.4 = 3.2 Hz), 7.28–7.34 (m, 2H, Ar), 7.46–7.48 (m,1H, Ar), 7.77–7.80 (m, 1H, Ar). 13C NMR, δ ppm: –1.75, 34.57, 73.06, 77.38, 109.49, 113.19, 119.77, 121.47, 123.11, 123.32, 134.87, 142.74, 143.79, 148.70, 163.37. 29Si NMR δ ppm: –9.68. GC-MS, m/z (%): 294 (M+, 100), 279 (M+ - Me, 16), 264 (M+ - 2Me, 21).
2.3.2.4 2-(5-Trimethylgermyl-2-furyl)-N-propylbenzimidazole (11)
Compound 11 was separated by crystallization from hexane after 8 h heating at 80 °C of starting substances. Yield: 42.4%. M.p. 82–84 °C. Anal. Calcd. for C17H22N2GeO: C, 59.54; H, 6.47; N, 8.17. Found: C, 59.63; H, 6.43; N, 7.98. 1H NMR δ ppm: 0.21 (s, 9H, GeMe3), 0.74 (t, 3H, Me, J = 7.6 Hz), 1.63–1.72 (m, 2H, CH2), 4.20 (t, 2H, CH2N, J = 7.4 Hz), 6.44 (d, 1H, H4 J3.4 = 3.4 Hz), 6.95 (1H, d, H3, J3,4 = 3.4 Hz), 6.99–7.01 (m, 2H, Ar), 7.10–7,12 (m,1H, Ar), 7.49–7.51 (m, 1H, Ar). 13C NMR, δ ppm: –1.91, 11.35, 23.62, 46.66, 109.50, 112.95, 119.54, 119.92, 122.49, 122.66, 135.56, 142.88, 144.30, 149.33, 163.73. GC-MS, m/z (%): 344 (M+, 75), 329 (M+ - Me, 100).
2.3.2.5 2-(5-Trimethylgermyl-2-furyl)-N-allylbenzimidazole (12)
Compound 12 was separated by column chromatography (hexane: EtOAc 5:1) after 12 h heating at 80 °C of starting substances. Yield: 37.5%. M.p. 98–100 °C. Anal. Calcd. for C17H20N2GeO: C, 59.89; H, 5.91; N, 8.22. Found: C, 60.06; H, 5.90; N, 8.06. 1H NMR δ ppm: 0.47 (s, 9H, GeMe3), 5.00–5.11 (m, 3H, CH2), 5.20–5.22 (m, 1H, CH2), 6.02–6.11 (m,1H, CH), 6.68 (d, 1H, H4, J3.4 = 3.4 Hz), 7.16 (d, 1H, H3, J3.4 = 3.4 Hz), 7.25–7.28 (m, 2H, Ar), 7.33–7.35 (m, 1H, Ar), 7.78–7.80 (m, 1H, Ar). 13C NMR, δ ppm: –1.87, 47.25, 109.62, 112.77, 117.20, 119.74, 122.63, 122.85, 132.48, 135.51, 143.17, 144.90, 148.98, 164.01. GC-MS, m/z (%): 342 (M+, 83), 327 (M+ - Me, 9).
2.3.2.6 2-(5-Trimethylgermyl-2-furyl)-N-propargylbenzimidazole (13)
Compound 13 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yields: 43.5%. M.p. 77–79 °C. Anal. Calcd. for C17H18N2GeO: C, 60.24; H, 5.35; N, 8.27. Found: C, 60.40; H, 5.33; N, 8.42. 1H NMR δ ppm: 0.45 (s, 9H, GeMe3), 2.26–2.28 (m, 1H, CH), 5.19–5.20 (m, 2H, CH2), 6.66 (d, 1H, H4, J3.4 = 3.6 Hz), 7.20 (m, 1H, H3, 3H, Ar), 7.22–7.28 (d, 1H, m, 2H, Ar), 7.39–7.43 (1H, m, Ar), 7.72–7.74 (1H, m, Ar). 13C NMR, δ ppm: –1.92, 34.51, 73.01, 109.43, 113.09, 119.85, 122.95, 123.14, 134.95, 143.03, 143.95, 148.58, 164.44. GC-MS, m/z (%): 340 (M+, 100), 295 (M+ - 3Me, 29).
2.3.2.7 2-(5-Triethylgermyl-2-furyl)-N-propylbenzimidazole (14)
Compound 14 was separated by column chromatography (hexane: EtOAc 5:1) after 10 h heating at 80 °C of starting substances. Yield: 47%. HRMS Calcd for: C20H29N2OGe (M+, 100) 387.1492. Found 387.1526. 1H NMR δ ppm: 0.97–1.14 (m, 15H, GeEt3, 3H, CH3), 1.87–1.96 (m, 2H, CH2), 4.48 (t, 2H, CH2N, J = 7.4 Hz), 6.70 (d, 1H, H4, J3.4 = 3.2 Hz), 7.25–7.27 (m, 1H, H3, 2H, Ar), 7.35–7.38 (m, 1H, Ar), 7.74–7.77 (m, 1H, Ar). 13C NMR, δ ppm: 7.75, 11.31, 23.65, 46.99, 110.88, 122.30, 123.89, 126.38, 134.98, 136.27, 141.99, 146.10, 168.03. GC-MS, m/z (%): 386 (M+, 35), 357 (M+ - Et, 100).
2.3.2.8 2-(5-Triethylgermyl-2-furyl)-N-allylbenzimidazole (15)
Compound 15 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yield: 41.4%. M.p. 78–80 °C. Anal. Calcd. for C20H26N2GeO: C, 62.72; H, 6.84; N, 7.31. Found: C, 62.63; H, 6.83; N, 7.18. 1H NMR δ ppm: 1.00–1.20 (m, 15H, GeEt3), 5.03–5.21 (m, 4H, CH2), 6.01–6.10 (m, 1H, CH), 6.70 (d, 1H, H4, J3.4 = 3.6 Hz), 7.22 (d, 1H, H3, J3.4 = 3.6 Hz), 7.27–7.39 (m, 3H, Ar), 7.77–7.81 (m, 1H, Ar). 13C NMR, δ ppm: 4.49, 8.90, 47.24, 109.66, 112.86, 117.03, 119.71, 121.14, 122.62, 122.80, 132.55, 135.54, 143.24, 144.63, 149.42, 162.29. GC - MS, m/z (%): 384 (M+, 100), 355 (M+ - Et, 56).
2.3.2.9 2-(5-Triethylgermyl-2-furyl)-N-propargylbenzimidazole (16)
Compound 16 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yields: 43.5%. M.p. 68–70 °C. Anal. Calcd. for C20H24N2GeO: C, 63.05; H, 6.35; N, 7.35. Found: C, 63.00; H, 6.34; N, 7.28. 1H NMR δ ppm: 1.04–1.16 (15H, m, GeEt3), 2.30–2.31 (t, 1H, CH), 5.29 (d, 2H, CH2), 6.72 (d, 1H, H4 J3.4 = 3.2 Hz), 7.28–7.43 (m, 1H, H3, 2H, Ar), 7.46–7.50 (m, 1H, Ar), 7.76–7.79 (m, 1H, Ar). 13C NMR, δ ppm: 4.48, 8.91, 34.51, 73.02, 77.43, 109.48, 113.18, 119.78, 121.18, 122.97, 123.11, 134.97, 143.07, 143.98, 148.99, 162.78. GC-MS, m/z (%): 382 (M+, 100), 353 (M+- Et, 6).
2.3.2.10 2-(5-Trimethylsilyl-2-thienyl)-N-propylbenzimidazole (17)
Compound 17 was separated by column chromatography (hexane: EtOAc 5:1) after 10 h heating at 80 °C of starting substances. Yield: 46%. M.p. 67–69 °C. Anal. Calcd. for C17H22N2SSi: C, 64.92; H, 7.05; N, 8.91. Found: C, 64.84; H, 6.88; N, 8.72. 1H NMR δ ppm: 0.33 (s, 9H, SiMe3), 0.99 (t, 3H, Me, J = 7.6 Hz), 1.86–1.96 (m, 2H, CH2), 4.30 (t, 2H, CH2N, J = 7.6 Hz), 7.22–7.26 (m, 3H, H4, Ar), 7.31–7.34 (m, 1H, Ar), 7.54 (d, 1H, H3, J3.4 = 3.6 Hz), 7.74–7.77 (m, 1H, Ar). 13C NMR, δ ppm: 3.0, 11.20, 23.97, 46.17, 109.65, 119.59, 122.39, 122.72, 127.51, 127.70, 128.28, 132.39, 135.89, 142.76, 147.30. 29Si NMR δ ppm: –5.85. GC- MS, m/z (%): 314 (M+, 100), 299 (M+ - Me, 93).
2.3.2.11 2-(5-Trimethylsilyl-2-thienyl)-N-allylbenzimidazole (18)
Compound 18 was separated by column chromatography (hexane: EtOAc 5:1) after 10 h heating at 80 °C of starting substances. Yield: 43.3%. M.p. 96–98 °C. Anal. Calcd. for C17H20N2SSi: C, 65.34; H, 6.45; N, 8.36. Found: C, 65.18; H, 6.47; N, 8.45. 1H NMR δ ppm: 0.35 (s, 9H, SiMe3), 4.97–4.98 (m, 2H, CH2), 5.03–5.31 (m, 2H, CH2), 6.06–6.15 (m, 1H, CH), 7.25–7.33 (m, 1H, H4, 3H, Ar), 7.53 (d, 1H, H3, J3.4 = 3.6 Hz), 7.80–7.82 (m, 1H, Ar). 13C NMR, δ ppm: –0.25, 46.81, 109.78, 117.41, 119.73, 122.66, 122.94, 128.58, 134.39, 135.96, 136.74, 143.00, 144.63, 147.83. 29Si NMR δ ppm: –5.97. GC-MS, m/z (%): 312 (M+, 83), 297 (M+ - Me, 52).
2.3.2.12 2-(5-Trimethylsilyl-2-thienyl)-N-propargylbenzimidazole (19)
Compound 19 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yields: 42.6%. M.p. 100–102 °C. Anal. Calcd. for C17H18N2SSi: C, 65.76; H, 5.84; N, 9.02. Found: C, 65.59; H, 5.82; N, 8.96. 1H NMR δ ppm: 1H NMR δ, ppm: 0.37 (s, 9H, SiMe3), 2.44 (s, 1H, CH), 5.07 (s, 2H, CH2), 7.20–7.38 (m, 1H, H4, 2H, Ar), 7.47–7.49 (m, 1H, Ar), 7.72 (d, 1H, H3, J3.4 = 3.2 Hz), 7.79–8.82 (m, 1H, Ar). 13C NMR δ ppm: –0.23, 34.43, 73.92, 77.18, 109.54, 119.95, 122.98, 123.26, 129.02, 135.39, 136.34, 142.95, 145.11, 147.31. NMR δ ppm 29Si NMR δ ppm: –5.85. GC-MS, m/z (%): 310 (M+, 100), 295 (M+ - Me, 42), 265 (M+ - 3Me, 12).
2.3.2.13 2-(5-Trimethylgermyl-2-thienyl)-N-propylbenzimidazole (20)
Compound 20 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yield: 44.2%. M.p. 58–60 °C. Anal. Calcd. for C17H22N2GeS: C, 56.87; H, 6.18; N, 7.80. Found: C, 56.93; H, 6.33; N, 7.57. 1H NMR δ ppm: 0.50 (s, 9H, SiMe3), 1.01 (t, 3H, CH3, J = 7.6 Hz), 1.89–1.98 (m, 2H, CH2), 4.32 (t, 2H, CH2N, J = 7.6 Hz), 7.21 (d, 1H, H4, J3.4 = 3.2 Hz), 7.27–7.29 (m, 2H, Ar), 7.34–7.40 (m, 1H, Ar), 7.56 (d, 1H, H3, J3.4 = 3.2 Hz), 7.77–7.80 (m, 1H, Ar). 13C NMR, δ ppm: –0.52, 11.33, 23.29, 46.23, 109.64, 119.72, 122.40, 122.64, 128.21, 133.22, 136.71, 143.04, 145.55, 147.63. GC-MS, m/z (%): 360 (M+, 51), 345 (M+ - Me, 100), 315 (M+ - 3Me, 5).
2.3.2.14 2-(5-Trimethylgermyl-2-thienyl)-N-allylbenzimidazole (21)
Compound 21 was separated by column chromatography (hexane: EtOAc 5:1) after 4 h heating at 80 °C of starting substances. Yield: 41.6%. M.p. 98–99 °C. Anal. Calcd. for C17H20N2GeS: C, 57.19; H, 5.65; N, 7.85. Found: C, 57.30; H, 5.50; N, 7.69. 1H NMR δ ppm: 0.50 (s, 9H, GeMe3), 4.97–4.99 (m, 2H, CH2), 5.03–5.31 (m, 2H, CH2), 6.06–6.15 (m, 1H, CH), 7.19–7.20 (d, 1H, H4, J3.4 = 3.6 Hz), 7.26–7.33 (m, 3H, Ar), 7.53–7.54 (d, 1H, H3, J3.4 = 3.6 Hz), 7.80–7.82 (m, 1H, Ar). 13C NMR, δ ppm: –0.52, 46.88, 109.78, 117.46, 119.79, 122.65, 122.90, 128.45, 131.98, 133.24, 136.03, 136.23, 143.13, 145.97, 148.00. GC-MS, m/z (%): 358 (M+, 60), 343 (M+ - Me, 100).
2.3.2.15 2-(5-Trimethylgermyl-2-thienyl)-N-propargylbenzimidazole (22)
Compound 22 was separated by column chromatography (hexane: EtOAc 5:1) after 8 h heating at 80 °C of starting substances. Yields: 42.4%. HRMS Calcd for: C17H19N2SGe (M+, 100) 357.0447. Found 357.0481. 1H NMR δ, ppm: 0.51 (s, 9H, GeMe3), 2.43–2.44 (t, 1H, CH), 5.06 (s, 2H, CH2), 7.24–7.25 (m, 1H, H4, J3.4 = 3.2 Hz), 7.30–7.33 (m, 2H, Ar), 7.46–7.48 (m, 2H, Ar), 7.72–7.73 (d, 1H, H3, J3.4 = 3.2 Hz). 13C NMR, δ ppm: –0.54, 34.44, 73.91, 77.21, 109.53, 119.91, 122.95, 123.19, 128.91, 129.73, 133.40, 135.91, 142.96, 146.43. GC-MS, m/z (%): 356 (M+, 100), 341 (M+ - Me, 67), 311 (M+ - 3Me, 52).
3 Results and discussion
Many synthetic methods for the synthesis of benzimidazoles are now available [11–30]. To achieve our final purpose, we planned a two-step strategy:
- • select the preferable reaction conditions to synthesize benzimidazole skeleton and;
- • modify and optimize the reaction conditions to fit our needs — reaction proceeds with retention of the silyl or germyl group.
Because of its simple operation and mild reaction conditions, we have chosen the reaction between o-phenylenediamine and silicon or germanium containing 2-furaldehydes or 2-thienyl-carbaldehydes in the presence of sodium hydrogen sulfite as a basic strategy. As a result, a new series of silyl(germyl)hetaryl substituted benzimidazoles (1–7) have been synthesized and studied for their in vitro cytotoxicity (Scheme 1).
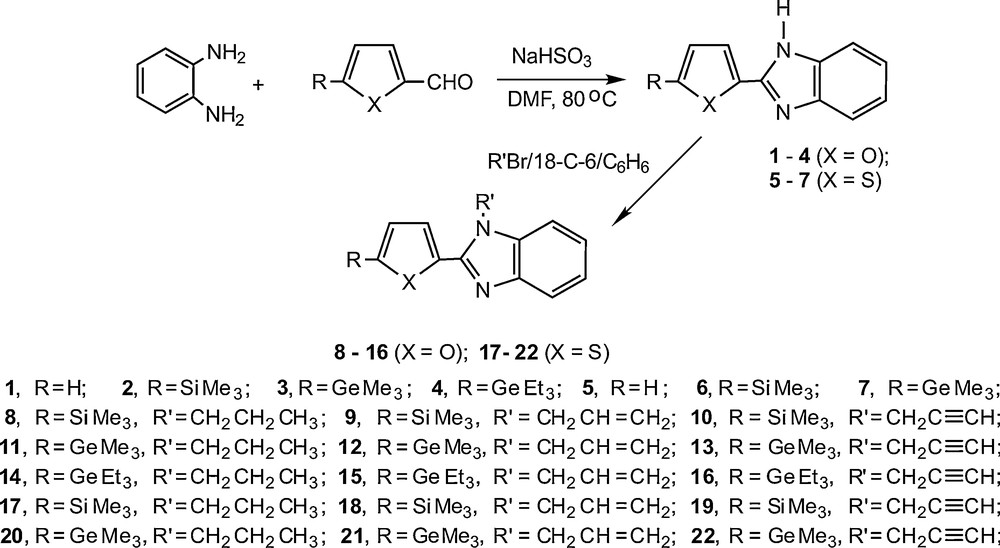
Synthesis of trialkylsilyl(germyl)hetarylsubstituted benzimidazoles and their benzimidazolinium salts.
Among the possible methods for synthesis of N-alkylsubstituted benzimidazoles phase-transfer catalyzed N-alkylation is one of the simplest and most convenient routes [38]. The alkylation of benzimidazoles by propyl-, allyl- or propargylbromide in a two-phase benzene-potassium hydroxide system in the presence of 18-crown-6 (18-C-6) used as phase-transfer catalyst (the molar ratio of benzimidazole:alkylating agent:18-C-6 is 1:1:0.03) affords the corresponding N-alkyl substituted benzimidazoles (8-22) in moderate yields (Scheme 1).
The most prominent benzimidazole in nature is N-ribosyldimethylbenzimidazole, which serves as an axial ligand for cobalt in vitamin B12. An antihistamine astemizole, proton pump inhibitors omeprazole, pantoprazole, lansoprazole, antihelmenthic mebendazole are some of the candidate drugs already in use [39]. It has been shown that N-alkylsubstituted benzimidazole derivatives, especially with silicon atom in the chain, increase the cytotoxic activity of benzimidazolium salts significantly in mouse melanoma B16 (IC50 0.001–0.008 μg ml−1) [40]. From the spectrum of activities reported so far in the literature, the 2-substitution of benzimidazole nucleus or N-substitution appears to influence activity and potency. On the other hand, heterocyclic compounds have occupied a prominent place among various classes of organic compounds by virtue of their diverse biological activities [41]. With no reports of silyl(germyl)furan or – thiophene substitution at position 2 in the benzimidazole system, our research has been concentrated to evaluate the bioefficiency of novel benzimidazoles and their salts incorporating silyl(germyl)furan or – thiophene rings at 2 position. The compounds designed by incorporating them are expected to provide improved biological activity.
The cytotoxicity of benzimidazoles derivatives 1–22 (in vitro) has been investigated on the following tumour cells: HT-1080 (human fibrosarcoma), MG-22A (mouse hepatoma) and normal mouse fibroblasts 3T3 to determine the effect of type of heterocycle, the substituent in the position 5 in heterocyle and N-substitution in benzimidazole on the antitumour activity. The experimental evaluation of cytotoxic properties is presented in Tables 1 and 2. A preliminary analysis of structure–activity relationship for the cytotoxic action clearly indicates the strong influence of the silicon and germanium substituent in heterocycle on cytotoxic effect in vitro (compounds 2–4, 6, 7). Precursors of these compounds (furan derivative 1 and thiophene derivative 5) were not cytotoxic to cancer cells studied (Table 1). The benzimidazoles 2, 4 showed high cytotoxic activity in cancer cells accompanied by high cytotoxic activity on normal cells 3T3 (IC50 3–5 μg ml−1). Compounds 3, 7 were less cytotoxic for normal cells, but even toxic. It means that the therapeutic index for these compounds is low.
Cytotoxicity (IC50 μg ml−1)a of benzimidazoles.
| Cell line | Method | Compound | ||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| HT-1080 | CV | 100 | 3 | 3 | 1 | 33 | 1 | 2 |
| MTT | 90 | 3 | 3 | 2 | 26 | 1 | 3 | |
| NO· | 4 | 300 | 150 | 100 | 43 | 200 | 200 | |
| MG-22A | CV | 32 | 2 | 1 | 1 | 8 | 2 | 2 |
| MTT | 29 | 3 | 2 | 1 | 10 | 3 | 3 | |
| NO· | 20 | 200 | 150 | 100 | 167 | 150 | 50 | |
| NIH 3T3 | NR | 213 | 5 | 10 | 3 | 100 | 5 | 13 |
| NIH 3T3 | LD50 (mg kg−1) | 829 | 195 | 286 | 171 | 621 | 191 | 317 |
a IC50 (mg mL-1) providing 50% cell killing effect (CV-crystal violet coloration, action on cell membranes; MTT–3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide coloration, influence on the activity of mitochondrial enzymes)
Cytotoxicity (IC50 μg ml−1) of N-substituted benzimidazoles.
| Cell line | Method | Compound | ||||||||||||||
| 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | ||
| HT- 1080 | CV | 1 | 3 | 2 | 3 | 1 | 3 | 3 | 1 | 1 | 7 | 3 | 3 | 2 | 1 | 2 |
| MTT | 1 | 3 | 1 | 10 | 1 | 3 | 3 | 1 | 1 | 4 | 3 | 3 | 2 | 1 | 2 | |
| NO· | 167 | 150 | 17 | 100 | 150 | 150 | 150 | 150 | 100 | 100 | 100 | 200 | 150 | 33 | 200 | |
| MG- 22A | CV | 4 | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 3 | 3 | 1 | 1 | 1 |
| MTT | 3 | 1 | 2 | 2 | 1 | 2 | 2 | 1 | 1 | 2 | 3 | 3 | 1 | 2 | 2 | |
| NO· | 200 | 150 | 28 | 150 | 150 | 150 | 200 | 150 | 150 | 150 | 100 | 150 | 150 | 67 | 200 | |
| NIH 3T3 | NR | 4 | 30 | 74 | 12 | 10 | 13 | 8 | 7 | 8 | 12 | 7 | 5 | 12 | 30 | 11 |
| NIH 3T3 | LD50 (mg kg–1) | 191 | 459 | 676 | 343 | 307 | 339 | 300 | 299 | 358 | 314 | 250 | 228 | 345 | 500 | 330 |
N-alkylation of new benzimidazoles by various alkyl, allyl, propargyl halides as a rule increases cytotoxic effect and leads for more active compounds (Table 2). The 2-(5-trimethylsilyl-2-furyl)-N-allylbenzimidazole (9), 2-(5-trimethylsilyl-2-furyl)-N-propargylbenz-imidazole (10) and 2-(5-trimethylgermyl-2-thienyl)-N-allylbenzimidazole (21) are the most promising compounds in this series of compounds: low toxicity (LD50 459–676 mg kg−1), high cytotoxicity on both cancer cell lines (IC50 1–4 μg ml−1) and lower cytotoxicity on normal fibroblasts (IC50 30–74 μg ml−1). All studied compounds exhibited moderate or low NO· generation ability. Inspection of the results presented in Tables 1 and 2 show that the generation of NO· by compounds does not correlate with their cytotoxicity.
Thus, introduction of silyl or germyl group at the heterocycle of heterylbenzimidazoles as well as N-alkylation of silyl(germyl)heterylbenzimidazoles significantly improves cytotoxicity against cancer cells and the cytoselectivity of new benzimidazoles.
Acknowledgements
This work has been carried out with the support of the Latvian Council for Science (projects 09.1556, 10.0030) and the European Social Fund “Support for Doctoral Studies at the University of Latvia”.
The authors thank Dr. S. Grinberga for recording the high-resolution mass spectra.
Vous devez vous connecter pour continuer.
S'authentifier