

1 Introduction
Development of an efficient and practical catalyst for organic transformation to synthesize valuable target compounds is one of the most important research areas in academia and industry [1,2]. Although homogeneous catalysts are finding enormous applications in diverse aspects of synthetic chemistry [3] in recent years, toxicity associated with such catalysts and their disposal and separation from the product is limiting their applications. Therefore, resin-bound catalysts are gaining in importance as surrogate catalysts in organic synthesis in recent years [4–14]. Use of resin-bound catalysts are marked by recyclability, ease of separation from the reaction mixture, higher safety features for potentially explosive reagents, and usage in continuous flow processes which are usually not displayed by their homogeneous counterparts.
The pyran ring is an important structural component of many biologically active natural compounds such as alkaloids, tocopherols, flavonoids, and anthocyanins [15–19]. Recently, functionalized pyrans have found many applications in the field of synthetic and medicinal chemistry [20–22]. Among the functionalized pyrans, 2-aminochromenes represent an important class of naturally occurring compounds having spasmolytic, diuretic, anticoagulant, anticancer, anti-HIV and antianaphylactic activities [23–27]. Especially, the 2-amino-4H-chromene derivatives with a nitrile functionality have found potential applications in the treatment of human inflammatory tumor necrosis factor (TNF)α-mediated diseases, such as rheumatoid and psoriatic arthritis [28–32]. Because of their high medicinal values, considerable efforts are being paid to develop synthetic protocols using inexpensive reagents and mild conditions in order to achieve good yields.
The synthesis of pyran annulated heterocycles is known to be catalyzed by acid, base and ammonium salts [33–39]. Although acid catalyzed synthesis of pyran annulated heterocycles can be accomplished at room temperature, it has the potential disadvantage of formation of undesired byproducts due to the competing Knoevenagel reaction of 4-hydroxycoumarin and malononitrile with aldehyde under acidic conditions. For example, Heravi et al. [33] studied the dihydropyrano[3,2-c]chromenes from the reaction of 4-hydroxycoumarin, aldehydes and ethyl cyanoacetate catalyzed by heteropoly acids and observed that selection of catalyst is very crucial to synthesize dihydropyrano[3,2-c]chromenes and avoid the formation of undesired bis-coumarins. For the base-catalyzed reaction, such cases are not reported, yet they need high temperature, possibly to avoid the reversibility of the Knoevenagel product (Scheme 1) which works as Michael acceptor for incoming nucleophile (in this case 4-hydroxycoumarin).
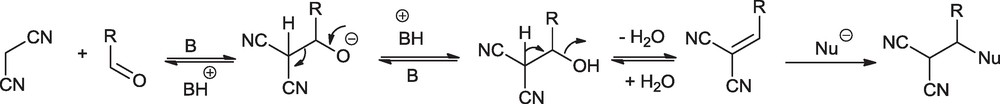
Reversibility of Knoevenagel reaction.
Khurana et al. [34] observed that the reaction of 4-hydroxycoumarin with 4-chlorobenzaldehyde and malononitrile in the presence of DBU did not give any product at room temperature, but the reaction worked extremely well at 100 °C. Khan et al. [35] also reported the same reaction in the presence of DMAP in ethanol at reflux temperature. Although these methods are extremely useful, requirement of high reaction temperature and difficulty of catalyst recovery are notable drawbacks. Therefore, the selection of an appropriate catalyst plays a critical role in overall efficiency of the protocol. In continuation of our interest [40–42] on development of environmentally compatible synthetic protocols employing environmentally benign and recyclable catalysts, we report synthesis of various pyran annulated heterocycles (Scheme 2) at room temperature using highly reusable ion-exchange polystyrene resin Amberlyst A21 as a surrogate solid base catalyst.
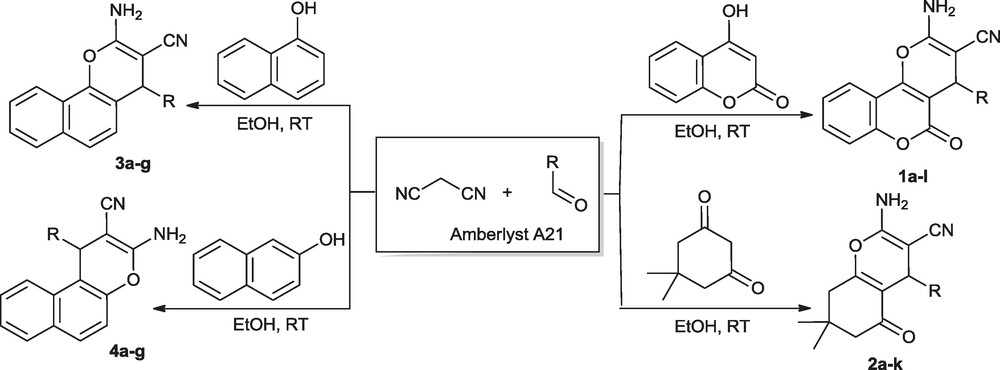
Synthesis of pyran annulated heterocycles.
2 Results and discussion
To start with, we opted to synthesize pyran annulated heterocycle starting from an equimolar ratio of 4-hydroxycoumarins, p-nitrobenzaldehyde and malononitrile. Due to its high catalytic activity [43–46] and selectivity [47,48], long lifetime, superior resistance to thermal, mechanical, and osmotic shock, excellent stability, and low leaching, we opted to use Amberlyst A21 as the base catalyst. The reaction was found to be complete upon stirring the mixture in ethanol at room temperature for half an hour. The resin was separated by filtration and washed thoroughly with warm (60 °C) ethanol. The combined filtrate was reduced in rotavapor and allowed to stand overnight to get the pure recrystallized product in 91% yield. Some more ion-exchange polystyrene resins such as Amberlite IRA 67, Amberlyst A26 OH, and Amberlite IRA 400 OH (Fig. 1) were also tested in order to evaluate and compare their catalytic activity for the said reaction. It is clear from the Fig. 1 that Amberlite IRA 67 also catalyzed the reaction, but took much higher reaction time (2 h) for complete conversion. The ion-exchange polystyrene resins containing the hydroxy counter ion were found to be less efficient and formed many side products besides the desired pyran derivative. Those reactions with ionic ion-exchange resins yielded much inferior yields even on stirring for 2 h. We could not observe any difference in yield and reaction time between reactions catalyzed by pretreated (as stated in Section 4.2) and untreated Amberlyst A21.
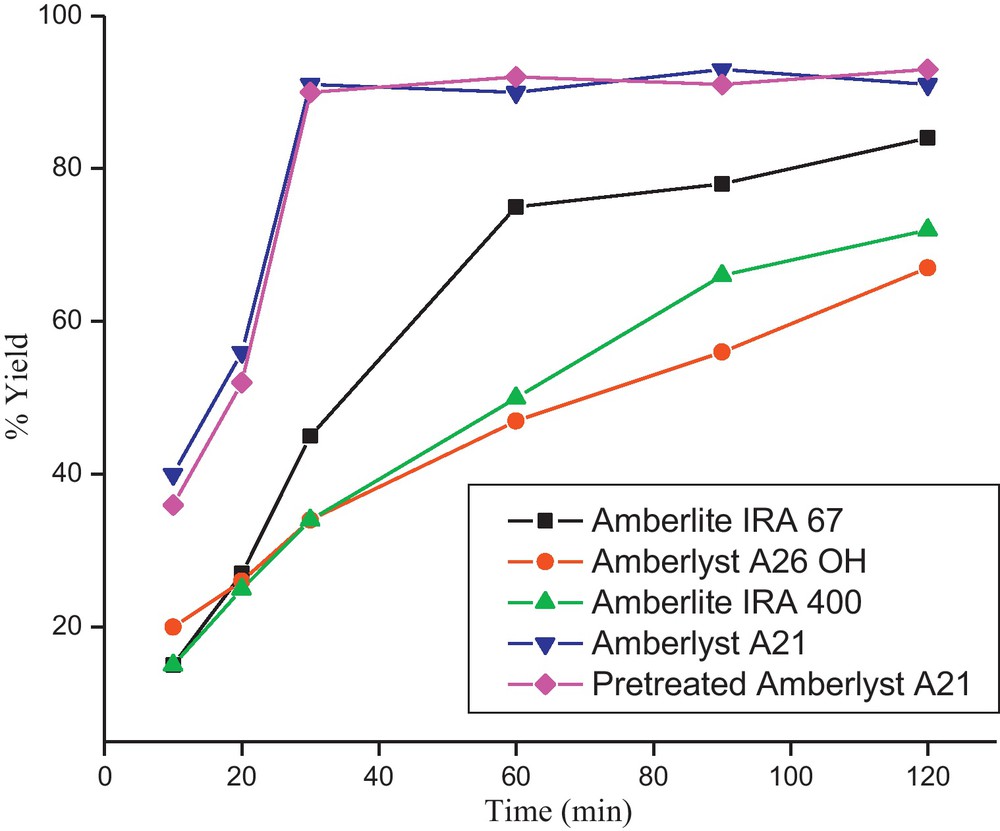
Catalyst optimization.
The catalyst loading was also optimized (Fig. 2) by monitoring the time required for complete conversion of the starting materials in said reaction at different catalyst loading, viz. no catalyst, 10 mg/mmol, 20 mg/mmol, 30 mg/mmol, 40 mg/mmol, and 50 mg/mmol of aldehyde. Although the reaction worked in all the cases, the conversion was not complete without catalyst. Under catalyst-free conditions, only 20% yield of the desired chromene was observed upon stirring for 6 h, while 10 mg/mmol and 20 mg/mmol of catalyst loadings yielded 70% and 90% yield, respectively, during the same reaction time. Interestingly, enhancement of catalyst loading to 40 mg/mmol and 50 mg/mmol did not reduce the reaction time appreciably and gave almost similar yields to that of optimum catalyst loading (30 mg/mmol) in 30 min.

Optimization of catalyst loading.
Since the catalytic activity of polystyrene resins depend on their swelling property in different solvents, we carried out the said reaction in solvents such as neat dichloromethane, chloroform, THF, DMF, methanol and water (Fig. 3) at fixed catalyst loading (30 mg/mmol of aldehyde). We observed that the reactivity of Amberlyst A21 was hardly dependent on the extent of swelling in a particular solvent. For example, the solvents such as dichloromethane, chloroform, THF and DMF can swell the catalyst better than that of ethanol, yet the yields in these solvents were much lower (maximum 40% in DMF). The fact that methanol was found to be the second preferred solvent (60% yield) for the said reaction, indicated that the use of protic solvent might play a crucial role on overall efficiency of the protocol. The reaction gave better yield in protic solvents as they can additionally provide the proton source. However, the reaction results in poor yield in water medium as the starting materials were not soluble at room temperature. Therefore, we concluded that the protic nature of the solvent and solubility of the starting materials played an important role in overall efficiency of the reaction.
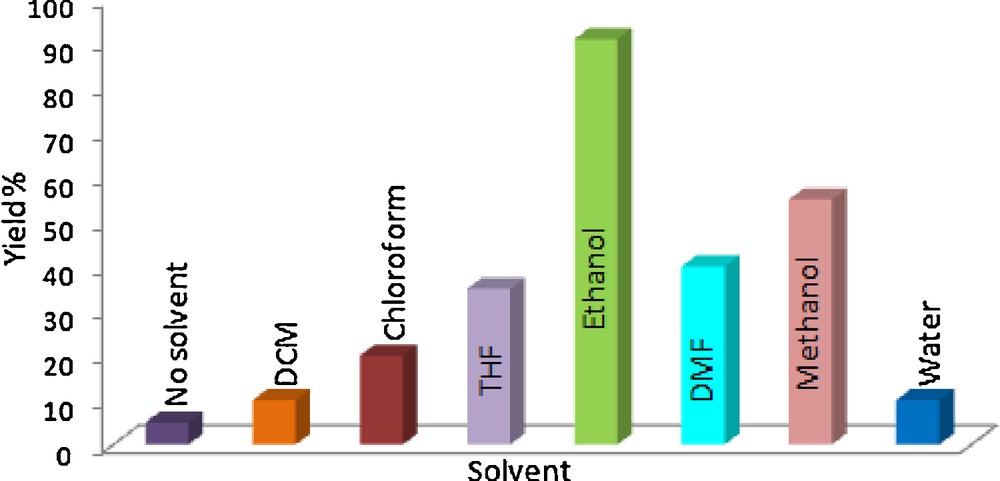
Study of solvent effect.
The recovered catalyst obtained from the residue after filtering off the reaction product in the model reaction was regenerated by washing successively with THF (5 mL) and EtOH (5 mL) thrice and drying at 100 °C for 2 h. It was observed that the efficiency of the catalyst did not change appreciably (Fig. 4) even upon using for five consecutive times in the model reaction.
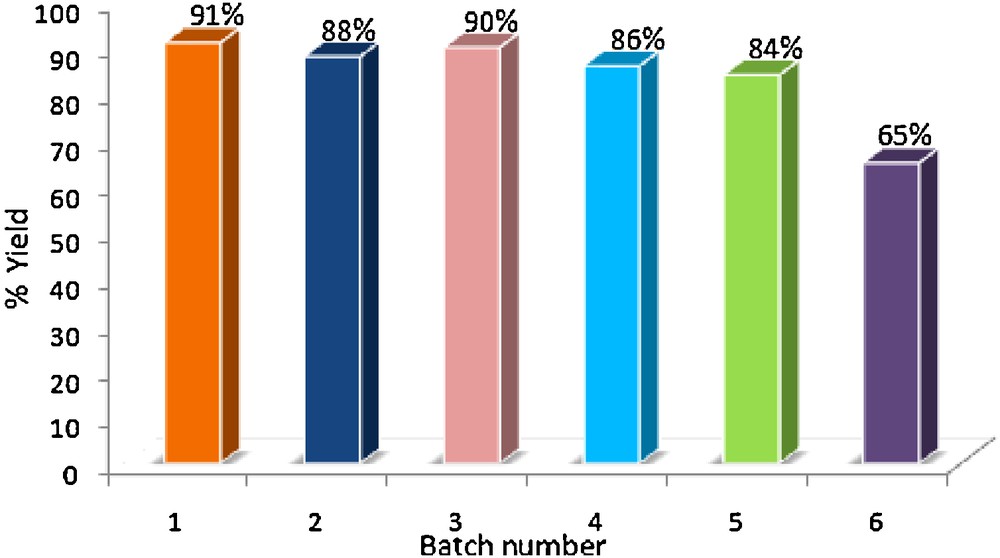
Catalyst recyclability.
Having optimized the reaction parameters such as catalyst, catalyst loading and solvent, the efficiency of the optimized protocol was tested for other aldehydes. In aromatic aldehydes, the nature of the substituents on the phenyl ring of the aldehydes did not have any impact on overall reaction yields, but the reaction time taken by more electrophilic aldehydes (entry 1c-d, Table 1) were comparatively shorter. Interestingly, only a limited number of reports are available for the synthesis of coumarin derived 2-aminochromenes from aliphatic aldehydes [34,39]. Aliphatic aldehydes (entry 1i-l, Table 1) also gave very good yields under our reaction conditions, albeit taking longer times than their aromatic counterpart.
Synthesis of 2-aminochromenes from 4-hydroxycoumarina,b.
1a (1.5 h, 87%) | 1b (2 h, 76%) | 1c (0.5 h, 91%) | 1d (0.5 h, 85%) |
1e (2 h, 87%) | 1f (2 h, 78%) | 1g (2.5 h, 82%) | 1hc (2 h, 90%) |
1i (4 h, 81%) | 1jc (5 h, 78%) | 1k (5 h, 75%) | 1lc (3 h, 90%) |
a Equimolar ratio of all the reactants with 30 mg Amberlyst A21/mmol of aldehyde at room temperature
b Isolated yields.
c New compound.
In order to extend the scope of our reaction protocol, we studied the synthesis of pyran annulated heterocycles derived from the condensation of aldehyde, malononitrile and dimedone in the presence of a catalytic amount (30 mg/mmol) of Amberlyst A21. It may be noted that 2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromenes show potent antibacterial activity [49] besides other biological activities shown by 2-amino-4H-pyrans. Interestingly, our protocol gave the desired products in excellent yields (Table 2) under similar reaction conditions irrespective of the nature of aldehyde.
Synthesis of 2-aminochromenes from dimedonea,b.
2a (0.5 h, 90%) | 2b (0.5 h, 92%) | 2c (1 h, 84%) | 2d (1 h, 87%) | 2e (1.5 h, 83%) | |
2f (1.5 h, 78%) | 2g (0.51 h, 80%) | 2h (4 h, 71%) | 2i (4 h, 81%) | 2jc (5 h, 78%) | 2kc (1.5 h, 88%) |
a Equimolar ratio of all the reactants with 30 mg Amberlyst A21/mmol of aldehyde at room temperature
b Isolated yields.
c New compound.
When our method was extended to the synthesis of 2-aminochromenes from α-naphthol under our reaction conditions by stirring a mixture of α-naphthol, m-nitrobenzaldehyde and malononitrile in ethanol in the presence of catalytic amount of Amberlyst A21 (30 mg/mmol of aldehyde), it generated the desired 2-aminochromene in very good yield (75%). The protocol was extended to synthesize other 2-aminochromenes (Table 3) derivatives also by varying the aromatic aldehyde and very good yield of desired products were obtained.
Synthesis of 2-aminochromenes from α-naphthola,b.
3a (3 h, 75%) | 3b (3 h, 78%) | 3c (6 h, 72%) | 3d (6 h, 71%) |
3ec (2.5 h, 77%) | 3f (4 h, 79%) | 3g (4 h, 76%) |
a Equimolar ratio of all the reactants with 30 mg Amberlyst A21/mmol of aldehyde at room temperature
b Isolated yields.
c New compound.
Likewise, almost identical results were obtained for β-naphthol (Table 4) upon stirring with m-nitrobenzaldehyde and malononitrile in ethanol in the presence of catalytic amount of Amberlyst A21 (30 mg/mmol of aldehyde) under similar reaction conditions. Here too, the electrophilicity of the aldehydes resulting from the +I or –I effect of substituents on the phenyl ring effected the overall reaction time for completion in spite of giving very good yield in all the cases. The products were successfully isolated and purified by recrystallization from ethanol without the use of chromatography.
3 Conclusion
Amberlyst A21, a neutral resin and a solid base, was proven to be an extremely effective catalyst for the synthesis of a diverse range of pharmaceutically important pyran annulated heterocycles. The efficiency of the catalyst was found independent of the swelling properties of various organic solvents, but on the protic nature and solubility of the reactants in a particular solvent. In comparison to ionic ion-exchange resins containing hydroxy counter ion, neutral ion-exchange resins were found to be more efficient for multicomponent reaction of aldehyde, malononitrile and 4-hydroxycoumarin to generate the desired product in almost pure form. Use of Amberlyst A21 found to have distinct advantages in the said synthesis over existing examples in the literature, in the form of recyclability of the catalyst and room temperature reaction conditions. Nevertheless, the requirement of no chromatographic purification involving hazardous organic solvents, short reaction time, and very good to excellent yield of the products are some of the major achievements of this reaction protocol that have potential to be extremely useful for the synthetic and medicinal chemists.
4 Experimental
IR spectra were recorded on a Perkin–Elmer FT-IR spectrometer. 1H NMR and 13C NMR spectra were recorded on a Bruker AMX 300 MHz/Bruker 400 MHz and 75 MHz/100 MHz spectrometer, respectively. Mass spectra were obtained from Waters ZQ 4000 mass spectrometer by the ESI method, while the elemental analyses of the compounds were performed on a Perkin–Elmer-2400 CHN/S analyzer. Melting points were determined in an open capillary and are uncorrected. TLC experiments were done using MERCK TLC aluminum sheets (silica gel 60 F254) and chromatograms were visualized by exposing in iodine chamber, UV-lamp or spraying with KMnO4 and heating.
4.1 Reactants and catalysts
All the starting compounds and the catalysts were purchased from Sigma-Aldrich Company, USA and used without any further purification. All the synthesized compounds were recrystallized from ethanol. Following heterogeneous catalysts were used: Amberlyst A21, IRA 67, Amberlyst A26 OH, and Amberlite IRA 400 OH.
4.2 Pretreatment of the catalyst, Amberlyst A21
The resin, Amberlyst A21 (500 mg) was taken in a round-bottom flask and millipore water added (5 mL). The suspension was mildly stirred for 2 h at 40 °C and then filtered. The residue was washed thoroughly with warm (40 °C) water until the effluent was cleared. The residue was then washed successively with ethanol (15 mL) and kept in an oven at 100 °C overnight. This pretreated Amberlyst A21 resin, obtained as above, was kept in a glass bottle and the catalytic activity was evaluated for the synthesis of pyran annulated heterocycles under our reaction conditions.
4.3 Typical procedure
A solution of p-nitrobenzaldehyde (0.151 g, 1 mmol) and malononitrile (0.066 g, 1 mmol) in ethanol (10 mL) was taken in round-bottom flask (50 mL) and 4-hydroxycoumarin (0.162 g, 1 mmol) and Amberlyst A21 (30 mg) were added while stirring mildly on a magnetic stirrer at room temperature. The progress of the reaction was monitored every 10 min by TLC. After completion of the reaction within 30 min, warm ethanol (60 °C) was added to the reaction mixture to dissolve the solid product and filtered. The residue of Amberlyst A21 was washed thoroughly with warm ethanol until no compound was detected in the residue. The combined ethanolic solution was concentrated under vacuum and allowed to stand in the refrigerator to get pure crystalline product in 91% (0.391 g, 0.91 mmol) yield.
4.4 Spectra of new compounds
4.4.1 2-Amino-5-oxo-4-(pyridine-3-yl)-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (1h)
IR (KBr): 552, 764, 1056, 1116, 1381, 1606, 1679, 1706, 2203, 2972, 3403, 3641, 3827 cm−1. 1H NMR (400 MHz, DMSO-d6): δ 4.49 (s, 1H), 7.29 (dd, J = 4.8, 8 Hz, 1H), 7.39–7.46 (m, 3H), 7.64–7.68 (m, 2H), 7.84 (dd, J = 0.8, 8 Hz, 2H), 8.40 (d, J = 4.4 Hz, 1H), 8.47 (d, J = 2 Hz, 2H). 13C NMR (100 MHz, DMSO- d6): δ 34.6, 56.9, 102.8, 112.9, 116.5, 119.0, 122.5, 123.8, 124.6, 133.0, 135.5, 138.7, 148.2, 148.9, 152.2, 153.8, 158.0, 159.6. MS (ES+) m/z 318 (M + H)+, 340 (M + Na)+. Elemental analysis for C18H11N3O3: calculated C 68.14, H 3.49, N 13.24; observed C 68.52, H 3.36, N 13.26.
4.4.2 2-Amino-5-oxo-4-pentyl-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (1j)
IR (KBr): 758, 976, 1049, 1195, 1268, 1321, 1394, 1606, 1672, 2203, 3018, 3297 cm−1. 1H NMR (400 MHz, DMSO- d6): δ 0.80 (t, J = 6.4 Hz, 3H), 1.04–1.22 (m, 6H), 1.52–1.58 (m, 1H), 1.70–1.75 (m, 1H), 3.24 (s, 1H), 7.31 (s, 2H), 7.44–7.48 (m, 2H), 7.67–7.81 (m, 2H). 13C NMR (100 MHz, DMSO- d6): δ 13.8, 21.9, 23.7, 30.7, 31.1, 33.5, 55, 104.2, 112, 116.5, 119.6, 122.1, 124.6, 132.7, 151.9, 154.1, 159.3, 159.9. MS (ES+) m/z 311 (M + H)+, 333 (M + Na)+. Elemental analysis for C18H18N2O3: calculated C 69.66, H 5.85, N 9.03; observed C 69.52, H 5.76, N 9.16.
4.4.3 Ethyl 2-amino-3-cyano-5-oxo-4,5-dihydropyrano[3,2-c]chromene-4-carboxylate (1l)
IR (KBr): 565, 1036, 1202, 1387, 1600, 1666, 1745, 2203, 2660, 2872, 2965, 3323, 3465, 3469, 3668, 3827 cm−1. 1H NMR (400 MHz, DMSO- d6): δ 0.95 (t, J = 7.2 Hz, 3H), 3.14 (s, 1H), 3.83–3.97 (m, 2H), 7.25–7.60 (m, 4H). 13C NMR (100 MHz, DMSO- d6): δ 14, 37.1, 51, 61.3, 99.9, 112.4, 116.8, 118.2, 122.2, 124.9, 133.5, 152, 153.9, 159, 159.9, 170.4. MS (ES+) m/z 313 (M + H)+, 335 (M + Na)+. Elemental analysis for C16H12N2O5: calculated C 61.54, H 3.87, N 8.97; observed C 61.52, H 3.76, N 9.06.
4.4.4 2-Amino-7,7-dimethyl-4-octyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (2j)
IR (KBr): 572, 724, 1036, 1155, 1228, 1381, 1460, 1527, 1613, 1666, 2196, 2932, 3191, 3310, 3390 cm−1. 1H NMR (400 MHz, DMSO- d6): δ 0.83 (t, J = 6.4 Hz, 3H), 0.99 (s, 3H), 1.02 (s, 3H), 1.19–1.47 (m, 14H), 2.14–2.49 (m, 4H), 3.14 (t, J = 4 Hz, 1H), 6.89 (s, 2H). 13C NMR (100 MHz, DMSO- d6): δ 13.9, 22, 24, 26.4, 28.5, 28.6, 28.9, 31.2, 31.6, 34.3, 50.1, 55.3, 112.6, 120.1, 159.8, 163.1, 169.2. MS (ES+) m/z 331 (M + H)+, 353 (M + Na)+. Elemental analysis for C20H30N2O2: calculated C 72.69, H 9.15, N 8.48; observed C 72.52, H 9.11, N 8.50.
4.4.5 Ethyl 2-amino-3-cyano-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-4-carboxylate (2k)
IR (KBr): 569, 727, 1136, 1230, 1374, 1434, 1606, 1672, 1725, 2196, 2932, 3174, 3310, 3386 cm−1. 1H NMR (400 MHz, DMSO- d6): δ 1.04 (d, J = 5.8 Hz, 6H), 1.14 (t, J = 7.2 Hz, 3H), 2.15 (d, J = 16 Hz, 1H), 2.31–2.36 (m, 2H), 2.56 (d, J = 17.6 Hz, 1H), 3.75 (s, 1H), 3.96–4.12 (m, 2H), 7.28 (s, 2H). 13C NMR (100 MHz, DMSO- d6): δ 14, 26.1, 28.6, 32.1, 35.7, 49.5, 55.9, 60.7, 108.7, 118.7, 159.6, 163.3, 171, 196. MS (ES+) m/z 291 (M + H)+, 313 (M + Na)+. Elemental analysis for C15H18N2O4: calculated C 62.06, H 6.25, N 9.65; observed C 62.02, H 6.21, N 9.67.
4.4.6 2-Amino-4-(pyridine-3-yl)-4H-benzo[h]chromene-3-carbonitrile (3e)
IR (KBr): 579, 764, 811, 1036, 1109, 1188, 1275, 1381, 1420, 1613, 1659, 2190, 2932, 3131, 3184, 3363 cm−1. 1H NMR (300 MHz, CDCl3 + DMSO- d6): δ 4,83 (s, 1H), 6.40 (s, 2H), 6.92 (d, J = 7.4 Hz, 1H), 7.16–7.20 (m, 1H), 7.42–7.52 (m, 4H), 7.71 (d, J = 8.1 Hz, 1H), 8.19 (d, J = 7.2 Hz, 1H), 8.41 (s, 1H), 8.49 (s, 1H). 13C NMR (75 MHz, DMSO- d6): δ 38.6, 115.7, 119.9, 120.6, 122.7, 123.4, 123.9, 125.3, 126.2, 127.1, 132.7, 135.3, 140.2, 142.9, 147.9, 148.6, 159.9. MS (ES+) m/z 300 (M + H)+, 322 (M + Na)+. Elemental analysis for C19H13N3O: calculated C 76.21, H 4.32, N 14.04; observed C 76.01, H 4.21, N 13.98.
4.4.7 Ethyl 2-amino-3-cyano-1H-benzo[f]chromene-1-carboxylate (4 g)
IR (KBr): 552, 618, 764, 830, 1036, 1096, 1195, 1241, 1301, 1427, 1593, 1659, 1732, 2196, 2495, 2793, 2932, 3211, 3330, 3396, 3443, 3641 cm−1; 1H NMR (300 MHz, CDCl3 + DMSO- d6): δ 1.18 (t, J = 6.9 Hz, 3H), 4.12 (q, J = 7.2 Hz, 2H), 4.86 (s, 1H), 5.45 (s, 2H), 7.14 (d, J = 9 Hz, 1H), 7.39–7.55 (m, 2H), 7.73–7.77 (m, 3H); 13C NMR (75 MHz, CDCl3 + DMSO- d6): δ 13.9, 39.2, 54.2, 61.6, 110.8, 116.6, 119.4, 122.4, 125.2, 127.5, 128.4, 129.8, 130.7130.8, 146.9, 161.0, 171.6. MS (ES+) m/z 295 (M + H)+, 317 (M + Na)+; Elemental analysis for C17H14N2O3: calculated C 69.38, H 4.79, N 9.52; observed C 69.21, H 4.71, N 9.31.
Acknowledgement
Authors thank University Grants Commission-Special Assistance Programme (UGC-SAP), North Eastern Hill University, Shillong for partial financial assistance. We thank the respected reviewers for useful observations and suggestions.
Vous devez vous connecter pour continuer.
S'authentifier