

1 Introduction
1,4,8,11-Tetraazacyclotetradecane (cyclam) based macrocycles form complexes of high thermodynamic and kinetic stability with transition metals [1–4]. N-functionalised derivatives are selective metal ion chelators and are commonly used as the chelating component (known as a bi-functional chelator or BFC) of targeted radiopharmaceuticals in nuclear medicine [5–8]. A bi-functional chelator contains a reactive site for attachment to a targeting group (such as an antibody or peptide) and, ideally, will fully retain the radiolabel under physiological conditions to allow localisation at the target site without transchelation occurring [9,10]. We are particularly interested in copper radioisotopes 60Cu, 61Cu, 62Cu and 64Cu, which are positron emitters with half-lives varying from 0.16 to 12.7 h that have been investigated for applications in positron emission tomography (PET) imaging [8,11–16]. 67Cu is a longer-lived β emitter (t1/2 = 61.9 h) that has been used in targeted radiotherapy [17,18]. Cross-bridged cyclam derivatives are promising candidates as copper radioisotope chelators for tumour targeting applications, due to the increased kinetic stability of the complexes [19–21].
The synthesis of cross-bridged macrocycles is generally a lengthy procedure, with a two-step (bis-substitution followed by reduction) reaction time of up to 32 days [22,23]. Initial mono-substitution of glyoxal cyclam with an alkyl halide is generally quite fast, with standard reaction times of 12–24 h [23–25]. However, one-pot bis-substitutions are usually more time consuming as the reactivity of the second exo-position after mono-alkylation is greatly reduced and therefore, occurs at a much slower rate [20,22,26–29].
In this work, two cross-bridged cyclam ligands were synthesised with an overall bis-substitution reaction time of less than 24 h, copper(II) complexes were produced and characterised by X-ray crystallography.
2 Results and discussion
2.1 Ligand synthesis
An investigation into the nucleophilic substitutions of substituted benzyl bromides by glyoxal bridged cyclam 3 using the bisaminal methodology was carried out [30]. The mono-substitution of benzyl bromide by the tetracyclic bisaminal generally proceeds in good yields with short reaction times of between 2 h and 3 days [23,25,31–33]. Not only does the mono-substitution of benzyl bromides by 3 occur rapidly, but also the precipitation of the quaternary ammonium salt signals the completion of the first substitution. The formation of one-step bis-subsitituted glyoxal cyclams generally involves the use of a large excess of the benzyl bromide and/or long reaction times, [20,23–25,34,35] with conditions and yields summarised in Table 1. However, not all bis-substituted compounds synthesised could successfully be reduced to form cross-bridged cyclam derivatives, this reduction was unsuccessful for the borane compound [24]. There are two patents published that report significantly shorter reaction times are possible indicating that further studies should be carried out which stimulated the interest in reducing reaction times [36,37].
Bis-substitution reactions of bisaminal 3.
| Reference | Substrate | Stoichiometry (2:X) | T | Time scale | Yield |
| [24] | MeI | 1:10 | 60 °Ca | 12 h | 86% |
| [24] | BH3–THF | 1:4 | 75 °C | 8 h | 86% |
| [20] | BzBr | 1:16 | r.t. | 14 d | 93% |
| This work | MeBzBr | 1:4 | r.t. | 16 h | 93% |
| This work | Br2BzBr | 1:4 | r.t. | 16 h | (38%)b |
a Sealed vessel.
b Mixture of mono-substituted and bis-substituted products. Yield of bis-substituted product by NMR.
The second exo-nitrogen position of 3 generally effects its nucleophilic substitutions at a slower rate than the first, and the appending of two benzyl arms can require reaction times between 10 and 16 days [20,34,38]. However, using 4-methylbenzyl bromide and 3,5-dibromobenzyl bromide, the bis-substituted bisaminal salts were produced within 16 h which is more in line with the results indicated in the patents [36,37]. These bis-substitutions are unusual due to the short time scale involved in the reactions, and the relatively low excess of substrate employed compared to other reported preparations.
In the case of the substitution of 4-methylbenzyl bromide, 4 was exclusively produced within 16 h, whereas the substitution of 3,5-dibromobenzyl bromide resulted in a mixture of 5 and mono-substituted cyclam (Scheme 1). The di-substitution makes it possible to convert these species to cross-bridged macrocycles using a reductive ring cleavage. Electronic effects may contribute to the stabilisation of the intermediates formed in the substitution reactions. It is also possible to achieve rapid bis-substitution with methyl iodide at elevated temperature using a five-fold excess of alkylation agent (Table 1).
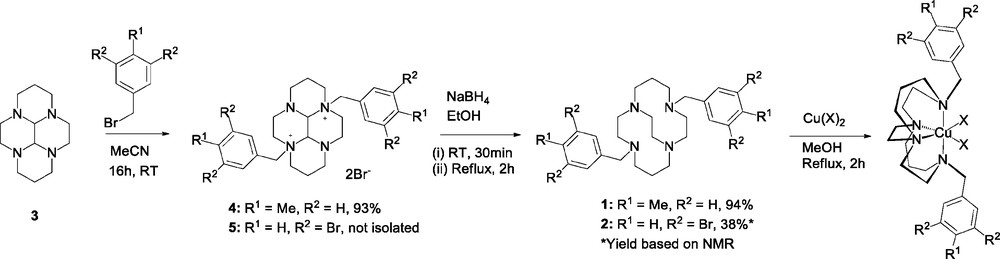
Synthesis of bis-substituted derivatives of glyoxal cyclam, reduction to form the cross-bridged macrocycle and copper(II) complex formation.
Benzyl pendent arms can be removed to produce the parent macrocyclic backbone (1,4,8,11-tetraazabicyclo[6.6.2]hexadecane), for example L1 and L3, to which the alternative pendent arm systems can be attached [20,34]. This route is well established, and can be adopted in any strategy requiring pendent armed cross-bridged cyclams. The published yield from bisaminal 3 to macrocycle L1 is 65% [20]. Once reduced to the parent macrocyclic backbone, L1, substitution reactions at the secondary amines can be used to add pendant arms, such as the acetate arms that further stabilise copper(II) complex formation in L2. Acetate or amine pendant arms, or click chemistry can be used for conjugation reactions, forming convalent links to targeting groups, such as peptides for binding to biological targets [20,34,39]. The yield may be improved upon for some pendent armed systems by appending the desired arms directly to the bisaminal to reduce the number of synthetic steps [32].
The bis-substitution by 3 to produce 4 and 5 was followed by a reductive ring cleavage step carried out using a procedure with a shorter timescale and higher temperature than is normally adopted for cross-bridged cyclam systems [31,33,40–42]: a 30-min stir at room temperature after the addition of sodium borohydride followed by a 2-hour reflux. This is in contrast to the room temperature 10–16-day stir more generally used for reduction to the cross bridge cyclam macrocycle [20,32,34,43]. Interestingly, this increased temperature and shortened time frame seemed not to have a detrimental effect on the production of the cross-bridged variants, 1 and 2. The synthesis of 1 returned an excellent yield (87%) from 3, while the production of 2 occurred alongside the side-bridged (piperazino cyclam) chelator due to the mixture of mono- and bis- substituted ammonium salts. This mixture was not separated and was used in the complex formation preparation.
The combination of the shorter reaction times for both steps means that 1 and 2 could be synthesised with an overall reaction time of less than 24 h, compared with times of up to 32 days in previous studies. However, the elevated temperature for the reduction may not be compatible with ester containing pendant arms and so, further studies may be required to determine the appropriate conditions when more sensitive functional groups are present.
2.2 Copper(II) complexation
Copper(II) complexes of cross-bridged cyclam form more kinetically stable complexes than many other macrocycles due to the structural rigidity [44]. However, longer timescale and higher temperature complexation conditions are generally required in comparison to non-bridged or less basic cyclams. The cross-bridged ligands are remarkably basic, binding a proton within the cavity of the ligand, forming hydrogen bonds between macrocyclic amines. Protons effectively compete with metal ions for occupation of the cavity [19].
This work uses the complexation conditions of Weisman et al. [20]. The ligand and one equivalent of copper(II) salt were dissolved in methanol, and heated under reflux for 2 hours, during which colour changes in the solution were often observed upon the addition of copper salt. The colour changes were significantly less rapid than the addition of copper(II) salt to side-bridged cyclam ligands [31]. This is due to the slower complex formation of copper(II) ions with cross-bridged ligands compared to the less rigid macrocyclic ligands. This can be a disadvantage in radiolabelling procedures where rapid complexation is preferred and temperature-sensitive biomolecules are often used.
2.3 X-ray crystal structures of [Cu1NO3]+ and [Cu2Cl2]
Crystals of sufficient quality for single crystal X-ray diffraction were grown by vapour diffusion of diethyl ether into methanolic solutions of the complex, and appeared after approximately 7 days. These crystal structures are shown in Figs. 1 and 2.
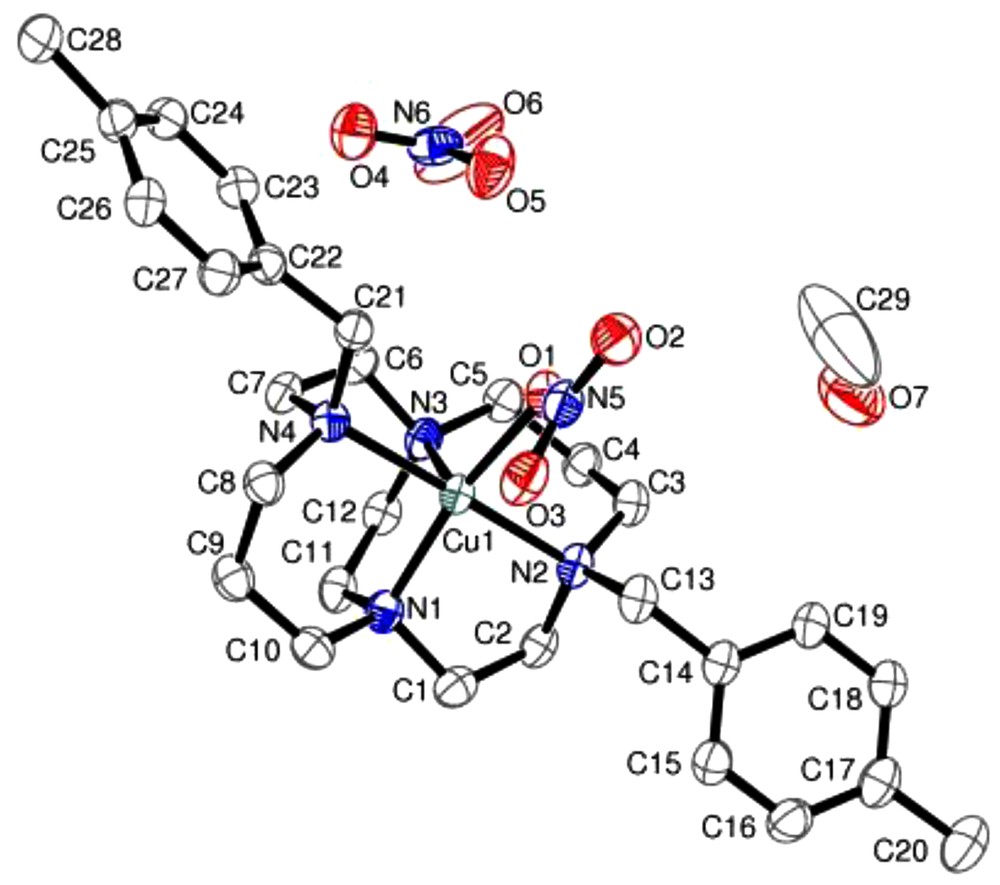
An ORTEP plot (50% probability ellipsoids) of the single crystal X-ray structure of [Cu1NO3]NO3·CH3OH.

An ORTEP plot (50% probability ellipsoids) of the single crystal X-ray structure of [Cu2Cl2]. The elongated N(2)–Cu(1)–N(4) axis is shown with dashed bonds.
The folded cis-V configuration is evident, [45] with the copper(II) ion sitting within the macrocyclic cavity. There is no interaction between the pendent arms of the macrocycle and the copper(II) ion. This may be due to the increased steric bulk of the aryl group of the pendent arms. The pendent arms of the two structures are directed away from the macrocycle, and are oriented at different angles to each other with respect to the axial Cu–N plane. The difference in orientation could be attributed to the bound counterions, the geometry they impart on the metal ion, and the steric repulsion along macrocyclic axes. There are also variations between the two structures in the symmetry of the crystal packing of the macrocycles within the crystal lattice (Supplementary data).
As there is no interaction between the copper(II) ion and the pendent arms, it is possible for counterions to occupy both vacant cis coordination sites. This is observed for [Cu2Cl2], in which the two chloride ligands bind to the copper(II) ion to complete the octahedral coordination sphere. In [Cu1NO3]NO3·CH3OH, the nitrate counterion completes a five coordinate geometry and the sixth coordination site is blocked by the position of O(3) of the nitrate. The nitrate anion is monodentate, with distortion from the intermediate position between the two equatorial nitrogen donors by 8.2°. The position of the nitrate donor results in a near linear N(1)–Cu(1)–O(1) angle at 168.55(11)°, while the corresponding N(3)–Cu(1)–O(3) angle is 153.1°. The Cu–O distances are significantly different, with the Cu–O(3) distance being 0.75 Å longer than the Cu–O(1) bond length, hence, not forming a bonding interaction. [46]
The constraint of the ethylene bridge restricts the angle between the equatorial nitrogen atoms and the copper(II) ion to 86.86(11)°. The Cu–N distances are comparable to those previously observed for similar constrained complexes. [35,47] There is a significant difference between the Cu–N(1) and Cu–N(3) bond distances involving the bridgehead nitrogens, which have previously been observed to be of comparable magnitude. [20,48,49] The longest of these two bonds is that of the N(3)–Cu interaction which occupies the apical position in the square based pyramidal geometry and demonstrates that there is sufficient flexibility in the macrocyclic chelator to accommodate the bond elongation preferred for copper(II).
[Cu2Cl2] is an octahedral complex, and it is expected that a Jahn–Teller type distortion will be observed along one of the ligand axes. In contrast to [Cu1NO3]NO3·CH3OH, the bridgehead nitrogen donor atoms form bonds of expected length, [44,48] as do the chloride counter anions [20]. The nitrogen donors, associated with the pendent arms, are the ligands that are extended along the z axis. The mean distorted Cu–N bond length of 2.475 Å shows elongation in the order of 0.4 Å from the other nitrogen donors in this example.
The angle at which the pendent arms are located with respect to the N(2)–Cu(1)–N(4) plane varies between the two structures. This is attributed to the location of the counterion(s) within the cavity. The bound nitrate counterion of [Cu1NO3]NO3·CH3OH is situated approximately above the centre of the cavity. As a result, the pendent arms are bent away from each other, as viewed along the N–Cu–N axis (Fig. 3). This minimises the steric repulsion between the aryl pendent arms and the nitrate counterion. The mean angle of bend of the pendent arms is 29.6° from the axial N–Cu–N plane bisecting the equatorial nitrogen atoms (Table 2)

[Cu1NO3]NO3·CH3OH and [Cu2Cl2] viewed along the N–Cu–N axis (ORTEP plots at 50% probability with the H-atoms removed for clarity).
Crystal data for the single crystal X-ray structures of [Cu2Cl2] and [Cu1NO3]NO3·CH3OH.
| [Cu2Cl2] | [Cu1NO3]NO3·CH3OH | |
| Formula | C26N4H34Cl2Br4Cu | C29N6H43O7Cu |
| Mr | 856.65 | 651.23 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/c | |
| a/Å | 14.207(2) | 8.7231(15) |
| b/Å | 13.9892(14) | 14.248(3) |
| c/Å | 16.133(3) | 14.316(2) |
| α/o | 90 | 117.139(12) |
| β/o | 113.877(12) | 100.121(13) |
| γ/o | 90 | 95.291(14) |
| Volume/Å3 | 2932.1(7) | 1528.3(5) |
| Z | 4 | 2 |
| Density (calc.)/Mg/m3 | 1.941 | 1.415 |
| μMoKα (mm−1) | 6.42 | 0.770 |
| T/K | 150 | 150 |
| θ range/o | 2.76–30.35 | 2.42–29.77 |
| Measured reflections | 35705 | 36891 |
| Unique reflections [Rint] | 8517 [0.1191] | 8901 [0.1169] |
| Completeness (θo) | 99.7% | 100.0% |
| Goodness-of-fit on F2 | 0.854 | 0.802 |
| R1, wR2 (I > 2σ(I)) | 0.0594, 0.1062 | 0.0510, 0.1075 |
| R1, wR2 (all data) | 0.1778, 0.1405 | 0.1491, 0.1406 |
| Extinction coefficient | 0.00128(14) | 0.0250(15) |
| Largest diff. peak and hole/e Å−3 | 0.970 and −0.999 | 0.725 and −1.232 |
As the two chloride ions are occupying the cis-equatorial sites in [Cu2Cl2], the position of the pendent arms with the least steric hindrance involves bisecting the angle between the equatorial ligands, as viewed down the N–Cu–N axis (Fig. 3). This results in the aryl groups being practically co-planar.
There remains a slight twist of the pendent arms away from the axial N–Cu–N plane, with a mean twist angle of 12.4°. The twist of the ethylene bridge causes the equatorial twisting of the chloride counterions in opposite directions to reduce steric repulsion, (Supplementary data, Fig. S1). The mean angle of twist of the four donors from the mean plane is 10.5°. As a result, in order to bisect the Cl–Cu–Cl angle, the pendent arms must twist away from the axial N–Cu–N plane, giving the orientation observed (Table 3).
Selected bond lengths (Å) and angles (o) for the single crystal X-ray structures of [Cu1NO3]+ and [Cu2Cl2].
| Bond lengths/angles [Cu1NO3]+ | Bond lengths/angles [Cu2Cl2] | |
| Cu(1)-N(1) | 2.038(3) | 2.071(7) |
| Cu(1)–N(2) | 2.096(3) | 2.455(8) |
| Cu(1)–N(3) | 2.154(3) | 2.091(7) |
| Cu(1)–N(4) | 2.131(3) | 2.495(8) |
| Cu(1)–O(1) | 1.989(3) | – |
| Cu(1)–Cl(1) | – | 2.322(2) |
| Cu(1)–Cl(2) | – | 2.302(2) |
| N(1)–C(11) | 1.503(4) | 1.488(11) |
| N(3)–C(12) | 1.473(5) | 1.493(11) |
| C(11)–C(12) | 1.534(5) | 1.505(11) |
| N(1)–Cu(1)–N(2) | 85.19(12) | 79.3(3) |
| N(1)–Cu(1)–N(3) | 86.86(11) | 83.2(3) |
| N(1)–Cu(1)–N(4) | 93.21(12) | 95.9(3) |
| N(2)–Cu(1)–N(3) | 93.18(11) | 92.0(3) |
| N(2)–Cu(1)–N(4) | 177.83(12) | 171.0(2) |
| N(3)–Cu(1)–N(4) | 85.27(11) | 79.8(3) |
| N(1)–Cu(1)–O(1) | 168.55(11) | – |
| N(1)–Cu(1)–Cl(1) | – | 92.47(18) |
| N(1)–Cu(1)–Cl(2) | – | 168.5(2) |
| N(2)–Cu(1)–O(1) | 88.40(11) | – |
| N(2)–Cu(1)–Cl(1) | – | 98.41(17) |
| N(2)–Cu(1)–Cl(2) | – | 89.94(18) |
| N(3)–Cu(1)–O(1) | 102.99(11) | – |
| N(3)–Cu(1)–Cl(1) | – | 167.8(2) |
| N(3)–Cu(1)–Cl(2) | – | 93.3(2) |
| N(4)–Cu(1)–O(1) | 93.42(11) | – |
| N(4)–Cu(1)–Cl(1) | – | 89.43(17) |
3 Conclusion
A rapid synthesis of two novel cross-bridged cyclam chelators (1 and 2) has been demonstrated with 1 isolated as a pure compound and 2 separated by complex formation and recrystallisation. A dramatic decrease in two-step reaction times is observed compared to the standard synthetic conditions to produce benzyl cross-bridged cyclams. Copper(II) complexes of these ligands were synthesised and characterised by X-ray crystallography showing an interesting variation in the coordination environment and the potential for distortion in the Cu–N bonds of the macrocyclic chelator. These structures show that despite the additional rigidity imposed by the ethylene bridge between non-adjacent nitrogens of the cyclam ring, there is still sufficient flexibility to elongate along the N–Cu–N axis. This distortion may be linked to the presence of two chloride ligands offering sufficient energetic benefit to offset the penalty of a less favourable conformation of the macrocyclic chelator. The rapid synthesis developed in this work could have a significant impact on the production of cross-bridged cyclam chelators for their use in radiopharmaceutical applications, such as PET imaging.
4 Experimental
All the reagents and solvents were purchased from Sigma-Aldrich Limited, Lancaster Chemicals Limited (Alfa-Aesar) or Fisher Chemicals Limited, and were used as supplied unless otherwise stated. All 1H-NMR and proton decoupled 13C-NMR spectra were collected on a JEOL JNM-LA400 spectrometer, at 400 and 100 MHz respectively, and referenced against residual solvent signals. Mass spectrometry was carried out by electrospray ionisation (ESI) with the data collected on a Finnigan LCQ spectrometer or by the EPSRC National Mass Spectrometry Service at the University of Swansea on a Waters ZQ4000 spectrometer. UV/visible spectra were obtained using an HP-Agilent 8453 diode array spectrometer.
The diffraction data set was collected on a Stoe IPDS-II imaging plate diffractometer using Mo Kα radiation (k = 0.71073 Å). The crystal was kept at 150 K during data collection using the Oxford Cryosystems Cryostream Cooler. The structures were solved using direct methods (SHELXS) and refined against F2 (SHELXL) [36]. H atoms were placed in idealised positions or located on the difference map, and refined using a riding model with C–H = 0.97A˚, N–H = 0.91A˚ and Uiso(H) = 1.2 times Ueq of the carrier atom. All non-H atoms were refined anisotropically. The WinGX package was used for refinement and production of data tables, and ORTEP-3 was used for structure visualisation [50,51].
4.1 Synthetic procedures
Glyoxal cyclam (3) (cis-perhydrotetraazapyrene) was synthesised in accordance with literature procedures [26].
3a,8a-bis(4-methylbenzyl)-decahydro-3a,5a,8a,11a-tetraazapyrenium dibromide 4
To a stirred solution of 3 (128 mg, 575 μmol) in acetonitrile (20 mL) was added 4-methylbenzyl bromide (333 mg, 1.80 mmol) and the solution was stirred at room temperature for 16 h. The solvent was removed and the residue was washed with diethyl ether (4 × 30 mL) to yield a white solid (316 mg, 93%). 1H-NMR (DMSO-d6): δ 7.54 and 7.36 (AB, 8H, J 8.0 Hz, H(Ar)), 5.31 (s, 2H, CH), 5.17 and 4.89 (AB, 4H, J 13.0 Hz, CH2–Ar), 4.27 (td, 2H, J 13.0, 5.0 Hz, CH2–N), 3.64 (td, 2H, J 13.0, 3.5 Hz, CH2–N), 3.48–3.24 (m, 10H, CH2–N), 2.98 (d, 4H, J 13.0 Hz, CH2–N), 2.85–2.80 (m, 2H, CH2–β–N), 2.37 (s, 6H, CH3), 1.80 (br d, 2H, J 15.0 Hz, CH2–β–N).
4,11-bis(4-methylbenzyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane 1
To a stirred solution of 4 (201 mg, 339 μmol) in ethanol (30 mL) was added slowly sodium borohydride (460 mg, 12.2 mmol), and the mixture was stirred for 30 min at room temperature, then heated under reflux for 2 h. After cooling to room temperature, water (10 mL) was added to decompose excess NaBH4, and the solvents were removed. Water (30 mL) was added to the residue, the solution was made basic (KOH, pH 14), and extracted with dichloromethane (4 × 30 mL). The combined organic extracts were dried, and the solvent was removed to yield a white solid (139 mg, 94%). 1H-NMR (CDCl3): δ 7.19–7.04 (m, 8H, H(Ar)), 3.93–3.87 (m, 1H, CH2–N), 3.71–3.61 (m, 5H, CH2–Ar x 4H, CH2–N x 1H), 3.23–3.04 (m, 5H, CH2–N), 2.93–2.71 (m, 7H, CH2–N), 2.45–2.26 (m, 6H, CH2–N), 2.30 (s, 6H, CH3), 1.68–1.22 (m, 4H, CH2–β–N). 13C-NMR (CDCl3): δ 137.4 (C(Ar)–CH2), 132.5 (C(Ar)–CH3), [129.7, 129.1] (C(Ar)), 57.7 (CH2–Ar), [57.5, 54.2, 53.3, 51.9, 50.3] (CH2–N), 24.1 (CH3), 21.0 (CH2–β–N). MS, m/z: 435 [M+H]+.
Mixture of 4-(3,5-dibromobenzyl)-1,4,8,11-tetraazabicyclo[10.2.2.]hexadecane and 4,11-bis(4,6-dibromobenzyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane 2
To a stirred solution of 3 (89.7 mg, 403 μmol) in acetonitrile (20 mL) was added 3,5-dibromobenzyl bromide (379 mg, 1.15 mmol) and the solution was stirred at room temperature for 16 h. Solvent was removed and the residue was washed with diethyl ether (4 × 30 mL) to yield a white solid (136 mg) (5), which was not isolated. To a stirred solution of this white solid (126 mg) in ethanol (30 mL) was added slowly sodium borohydride (136 mg, 3.59 mmol), and the mixture was stirred for 30 min at room temperature, then heated under reflux for 2 h. After cooling to room temperature, water (10 mL) was added to decompose the excess NaBH4, and the solvents were removed. Water (30 mL) was added to the residue, the solution was made basic (KOH, pH 14), and extracted with dichloromethane (4 × 30 mL). Combined organic extracts were dried, and solvent was removed to yield a colourless oil (86.4 mg of the product mixture, 38% bis-substituted based on NMR). 1H-NMR (CDCl3): δ 7.58–7.56 (m), 7.51 (m), 7.41 (d, J 2.0 Hz), 7.38 (d, J 2.0 Hz), 3.54 (s), 3.29–3.12 (m), 2.91–2.88 (m), 2.74–2.68 (m), 2.60–2.47 (m), 2.43–2.37 (m), 2.28–2.25 (m), 1.88–1.86 (m), 1.71–1.67 (m), 1.59–1.20 (m). 13C-NMR (CDCl3): δ 143.6, 142.5, 132.7, 131.2, 130.5, 130.4, 122.8, 122.5, 59.1, 58.6, 56.3, 56.2, 55.7, 55.5, 55.5, 53.9, 53.0, 50.7, 50.2, 49.3, 49.3, 48.4, 48.1, 48.1, 25.0, 23.5, 22.0.
[Cu1(NO3)]NO3
To a stirred solution of 1 (49.7 mg, 114 μmol) in methanol (10 mL) was added dropwise a solution of copper(II) nitrate trihydrate (36.1 mg, 149 μmol) in methanol (5 mL). The blue solution was heated under reflux for 2 h. The solvent was removed, and the residue was purified by size exclusion chromatography (methanol) to yield a blue solid (62.9 mg, 89%). MS, m/z: 497 [M]+. HRMS: calcd for C28H42N4Cu1: 497.2700; found 497.2703. The single crystal X-ray structure of [Cu1NO3]NO3 was obtained. The crystals were grown by vapour diffusion of diethyl ether at room temperature into solutions of [Cu1(NO3)]NO3 (3 mg) in methanol (0.5 mL) over a period of ca. one week.
[Cu2Cl2]
To a stirred solution of a mixture containing ca. 40% 2 (39.9 mg) in methanol (10 mL) was added dropwise a solution of copper(II) chloride dihydrate (14.4 mg, 84.5 μmol) in methanol (5 mL). The blue solution was heated under reflux for 2 h. The solvent was removed, and the residue was purified by size exclusion chromatography (methanol) to yield a green solid (42.2 mg). MS, m/z: 572 [M–CH2C6H3Br2+Cl]+. The single crystal X-ray structure of [Cu2Cl2] was obtained. Crystal growth experiments yielded a small amount of pure [Cu2Cl2] and the crystals were grown by vapour diffusion of diethyl ether at room temperature into a solution of a mixture containing [Cu2Cl2] (5 mg) in methanol (0.5 mL) over a period of ca. one week.
Acknowledgements
We acknowledge the EPSRC for funds, which enabled the purchase of the Stoe IPDS-II diffractometer. We thank the University of Hull for the provision of a scholarship for BPB and the EPSRC for the provision of a scholarship for JDS. We acknowledge the use of the EPSRC's National Mass Spectrometry Service at Swansea, and Chemical Database Service at Daresbury [52].