

1 Introduction
Metal-binding peptides are interesting candidates to model metalloprotein active sites and biological metal transporters or to design metal-based probes. In particular, the application of the unique magnetic and spectroscopic properties of trivalent lanthanide ions (Ln3+) [1–3] to magnetic resonance imaging [4,5] and optical imaging of cells [6–8] is of rising interest. The lanthanide series offers a variety of metal ions with diverse physical properties: for instance Eu3+ and Tb3+ complexes find applications as luminescent sensors because of their long-lived luminescence and Gd3+ complexes are commonly used as magnetic resonance imaging contrast agents, thanks to the high spin value and long electronic relaxation time of the Gd3+ ion.
Therefore, Ln-binding peptides have been investigated by several research groups to benefit both from Ln-based spectroscopic properties and the biological peptide scaffold. Indeed, the characteristics of the peptide scaffold can give rise to interesting features, such as high solubility in water, versatile insertion in proteins and most importantly selective recognition of biological partners. Ln-complexing peptides or proteins have been elaborated de novo or starting from naturally occurring Ca2+ binding loops as Ca2+ and Ln3+ have similar ionic radii. For instance, Ln-binding tags developed by Imperiali et al. are short peptides optimized for tight Ln3+ binding, i.e. nanomolar affinity and water exclusion from the coordination sphere in order to obtain efficient sensitized terbium luminescence [9]. An asset of Ln-binding peptides is their versatile introduction into protein sequences either by peptide synthesis or bioengineering, two methods to append a probe for various applications.
However, for either in vitro or in vivo applications, these short Ln-binding sequences are inserted in more complex architectures for targeting or detection, and therefore the affinity for the lanthanide ion must be sufficient to avoid release of the cation and its coordination in other metal binding-sites such as Ca2+-binding EF motifs. Therefore, the Ln3+ complex stability is a key-parameter in the design of bioprobes [7,10]. Lanthanide ions possess characteristic 4fn open-shell configuration and the 4f orbitals are considered as core orbitals, which are little involved in covalent interactions upon chemical bonding [1,3]. Consequently, Ln3+ behave as hard Lewis acids in the Hard and Soft Acids and Bases Theory [11] and bind most strongly to hard bases such as oxygen. They display large and variable coordination numbers – from 8 to 12 – with coordination geometries mostly imposed by steric hindrance. Factors controlling Ln3+ complexation have been extensively studied, in particular with multidentate ligands of the polyaminocarboxylate family, which carry large numbers of donor atoms (N and O) close or equal to the high coordination numbers of Ln3+ [12–14]. For instance, the acyclic diethylenetriamine-N,N′,N′′-pentaacetic acid (DTPA) or the macrocyclic 1,4,7,10-tetraazacyclododecane- N,N′,N′′,N′′′-tetraacetic acid (DOTA) are both octadentate ligands with very high affinities for Ln3+ (K > 1022) [15]. Chelate ligands with exclusively oxygen donors may also demonstrate very large affinities as shown by Raymond et al., who developed polydentate molecules containing 2-hydroxyisophthalamide or 1-hydroxypyridin-2-one [16–18].
In this context, the above-mentioned peptides show an intrinsic limitation of the stability of their Ln3+ complexes if only natural amino acids are used. Indeed, the latter bear only simple oxygen ligands with a moderate affinity for Ln3+ in comparison to synthetic multidentate ligands such as polyaminocarboxylates. Therefore, the design of high affinity Ln-binding peptides is a challenging question, which we have chosen to address by using unnatural amino acids carrying high affinity chelating groups. Recently, we developed short peptide sequences containing two unnatural amino acids bearing tridentate, tetradentate or pentadentate polyaminopolycarboxylate chelating groups [19–22]. This strategy allowed us to obtain Ln-binding peptides of significantly enhanced affinity for lanthanide ions in comparison with peptides derived from natural amino acids.
In this short review, we will first present Ln-binding peptides based on natural amino acids and then introduce approaches using unnatural amino acids to modulate their properties such as luminescence or affinity, two relevant properties for the design of bioprobes.
2 Peptides built with natural amino acids
Trivalent Ln3+ ions have ionic radii ranging from 1.216 Å (La3+) to 1.032 Å (Lu3+) for a coordination number of nine, which are very similar to that of the calcium Ca2+ ion (1.18 Å), [23] present at significant concentration in vivo (up to ∼ 1 mM). Therefore, Ln3+ can substitute Ca2+ in proteins since they show a larger affinity for Ca2+ natural binding sites than Ca2+ itself, due to their higher effective charge [24]. Furthermore, Eu3+ and Tb3+ ions can be detected by luminescence if some aromatic protein residues such as tryptophan or tyrosine are present, thanks to the antenna effect (Fig. 1). Indeed Ln3+ ions are poorly luminescent because their direct absorption is very weak since 4f transitions are parity-forbidden. In order to circumvent the low extinction coefficients of Ln3+, a chromophore-containing group can be used as an antenna, which efficiently absorbs incident light and then transfers this excitation to the metal ion, which can then deactivate by undergoing its typical luminescent emission.
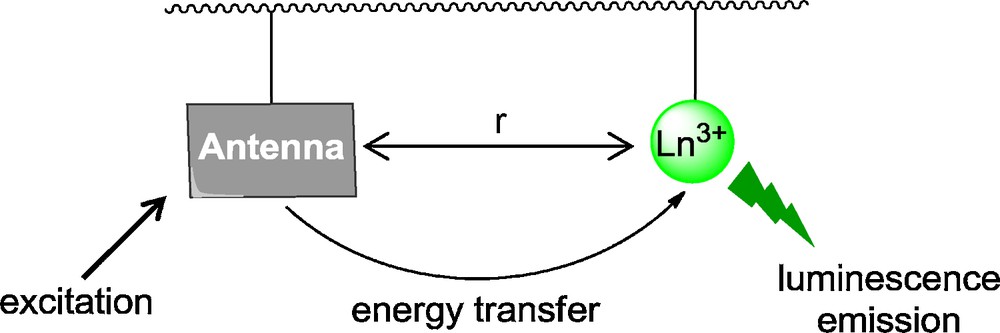
Illustration of the antenna effect.
Hence, Eu3+ and Tb3+ ions, which are emitting in the visible range, have been used to probe divalent metal ion binding sites in proteins [25] such as parvalbumin, [26,27] thermolysin, [28] or glutamine synthetase [29]. Distances (r in Fig. 1) between intrinsic protein fluorophores – i.e. the indole group of tryptophan or the phenol group of tyrosine – and bound metal ions could be measured thanks to Ln3+ luminescence. These natural Ca2+ binding sites, which show a significant affinity for Ln3+ ions were the starting point for the design of Ln-binding peptides.
2.1 Ln-binding peptides inspired from Ca2+ binding loops
Common Ca2+ binding loops in proteins such as calmodulin have 12 amino acids and the metal ion is coordinated by the O-donors of natural amino acid side-chains such as carboxylates of aspartates (Asp) or glutamates (Glu), phenolates of tyrosines (Tyr), amides of asparagines (Asn) and glutamines (Gln) or amides of the backbone peptide linkages. Small peptides based on these Ca2+ binding loops have been the starting point for the design of Ln3+–peptide complexes [30–32]. Szabo et al. designed 14-mer synthetic peptide loops from proteins of the calmodulin family to complex Tb3+ [33,34]. The consensus sequence DKDGDGYIDFEE was obtained by examination of more than 200 species variant loops. It was demonstrated that peptides bearing a too large number of negatively charged amino acid may induce electrostatic repulsion and decrease the affinity for the metal ion. Therefore, one charged Asp (D) residue was replaced by a neutral Asn (N) residue (in position 3) to give the peptide PN, which shows a micromolar affinity for La3+ (Table 1) [32]. Aromatic residues such as tryptophan or tyrosine were introduced in the binding loop to benefit from the antenna effect and to sensitize Tb3+ luminescence (Fig. 1). Position 7 for the aromatic Tb3+ sensitizer in the peptide sequence is optimal for luminescence enhancement since the aromatic ring in the closest to the metal ion, only 5 Å [33].
Ln-binding peptides derived from natural amino acids. q is the number of water molecules coordinated to the Ln3+ ion in the 1:1 complex, when available. Kd is the dissociation constant of the 1:1 complex measured at pH 7.
| Name | Sequence | q | Ln3+ | Kd (μM) | Reference |
| PN | Ac-DRNADGYIDAEEL-NH2 | nd | La | 4 | [32] |
| P3W | TERRRQQL−DKDGDGTIDERE−IKIWFQNKRAKIK (Ln-binding domain in bold, DNA-binding domain in italics) | 2 | Eu | 1.2 | [41] |
| LBT1 | Ac-GDYNKDGWYEELEL | nd | Tb | 8 | [47] |
| LBT | YIDTNNDGWYEGDELLA | 0 | Tb | 0.057 | [45,46] |
| LBTSS | nd | Tb | 0.002 | [45] | |
| pY125 | Ac-DPDNEApYEMPSEEG | nd | Tb | 0.31 | [53] |
| LF4 | AYTDDPSDTFSTPEQLAKELLQEHGI | nd | Eu | 28 | [55] |
| PA | c(DREPGEWDPG) | 2 | Eu | 0.16 | [57] |
Since then, many short peptide sequences derived from Ca2+ binding proteins have been studied [35–39]. Among them, chimeric peptides such as P3W (Table 1), which have a transcription factor helix-turn-helix (HTH) DNA-binding domain and a topologically equivalent Ca-binding EF-hand motif, may be considered as artificial endonucleases [40]. P3W was shown to bind Ln3+ ions with a micromolar affinity [41] and to promote DNA hydrolysis thanks to the presence of the metal ion [42,43]. Moreover, the Gd3+ complex of P3W showed a good MRI contrast efficiency, named relaxivity, which is even highly enhanced when the complex binds to DNA due to the larger molecular weight of the peptide-DNA adduct [44].
All Ln-binding peptides derived from Ca2+ binding loops demonstrated micromolar affinity for Ln3+. This is why these short peptide sequences and structures needed further optimization, specifically for intense luminescence and higher binding affinity.
2.2 Optimized Ln-binding tags
Ln-binding tags made up entirely of natural and encodable amino acids have been optimized for Tb3+ binding and luminescence by Imperiali et al. [45,46]. Prototype sequences were based on calcium-binding motifs of EF-hand proteins, which provide six coordinating residues [47]. Libraries of peptides were designed starting from the 14-mer LBT1 (Table 1) by varying C-terminal ligating residues, adding flanking hydrophobic residues and randomizing sensitizing residue and non-coordinating residue position [45]. The linear sequence of LBT (Table 1) was then selected by screening luminescent Tb3+ complexes. It demonstrates an affinity for Tb3+ in the nanomolar range, which represents a significant stabilization with respect to the starting prototype LBT1 [46]. The crystallographic structure of the Tb3+ complex of LBT reveals that Tb3+ is bound in an eight-coordinate complex without any inner-sphere water molecule, which leads to intense luminescence properties of the latter ion. Besides introduction of a disulfide bond in optimal positions in the cyclic peptide, LBTSS (Table 1) provides an impressive 30-fold increase in Tb3+ affinity compared to the best linear sequence of LBT. The expression of LBT in proteins has proven its efficiency for the determination of protein concentration by luminescence, [47] the structural resolution of biomolecules by solution paramagnetic NMR or X-ray diffraction [48–50] and the study of protein interactions thanks to luminescence resonance energy transfer [51]. More recently, this sequence could also be introduced into loop regions of proteins without significantly changing the protein structure [52].
2.3 Phosphorylated peptides
The influence of peptide's phosphorylation on Ln3+ complexation was studied for α-synuclein (α-syn) fragment 119-132 because its sequence, in particular the carboxylate groups arrangement, is similar to Ca2+ binding loops [53,54]. The type and location of the phosphorylated amino acid (Tyr or Ser) have a dramatic influence on the metal-binding properties of the peptides. The largest stability constant for 1:1 Tb3+ binding was obtained for the peptide pY125 (Table 1), with a phosphorylated tyrosine. Besides, comparing the phosphorylated peptide with the unphosphorylated version demonstrates that phosphorylation increases the affinity for metal ions.
2.4 Lanthanide-fingers
Other native sequences than Ca2+ binding loops may be modified to generate Ln-binding peptides. A “lanthanide-finger” was recently designed by replacing the cysteine and histidine residues involved in the metal coordination of a compact zinc finger (Cys2His2) by amino acids with O-donors. The cysteines and histidines were substituted by aspartic acids and glutamic acids, respectively, to generate a tetracarboxylate peptide ligand (Asp2Glu2). LF4 demonstrate the largest affinity for Ln3+ with a micromolar affinity for Eu3+ (Table 1) [55].
2.5 De novo designed peptides
Another approach consists in preorganizing the side-chains of coordinating amino acids in de novo designed peptides to afford well-defined Ln3+ coordination sites. This methodology was applied in de novo designed helical coiled coils: metal ion binding sites namely glutamates were incorporated into heptad positions which commonly contain hydrophilic side-chains and binding of La3+ was demonstrated to enhance the stability of the overall supramolecular peptide assembly, even though the binding of La3+ appeared to be weak (in the mM range) [56].
More recently, the cyclodecapeptide PA (Table 1) was tested as a novel peptide architecture to coordinate Ln3+. Indeed PA is particularly adapted to metal coordination since its β-sheet structure allows the preorientation of four amino acid side-chains of glutamic or aspartic acids to coordinate the Ln3+ ion. The solution NMR structure of the free peptide demonstrates that the four carboxylic acid groups are oriented in the same half-space of the β-sheet peptide scaffold, which is perfectly suitable to coordinate metal ions [57]. A mononuclear Ln3+ complex with two water molecule in the Ln3+ coordination sphere is specifically formed with an affinity constant in the same range or even larger than EF-hand motifs, emphasizing the relevance of this de novo approach using cyclic templates. Interestingly, the corresponding Gd3+ complex combines the relaxation properties of Gd3+ with the high hydrophilicity of the peptide scaffold to provide large second-sphere contributions to water relaxation [57,58].
Peptide sequences built exclusively from natural amino acids exhibit affinities for Ln3+ in the micromolar range or in the low nanomolar range for the best Ln-binding tags, which structure is perfectly suited to Ln3+ coordination. Indeed, these natural residues bind metal ions with carboxylates, phenolates, amides or phosphates, which show a moderate affinity for Ln3+ ions. Therefore, all these peptides with O-donors have a modest affinity for lanthanide ions in comparison with synthetic multidentate ligands such as polyaminocarboxylates [15]. As mentioned before, the stability of the lanthanide complex is a key-parameter for the design of bioprobes in particular to avoid release of the cation and its coordination in other metal binding-sites. Therefore, non standard amino acids are increasingly used for the design of peptide ligands to introduce strong donor groups for metal complexation or to improve the sensitization of metal ion emission.
3 Unnatural amino acids to better Ln-binding peptide's properties
3.1 Unnatural amino acids for Ln3+ sensitization
Time-resolved luminescence of lanthanides is particularly attractive for applications of these ions as luminescent bioprobes, since it allows to eliminate the background natural fluorescence. The aromatic groups of tyrosine and tryptophan (Fig. 2) are Tb3+ sensitizers as exemplified by lanthanide–peptide complexes presented in the previous section. The overall quantum yield, , is a key-parameter since it characterizes the luminescence efficiency of the complex: it is equal to the ratio between the number of emitted photons divided by the number of absorbed photons. Two important factors contribute to the overall quantum yield: the metal-centered light emission or intrinsic quantum yield, , and the sensitization efficiency, (Eq. 1) [59–62].
| (1) |

Ln3+ sensitizing natural and non natural amino acids.
The intrinsic quantum yield, , reflects the extent of non-radiative deactivation processes, which compete with luminescence of the lanthanide ion. These deactivation processes may occur both in the inner- and outer-coordination sphere of the metal ion and are for instance vibration-induced processes or photo-induced electron transfer deactivations. Vibrations of bound ligands such as OH, NH or CH are particularly efficient non-radiative deactivation processes. Hence, to get efficient Ln3+ emission, water molecules should be excluded from the coordination sphere to get low hydration numbers, which may be evaluated from Ln3+ luminescence lifetimes in light water and heavy water according to empirical equations derived from series of complexes [63–65].
The sensitization efficiency is highly dependent on the energy gap between the sensitizer excited state (donor) and the Ln3+ emitting level (acceptor). It is generally considered that a difference of 2500–3500 cm−1 between these two energy levels is ideal to optimize the energy transfer and minimize potential back-transfer [10].
Another key-parameter is the donor-acceptor distance, with a 1/r6 relationship for a dipole-dipole mechanism [26,29,66]. The distance between the Trp indole sensitizer and the accepting Ln3+ ion measured in the X-ray structure of the Tb3+-LBT complex is 7 Å [46]. Since the aromatic indole group is not directly coordinated to the metal, the sensitizer to metal distance is quite long and the energy transfer is clearly a limiting parameter in the quantum yields of these complexes. But, even though the energy transfer from Trp to Tb3+ is not optimum, the latter ion is significantly sensitized upon binding to the peptides, which Tb3+ complexes may be detected in the low nanomolar range.
The sensitization of Eu3+ by Trp and Tyr is far less efficient because of photo-induced electron transfer, which highly contributes with this readily reduced Ln3+ ion [27,67]. This mechanism quenches the aromatic group fluorescence without sensitizing the red-emitting Eu3+ ion.
To expand the potential applications of Ln-binding peptides, in particular to foresee their in vivo use, artificial chromophores (Fig. 2) with higher excitation wavelengths were introduced in Ln-binding tags [68] or in peptide sequences derived from the parvalbumin Ca2+ binding loop [38]. Carbostyril 124 (cs124) was demonstrated to sensitize Tb3+ luminescence upon excitation at 337 nm, with a better efficiency than Trp, without affecting the affinity and structure of the complex [68]. Moreover, whereas Trp-containing peptides are not able to sensitize Eu3+, the two lower energy chromophores acridone (Acd) [68] and naphthalimide (Naph) [38] introduced in short peptide sequences were successfully used to populate the Eu3+ excited state.
Unnatural amino acids may also be used to increase the peptide's affinity for Ln3+ ions as illustrated in the next sections.
3.2 Unnatural amino acids to enhance peptide affinity for Ln3+ ions
Some metal ions such as Zn2+, Cu+, Cu2+, Pb2+ or Hg2+ display large affinities for natural binding groups in proteins, such as the soft sulfur donors of cysteines or methionines or the borderline nitrogen donor of histidines. Therefore, efficient binding peptides of these metal ions can readily be obtained by incorporating cysteines, histidines or methionines in the sequences. Some of these metal-binding peptides are inspired from natural binding sites like metal transporters [69–77] or zinc fingers, [78–80] others are de novo designed highly-organized peptide architectures such as triple coiled coil assemblies [81,82] or pseudopeptides with a chemical scaffold to append several peptide units [83–86].
On the contrary, hard Ln3+ cations do not show such large affinities for natural binding sites as illustrated in Section 1. Therefore, high affinity Ln-binding groups were appended to synthetic unnatural amino acids to obtain more stable Ln-binding peptides. γ-carboxyglutamic acid (Gla), which contains two carboxylate substituents on the γ carbon (the side chain is a malonate type group, Fig. 3), was introduced in peptide chains to amplify the effects of interhelical repulsions and metal-binding on coiled coil structures. Whereas the apo form is destabilized by strong electrostatic repulsions, the folded form is stabilized by Ln3+ binding. Nevertheless, despite the presence of several bidentate Gla amino acids, the Ln3+ complex stability remains in the micromolar range [87].
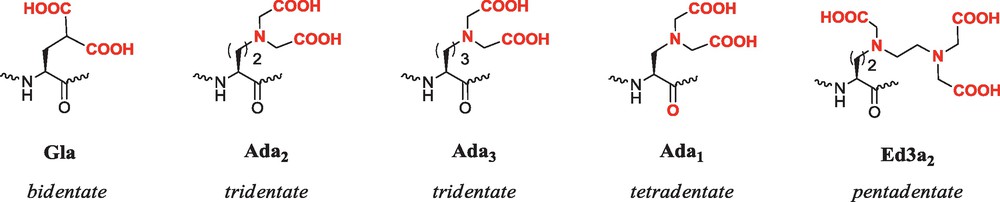
Unnatural chelating amino acids (coordinating groups are represented in bold red).
This led us to consider using synthetic amino acids with a larger affinity for Ln3+, i.e. with appended polydentate chelating groups of the polyaminocarboxylate family [4]. A related strategy is to functionalize the peptide or the protein with a Ln3+ chelate to get peptide/protein conjugates [88–91]. The latter benefit simultaneously from the stability of the Ln3+ chelates and from peptide-based properties such as biomolecular recognition. In these conjugates, the binding site is appended at the periphery of the biomolecule and no efficient coupling between the properties of the chelating appendage and that of the peptide substructure is thus expected.
On the contrary, we chose to incorporate the Ln3+ binding site embedded in the peptide structure in order to intimately couple the metal ion and the peptide scaffold properties. Among metal-chelating amino acid side-chains, aminodiacetate groups have first attracted our attention [92–94]. The corresponding synthetic residues are referred to as Adan, n being the length of the alkyl chain separating the peptide backbone from the aminodiacetate nitrogen (Fig. 3). Ada2 and Ada3 are tridentate ligands, whereas the shortest analogue Ada1 may act as a tetradentate donor as the coordination of the backbone carbonyl is favored due to the formation of a six-membered chelate ring. More recently the novel pentadentate amino acid Ed3a2 (Fig. 3), which carries an ethylenediamine triacetate side-chain was synthesized to increase the denticity of the peptides.
The chelating unnatural amino acids Adan and Ed3a2 were synthesized with protecting groups compatible with Fmoc/tBu strategy in solid phase peptide synthesis, i.e. in the form Fmoc-aa(tBu)x-OH. The latter display normal reactivity in the on-resin synthetic steps, which allowed us to synthesize hexapeptides in the 50-mg scale with good yields.
4 Efficient Ln-binding peptides with two synthetic chelating amino acids
4.1 Design of hexadentate peptides
To design peptides with a Ln-ligating site embedded in the peptide structure, we first inserted two Adan unnatural amino acids in the sequence to get potentially hexadentate ligands. However, the mere inclusion of chelating amino acids in an arbitrary peptide sequence is insufficient to achieve highly stable mononuclear complexes because the chelating side-chains would show independent behavior. To overcome this difficulty, the peptide backbone was used as a non-innocent spacer between the two chelating groups and chosen to favor a β-turn containing structure. In such a turn, a hydrogen bond is usually present and residues in position i and i+3 are brought in close proximity, which can be used to bring closer the two aminodiacetate side-chains in a disposition suitable for metal chelation. The XProGlyX sequence was chosen as a turn inducer between the two Adan residues. Indeed, being cyclic, l-proline is unique among amino acids to promote changes in protein backbone direction and to restrict the conformational space accessible to the peptide. In contrast, glycine possesses enough conformational freedom to comply with the backbone dihedral angle requirements of the type II β-turn structure and also provides sufficient conformational elasticity for efficient metal coordination. Finally, even though this sequence is not usually considered as a strong turn inducer in linear peptides, it is frequently encountered in type II β-turns in proteins [95].
The two hexapeptides P22 and P33 that incorporate two Adan chelating amino acids were first designed with a ProGly peptide spacer (Fig. 4 and Table 2). A tryptophan residue was also inserted in the sequence to benefit from the antenna effect to sensitize Tb3+ luminescence. The comparison of the Ln3+ complexation properties of the two latter peptides evidences a dramatic effect of the Adan side-chain length [19]. Indeed, the most flexible peptide P33 with propyl spacers between the peptide backbone and the chelating aminodiacetate groups forms a mononuclear hexadentate complex of moderate stability, in which the two chelating moieties behave as nearly independent subunits. This also leads to the formation of polymetallic species in excess of metal ion and therefore to uncontrolled speciation of metal complexes. On the contrary, the more compact peptide P22 acts as an efficient hexadentate ligand of Ln3+ ions with picomolar affinity in water, which does not give any polymetallic species. The TbP22 complex shows a significant stabilization of two orders of magnitude, in comparison with ligands bearing independent aminodiacetate moieties, even though the two coordinating nitrogen atoms are separated by 14 atoms. This substantial stabilization is assigned to peptide secondary structure elements evidenced in the solution NMR structure of LaP22: an expected type II β-turn with a well-defined H-bond and an amazing hydrophobic Trp(indole)–Pro interaction. Therefore, P22 demonstrates that the peptide backbone can be used as a non-innocent scaffold with a defined conformation and therefore as an asset to the Ln3+ complex stability.
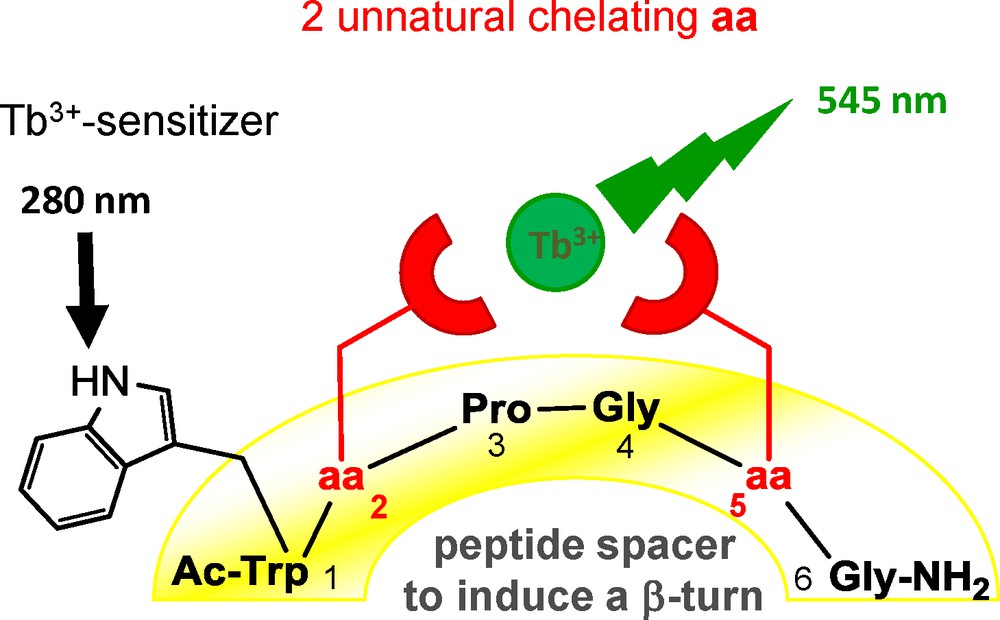
Peptide sequences incorporating two synthetic amino acids.
Ln-binding peptides derived from unnatural amino acids. q is the number of water molecules coordinated to the Ln3+ ion in the 1:1 complex. KdpH7 and Kd are the dissociation constant of the Tb3+ 1:1 complex measured at pH 7 and independent on the pH, respectively. The formation of polymetallic complexes in excess of metal ion is also indicated.
| Name | Sequence | q | KdpH7 (M) | Kd (M) | Polymetallic | Reference |
| P22 | Ac-W Ada2 PG Ada2 G-NH2 | 3 | 10−9.1 | 10−12.1 | No | [19] |
| P33 | Ac-W Ada3 PG Ada3 G-NH2 | 3 | 10−5.4 | 10−9.9 | Yes | [19] |
| P11 | Ac-W Ada1 PG Ada1 G-NH2 | 0-1 | 10−10.3 | 10−10.8 | Yes | [20,22] |
| P12 | Ac-W Ada1 PG Ada2 G-NH2 | 0 | 10−9.5 | 10−11.0 | No | [22] |
| P21 | Ac-W Ada2 PG Ada1 G-NH2 | 3 | 10−9.0 | 10−10.5 | Yes | [22] |
| PHD2 | Ac-W Ed3a2 PG Ada2 G-NH2 | 0 | 10−12.7 | 10−16.2 | No | [21] |
| PHD5 | Ac-W Ada2 PG Ed3a2 G-NH2 | 0 | 10−12.7 | 10−16.2 | Yes | [21] |
| PHD’ | Ac-S Ed3a2 GW Ada2 A-NH2 | 0-1 | 10−11.9 | 10−15.4 | Yes | This work |
4.2 Higher denticity hexapeptides
Higher denticity Ln-binding peptides were designed in order to increase the complex stability and also to dehydrate the Ln3+ ion so as to enhance the luminescence properties by decreasing the vibrational non-radiative deactivations from water molecules O–H. To further enhance the denticity of Ln-binding peptides, we used the shorter side-chain analogue Ada1 in peptides P11, P12 and P21 (Table 2). Indeed, Ada1 may act as a tetradentate donor thanks to the coordination of its backbone carbonyl in a six-membered chelate ring. This attempt was successful in decreasing the hydration state of the Ln3+ ion (qTb = 0 in TbP12 and q = 0-1 in TbP11) but did not lead to greater stability of the Ln3+ complexes (Table 2) [20,22]. Even though the carbonyl function of Ada1 in position 2 was involved in the metal coordination by the two peptides P12 and P11, no significant stabilization was observed due to this additional donor (Kd in Table 2). Indeed, supplementary carbonyl binding groups are neutral donors, which do not provide significant stabilization of Ln3+ complexes in water, as evidenced before with neutral tripodal N,O ligands [96]. But, interestingly Ada1 is less basic than Ada2, thanks to the withdrawing effect of the peptide backbone, which is greater via the short Ada1 side-chain; this decreases the overall basicity of Ada1-based peptides and reverses the order of the complexes stabilities at pH 7 (KdpH7, Table 2).
To get a major effect on both the Ln3+ ion dehydration and the complex stability, the novel chelating amino acid Ed3a2, with a pentadentate polyaminocarboxylate side-chain and thus an extra-aminoacetate group, was inserted in place of one of the two tridentate residues in P22 [21]. The two hexapeptides PHD2 and PHD5 (Table 2) are only differing in the position of the pentadentate residue and are able to bind Ln3+ in an octadentate coordination mode. The mononuclear Tb3+ complexes demonstrate low femtomolar stability in water, i.e. a gain of four orders of magnitude with respect to the first generation hexapeptide P22. The octadentate coordination of Tb3+ by PHD2 and PHD5 leads to total dehydration of the metal ion in the mononuclear complexes with long luminescence lifetimes (> 2 ms). This results in an improvement of the luminescence properties with respect to the trisaquo TbP22 complex of a factor 4.
The position of the higher denticity amino acid Ada1 or Ed3a2 in the hexapeptide sequence appears to be critical for the control of the metal complex speciation. Indeed, the peptides PHD2 and PHD5 or P12 and P21 only differ by the positions of the two chelating amino acids with respect to the Pro-Gly spacer and show different behaviors regarding speciation. Whereas PHD5 and P21 promote the formation of polymetallic species in excess of Ln3+, PHD2 and P12 form exclusively the mononuclear complex. Hence, it appears that positioning the higher denticity amino acid, either Ada1 or Ed3a2 in position 5 after the Pro-Gly spacer favors an independent coordination of the two chelating moieties. Therefore, to control speciation and stabilize the higher coordination mode that involves the two chelating arms of the hexapeptides, the higher denticity amino acid has to be introduced in position 2, prior to the cyclic proline residue, which probably constrains more efficiently the peptide backbone. The position of the two chelating amino acids with respect to the Pro-Gly spacer is thus a key-parameter for the control of Ln3+ complexes speciation.
4.3 How does the sequence affect the structure of the complexes?
The influence of the sequence onto the complex's structure could be investigated by solution NMR for two complexes. Indeed, LaP22 and LuP11 showed numerous nuclear Overhauser correlations in their NOESY spectra, which allowed their solution structure's calculation. The difference in the Ln3+ coordination mode by P22, which is hexadentate and P11, which is heptadentate, modify deeply the structures of the peptides in their complexes (Fig. 5).
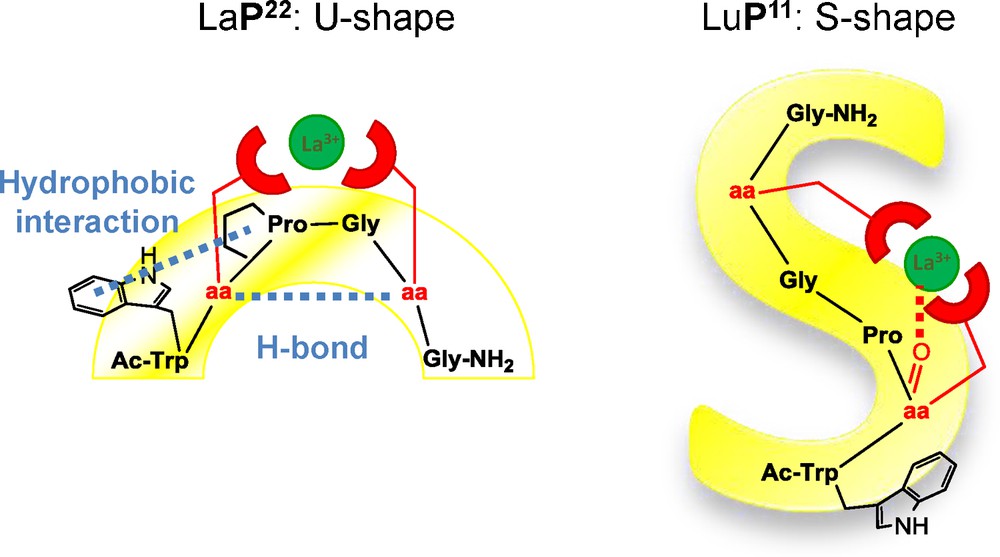
Structures of LaP22 and LuP11 from the solution NMR data.
In LaP22, the Ada2PGAda2 motif forms a type II β-turn showing a perfect H-bond between the carbonyl of Ada22 and the NH group of Ada25 and a hydrophobic interaction between the aromatic group of the tryptophan residue and the aliphatic proline ring. The peptide backbone adopts a U-shape structure with an “upper face” devoted to La3+ coordination and a “lower face” where the hydrophobic interaction between the Trp indole group and the proline cycle takes place. The La3+ ion is located 8.7 Å from the center of the sentizing indole group [19]. These two structural stabilization elements due to the peptide scaffold are responsible for the high stability of LnP22 complexes as mentioned in Section 3.1.
On the contrary, LuP11 revealed an extended S-shape structure due to the extra coordination of the amide carbonyl of Ada1(2), which prevents the formation of the β-turn and drives the Ln3+ ion in the proline plane and closer to the Trp indole sensitizer (5.7 Å) [20].
Although no structure could be calculated for the other complexes, their photophysical data give insights into the backbone arrangements and the metal coordination. For instance, LnP21 and LnP22 complexes point to similar structures with a triply hydrated Ln3+ ion and a U-shape conformation of the peptide backbone. Indeed, both Tb3+ complexes show a metal hydration state of 3 and an increase of Trp fluorescence upon Tb binding, which indicate similar Trp environment in the Ln3+ complexes. On the contrary, LnP12 and LnP11 complexes exhibit dehydrated Ln3+ ions, a decrease of Trp fluorescence upon Tb binding and short Eu3+ luminescence lifetimes in D2O, indicative of deactivation by CH groups of the ligand. All these characteristics allow us to conclude to the coordination of the carbonyl group of Ada1(2) inducing a S-shape conformation of the peptide backbone, as evidenced previously in the NMR structure of LuP11 [22].
Even though P11 and P12 are heptadentate Ln3+ ligands due to the extra-coordination of the backbone carbonyl of Ada1(2), they do not benefit from the secondary structure elements found in the U-shape structure of LnP22. Therefore, TbP11 and TbP12 complexes are less stable than TbP22 (Kd in Table 2).
4.4 Role of the spacer between the two chelating amino acids
Finally, the role of the Pro-Gly spacer was studied by designing a high denticity peptide similar to the most efficient compound PHD2, but with a Gly-Trp spacer, which cannot induce a turn in the peptide backbone (PHD’ in Table 2). The synthesis and complexing properties of PHD’ are reported in the supplementary material. A mononuclear complex TbPHD’ of significantly lower stability than TbPHD2 is formed. In addition, PHD’ gives polymetallic complexes in excess of metal. Whereas PHD2 and PHD5 are able to promote the total dehydration of Tb3+ ions in the octadentate complexes (q = 0), coordination by PHD’ leave some access of water to the metal center (q = 0.5).
These data evidence the crucial role of the turn inducing effect of the Pro-Gly spacer in the stabilization of the mononuclear complex with simultaneous coordination of the two chelating amino acid side chains and in promoting a perfect Tb3+ octadentate coordination with no access of water to the metal center.
5 Conclusion
Lanthanide-binding peptides have already proven their usefulness in the characterization of biomolecules. In particular, Ln-binding tags developed by B. Imperiali et al., based on natural amino acids, were inserted in proteins by bioengineering to infer into their structures and functions. Our contribution to the field is the design of peptides with higher affinities for Ln3+ thanks to the use of non natural chelating amino acids, which are rationally arranged in short peptide sequences. We focused on polyaminocarboxylate side chains as they are known to efficiently chelate Ln3+ ions. Synthetic peptides with two unnatural chelating residues were obtained by conventional Fmoc/tBu solid phase peptide synthesis in the 50-mg scale starting from the protected unnatural amino acids, which syntheses were optimized in the laboratory.
We demonstrated that a subtle interplay between the basicity, the compactness of the ligand and the secondary structure of the peptide backbone can tune Ln-binding peptide's properties. Our approach leads to lanthanide-peptide complexes of enhanced stability, with Ln3+ binding sites embedded in the peptide structure in order to intimately couple the properties of the metal ion and the peptide scaffold. Indeed, this coupling was observed in LnP22 complexes, which picomolar stability in water is partly due to typical structural elements of the peptide scaffold.
The design of a novel pentadentate chelating amino acid Ed3a2 and its insertion in P22 sequence in place of one Ada2 was particularly efficient: octadentate Tb3+ complexes with impressive femtomolar stability were formed in water. The octadentate coordination provides totally dehydrated complexes with better luminescence properties. The pentadentate amino acid position is demonstrated to be critical: it has to be inserted in position 2, to control the speciation and avoid the formation of polymetallic complexes. The short Ln-binding peptide PHD2 could be inserted in more complex peptide or protein sequences to investigate biologically-relevant functions. In particular, the greater stability of these Ln-peptide complexes will prevent the de-coordination of the Ln3+ ion from the probe when interacting with biological molecules, which bear potential Ln3+-binding sites such as proteins with metal-binding EF hands or DNA sequences.
The synthesis of these novel Ln-binding peptides is straightforward, which will allow their simple insertion in more complex peptide architectures. Current developments are aiming at changing the Ln sensitizing unit and at coupling the most interesting compounds, i.e. P22 and PHD2, with DNA recognition units.
Acknowledgements
We deeply thank Federico Cisnetti who contributed a lot at the beginning of our project on Ln-binding peptides, and Yves Chenavier for his significant contribution for the synthesis of the unnatural amino acids. Laetitia Ancel held a doctoral fellowship from la Région Rhône-Alpes.