

1 Introduction
3a-Hydroxytetrahydropyrrolo[2,3-b]indole is the structural motif characteristic of many compounds of high biological importance, such as notoamide D – obtained from a culture of marine-derived fungus Aspergillus sp. and which demonstrated noticeable cytotoxity on HeLa and L-1210 tumor cells [1] –, gypsetin – derived from dermatophyte Nannizzia gypsea and which confirmed acyl-CoA acyltransferase inhibitory activity [2] – or himastatin – a new anti-tumor antibiotic from Streptomyces hygroscopicus [3]. Among the several possible methods for the synthesis of this system [4], oxidative cyclization of indole derivatives seems to be the simplest one [5]. An example of such reaction is the oxidation of melatonin 1 with singlet oxygen, in the presence of photosensitizers, such as Bengal rose at −78 °C, to produce cyclic 3-hydroxymelatonin 2 [6] (Scheme 1).

Oxidation of melatonin 1 with singlet oxygen.
We have recently demonstrated that compounds possessing a carbamate substituent at position 5 of the 3a-hydroxytetrahydropyrrolo[2,3-b]indole skeleton exhibit a high inhibitory activity against human cholinesterases [7]. This type of compounds represents an interesting synthetic target, poorly explored so far.
It was reported that the oxidation of carbamate melatonin derivatives with singlet oxygen results in the cyclization to 3a-hydroxytetrahydropyrrolo[2,3-b]indole system [7]. Unfortunately, this method turns out to be time-consuming and merely effective due to a low degree of conversion.
2 Results and discussion
We have, therefore, investigated a series of reactions of melatonin 1 and its derivatives with different oxidative reagents, in order to find an easy and more efficient method for the synthesis of the desirable tricyclic system. Unfortunately, we found that the oxidation with H2O2, NaOCl, MnO2, SeO2, CrO3, Oxone®, or Sharpless epoxidating reagent was, depending on the conditions used, either completely ineffective, or resulted in the decomposition of the substrate to unidentifiable products. Attempts to oxidize melatonin 1 with peracids seemed to be the most promising.
In order to obtain the 3a-hydroxytetrahydropyrrolo-[2,3-b]indole skeleton substituted at position 5 with a carbamate group, we subjected 3-[2-(acetylamino)ethyl]-1H-indol-5-yl carbamates to a reaction with performic acid, freshly prepared by adding H2O2 to concentrated formic acid according to the known procedure [8] (Scheme 2).
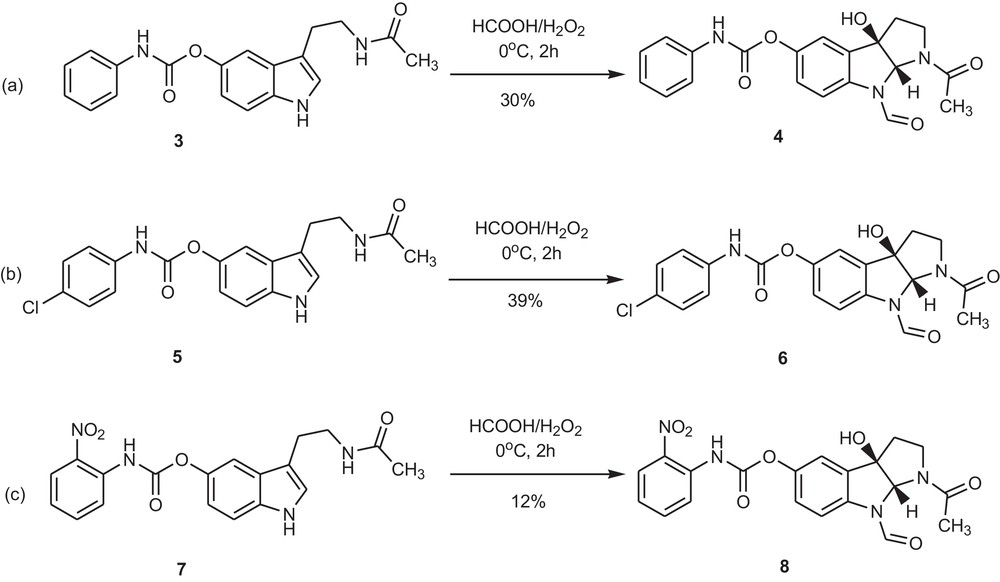
Oxidation of 3-[2-(acetylamino)ethyl]-1H-indol-5-yl carbamates 3, 5 and 7 with performic acid.
We were pleased to observe the fast consumption of the substrate as well as the formation of a single product, easy to isolate. The oxidation of 3-[2-(acetylamino)ethyl]-1H-indol-5-yl phenylcarbamate 3 resulted in the desired tricyclic product 4, with undisturbed carbamate moiety. An additional formulation of the N8 atom occurred, which can be explained by the already documented formulating ability of performic acid [9]. Postulated structure of the product was confirmed by X-ray analysis (Fig. 1).

ORTEP diagram from X-ray analysis of compound 41.
In the case of carbamates 5 and 7, we obtained similar results. The reaction yields were not high but still satisfying in comparison with the methods used so far. This disadvantage was in addition compensated by the simplicity of the reaction and purification procedure and the low cost of the reagents used. The substrates may be easily recovered and recycled. The yields calculated on a basis of the consumed substrates were almost quantitative.
Hoping to have in hand a general method for the synthesis of tricyclic carbamates, we decided to broaden the spectrum of the investigated substituents at the aromatic ring of the phenylcarbamate moiety. Therefore, we used the previously optimized methodology to oxidize o-isopropylphenyl-, p-isopropylphenyl- and o-methoxyphenyl-carbamates (Scheme 3).

Oxidation of 3-[2-(acetylamino)ethyl]-1H-indol-5-yl carbamates 9, 11 and 13 with performic acid.
Similarly to our previous results, we observed the formation of a single product with the yield of around 30%. In the NMR spectra, we identified signals characteristic of the tricyclic system but, surprisingly, no signal from the expected formyl group was present. The mass spectrometry analysis confirmed the absence of the formyl substituent in all the three cases mentioned. For the products possessing p-isopropyl substituents, we succeeded to obtain a monocrystal suitable for X-ray analysis and we were surprised to find out that the carbamate bond hydrolyzed and the released group formed a urea bond with the N8 atom of the tricyclic system (Figs. 2 and 3). Detailed spectral analysis further confirmed the proposed structure of the products 10, 12 and 14.

ORTEP diagram from X-ray analysis of compound 102. For interpretation of colors, see the web version of this article.

ORTEP diagram from X-ray analysis of compound 123. For interpretation of colors, see the web version of this article.
Surprised by the difference in the results obtained for carbamates 3, 5, 7 and 9, 11, 13, we repeated all the reactions several times, changing the concentration of the oxidizing agent and we gained a conviction that it does not affect the type of product. We can then assume that the structure of the product depends only on the substrate used. Unexpected influence of the substituent at the phenyl ring on the reaction course can be attributed to the presence of a conjugated bonds system, which covers a considerable part of the molecule of the substrate and can generate unexpectedly strong long-distance electronic effects. In order to rationalize the differences in the reactivity of various carbamates with performic acid, we initially concentrated on the thermodynamic aspects of this reaction. The complete set of free enthalpy (ΔG0) values for all the reactions leading to the products, both observed and hypothetical (Fig. 4) were calculated using the Spartan 08 software [10]. Ab initio calculations using density functional method at B3LYP level with 6–31 + G* function basis were performed. The structure of the molecules of the products and substrates were optimized and their free enthalpies were calculated. The results are summarized in Table 1.

The structures of the alternative products 15, 16, 17 used for calculations.
Comparison of free energy values for reactions leading to formed and hypothetical products.
| Entry | Substituent | Reaction | ΔG0 [kJ/mol] |
| 1 | p-Cl | G0(6)–G0(5) | –66.8 |
| 2 | p-Cl | G0(15)–G0(5) | –76.0 |
| 3 | o-NO2 | G0(8)–G0(7) | –75.3 |
| 4 | o-NO2 | G0(16)–G0(7) | –57.0 |
| 5 | o-OCH3 | G0(14)–G0(13) | –67.1 |
| 6 | o-OCH3 | G0(17)–G0(13) | –75.2 |
These results of calculations indicate that due to rather minute differences in ΔG0 values, the thermodynamic influence is not significant and apparently the reaction is governed by kinetic factors.
The course of the reaction, leading to unexpected products listed in Scheme 3, might be explained by a process starting with the protonation of the carbamate oxygen atom followed by the formation of the reaction intermediate whose sandwich-like dimeric structure allows the simultaneous transfer of both carbamate units between the molecules and the subsequent formation of the observed product. The example of such reaction sequence for compound 13 is shown in Scheme 4. We are aware that such a process might be highly unfavourable, leading to a dimeric structure of an extremely high energy and the real reaction course is probably a multi-step process but we hoped that our model would at least illustrate the tendency to the appropriate bonds forming/breaking sequence.

The postulated mechanism for the formation of product 14.
We, therefore, compared two alternative processes of the dissociation of the hypothetical dimeric structures: the first one consisting in the dissociation of the C–N bonds (shown in red in Scheme 4) leading to a “normal” product without rearrangement, and the second one consisting in the dissociation of the phenolic C–O bond (marked in blue in Scheme 4) leading to “rearranged” products.
To establish the energy of each process, we performed an ab initio optimization of the structures of the respective dimers (Fig. 5) and the corresponding monomers and then, we compared the energy differences:
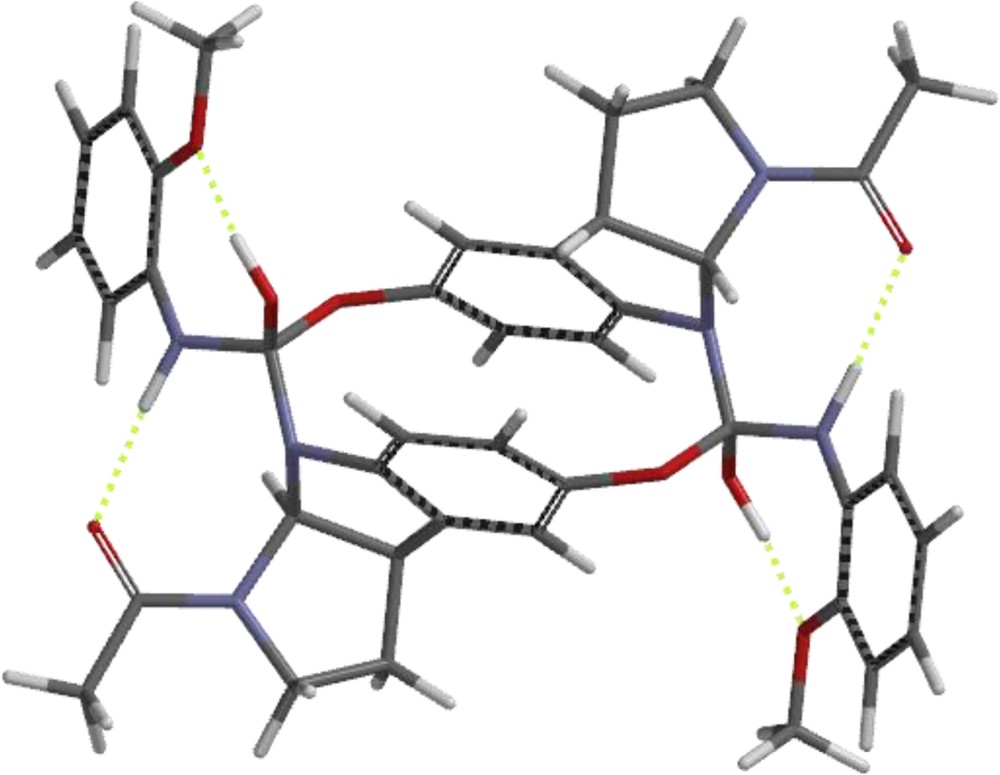
The optimized structure of postulated intermediate 13a. For interpretation of colors, see the web version of this article.
Indeed, very high ΔE values of the processes encompass not only bond energies but also repulsion and deformation energies. The last column in Table 2 denoted as ΔΔE contains the differences between dissociation energies listed in columns 2 and 3. These values show a possible gain in energy when a new C–N bond is formed in a product with a simultaneous dissociation of the existing C–O bond. In all instances where such value is positive, the process is unfavourable whereas negative value corresponds to a net energy gain. The comparison of the energies listed in Table 2 leads to the conclusion that the rearrangement process, i.e. the breaking the C–O bond, is preferred for intermediate 13a, whereas for the two others (5a and 7a), the preferred one is the dissociation or more exactly the non-formation of C–N bonds. These results are in agreement with the experimental observations and seem to confirm a plausible mechanism for the observed rearrangement.
The comparison of energies (in kJ/mol) of alternative dissociation processes.
| Intermediate | ΔE | ΔΔE | |
| C–N dissociation | C–O dissociation | ||
| 5a | –6199.0 | –6252.4 | 53.4 |
| 7a | –6202.2 | –6223.2 | 21.0 |
| 13a | –6358.6 | –6041.8 | –316.8 |
3 Conclusion
In summary, we found that certain melatonin derivatives possessing a carbamate moiety, when treated with performic acid form the corresponding 3-hydroxytetrahydropyrrolo[2,3-b]indoles. An unusual migration of carbamate groups was observed in some cases, and the reaction appeared to be strongly dependent on the type of substituent in the phenyl ring of the phenylcarbamate group. Theoretical calculations suggest that the reaction is kinetically controlled.
4 Experimental
Anhydrous solvents were used for all the reactions. TLC analyses were performed on silica gel plates (Merck Silica Gel 60 F254) and made visual using a UV lamp and I2 vapor. The melting points were determined on a Boetius hot-plate microscope and are uncorrected. Column chromatography was carried out at atmospheric pressure using Merck Silica Gel 60 (100–200 mesh) or Al2O3 using mixtures of cyclohexane–ethyl acetate or diethyl ether–methanol as eluents. The NMR spectra were recorded on a Varian Unity Plus spectrometer operating at 200 MHz or 500 MHz for 1H NMR and at 50 MHz or 125 MHz for 13C NMR. The spectra were measured in CDCl3, CD3OD and DMSO-d6 and chemical shifts (δ) are reported in ppm relative to TMS. Mass spectra were obtained using Quattro LC Micromass and LCT Micromass TOF HiRes apparatus. 3-[2-(acetylamino)ethyl]-1H-indol-5-yl carbamates 3, 5, 7, 9, 11 and 13 were prepared according to the known procedure [7]. The X-ray data were collected using Oxford Diffraction Xcalibur R single crystal diffractometer. Data were collected at room temperature by the application of Cu Kα monochromatic radiation. The structures were solved using SHELXS-93 program [10] and then refined by the application of SHELXL-93 software [10]. Full structural data have been deposited with the Cambridge Crystallographic Data Centre under respective numbers CCDC 9103371, CCDC 9103382, and CCDC 9103393.
4.1 Oxidation of N-acetylserotonin carbamates with performic acid. General procedure
Performic acid was freshly prepared by adding 30% H2O2 (0.5 mL) to 98% formic acid (9.5 mL) and stirring at room temperature for 2 h.
3-[2-(acetylamino)ethyl]-1H-indol-5-yl carbamate was dissolved in 98% formic acid (2 mL). The mixture was cooled to 0 °C and performic acid (1.1 equiv) was added. The mixture was stirred at 0 °C for 2 h and washed with 10% aqueous Na2SO3 solution (10 mL). Na2CO3 (2 g) was added successively. The mixture was extracted with CHCl3 (2 × 15 mL). The organic layer was dried over anhydrous MgSO4 and concentrated.
4.1.1 1-Acetyl-8-formyl-3a-hydroxy-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl phenylcarbamate 4
The residue was purified by column chromatography (silica gel, 0–5% MeOH–Et2O) to give a white solid; yield: 168 mg (30%); mp 154–157 °C. 1H NMR (500 MHz, CD3OD): δ = 2.09 (s, 3 H, COCH3), 2.46–2.57 (m, 2 H, CH2), 3,18 (m, 1 H, CHH), 3.87 (t, 1 H, J = 9.0 Hz, CHH), 5.87 (s, 1 H, H-8a), 7.07 (t, 1 H, J = 7.0 Hz), 7.23 (dd, 1 H, J1 = 2.0 Hz, J2 = 9.0 Hz), 7.32 (t, 2 H, J = 8.0 Hz), 7.38 (d, 1 H, J = 2.0 Hz), 7.51 (d, 2 H, J = 7.5 Hz), 8.02 (d, 1 H, J = 8.5 Hz), 8.96 (s, 1 H, CHO). 13C NMR (125 MHz, CD3OD): δ = 21.01, 36.41, 46.84, 80.65, 85.18, 117.44, 117.98, 118.70, 123.37, 123.70, 128.81, 134.84, 138.57, 138.63, 148.48, 152.94, 163.31, 172.27. HRMS (ESI): m/z [M+Na]+ calcd for C20H19N3O5Na: 404.1223, found: 404.1226. X-ray analysis1.
4.1.2 1-Acetyl-8-formyl-3a-hydroxy-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl (4-chlorophenyl)carbamate 6
The residue was purified by column chromatography (silica gel, 50–100% AcOEt–cC6H12) to give a white solid; yield: 53 mg (39%); mp 177–180 °C. 1H NMR (500 MHz, DMSO-d6): δ = 2.01 (s, 3H, COCH3), 2.39–2.50 (m, 2 H, CH2), 3.04 (m, 1 H, CHH), 3.82 (t, 1 H, J = 9.6 Hz, CHH), 5.76 (s, 1 H, H-8a), 6.36 (s, 1 H), 7.22 (dd, 1 H, J1 = 2.4 Hz, J2 = 8.7 Hz), 7.39 (d, 2 H, J = 9.0 Hz), 7.40 (s, 1 H), 7.54 (d, 2 H, J = 9.0 Hz), 7.89 (d, 1 H, J = 9.0 Hz), 8.87 (s, 1 H, CHO). 13C NMR (125 MHz, DMSO-d6): δ = 22.11, 36.60, 46.42, 79.95, 84.72, 116.44, 118.08, 119.97, 123.22, 126.68, 128.78, 135.21, 137.60, 138.29, 147.17, 151.76, 162.25, 170.19. HRMS (ESI): m/z [M+Na]+ calcd for C20H18N3O5ClNa: 438.0833, found: 438.0835.
4.1.3 1-Acetyl-8-formyl-3a-hydroxy-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl (2-nitrophenyl)carbamate 8
The residue was purified by column chromatography (silica gel, AcOEt) to give a yellow oil; yield: 13 mg (12%). 1H NMR (200 MHz, CDCl3): δ = 2.05 (s, 3 H, COCH3), 2.52 (m, 2 H, CH2), 3.14–3.28 (m, 1 H, CHH), 3.70–3.79 (m, 1 H, CHH), 3.95 (br.s, 1 H, OH), 5.88 (s, 1 H, H-8a), 7.18–7.32 (m, 3 H), 7.68 (t, 1 H, J = 7.8 Hz), 8.04 (d, 1 H, J = 8.4 Hz), 8.27 (dd, 1 H, J1 = 1.6 Hz, J2 = 8.7 Hz), 8.51 (d, 1 H, J = 7.6 Hz), 8.87 (s, 1 H, CHO). 13C NMR (50 MHz, CDCl3): δ = 22.15, 37.12, 46.68, 80.20, 85.51, 117.30, 117.94, 120.80, 123.15, 123.89, 126.00, 133.96, 134.49, 136.06, 136.49, 138.98, 147.35, 151.39, 162.98, 170.62. HRMS (ESI): m/z [M+Na]+ calcd for C20H18N4O7Na: 449.1073, found: 449.1076.
4.1.4 1-Acetyl-3a,5-dihydroxy-N-(2-isopropylphenyl)-2,3,3a,8a-tetrahydropyrrolo[2,3-b]indole-8(1H)-carboxamide 10
The residue was purified by column chromatography (silica gel, 0–5% MeOH–Et2O) to give a white solid; yield: 92 mg (31%); mp 265–266 °C. 1H NMR (500 MHz, CD3OD): δ = 1.22 (d, 6 H, J = 7.8 Hz, (CH3)2CH), 2.17 (s, 3 H, COCH3), 2.45 (m, 2 H, CH2), 3.14 (m, 2 H, (CH3)2CH, CHH), 3.92 (m, 1 H, CHH), 5.89 (s, 1 H, H-8a), 6.78 (dd, 1 H, J1 = 2.0 Hz, J2 = 8.5 Hz), 7.18 (m, 2 H), 6.87 (d, 1 H, J = 2.5 Hz), 7.30 (dd, 1 H, J1 = 2.0 Hz, J2 = 7.5 Hz), 7.34 (dd, 1 H, J1 = 2.0 Hz, J2 = 7.5 Hz), 7.45 (d, 1 H, J = 9.0 Hz). 13C NMR (125 MHz, CD3OD): δ = 22.08, 24.13, 30.74, 38.21, 48.66, 84.14, 87.55, 110.84, 117.89, 120.36, 126.78, 126.99, 127.35, 128.20, 134.51, 136.29, 137.83, 145.40, 155.53, 157.75, 174.15. HRMS (ESI): m/z [M+Na]+ calcd for C22H25N3O4Na: 418.1743, found: 418.1746. X-ray analysis2.
4.1.5 1-Acetyl-3a,5-dihydroxy-N-(4-isopropylphenyl)-2,3,3a,8a-tetrahydropyrrolo[2,3-b]indole-8(1H)-carboxamide 12
The residue was purified by column chromatography (silica gel, 0–5% MeOH–Et2O) to give a white solid; yield: 65 mg (24%); mp 166–169 °C. 1H NMR (200 MHz, CD3OD): δ = 1.22 (d, 6 H, J = 7.8 Hz, (CH3)2CH), 2.17 (s, 3 H, COCH3), 2.40 (m, 2 H, CH2), 2.85 (quin, 1 H, J = 6.4 Hz, (CH3)2CH), 3.08 (m, 1 H, CHH), 3.87 (m, 1 H, CHH), 5.81 (s, 1 H, H-8a), 6.78 (dd, 1 H, J1 = 2.4 Hz, J2 = 8.6 Hz), 6.86 (d, 1 H, J = 2.6 Hz), 7.15 (d, 2 H, J = 8.4 Hz), 7.40 (d, 2 H, J = 8.6 Hz), 7.47 (d, 1 H, J = 8.6 Hz). 13C NMR (50 MHz, CD3OD): δ = 22.30, 24.69, 34.96, 38.27, 48.72, 84.03, 87.63, 110.99, 118.02, 120.50, 121.12, 127.75, 134.81, 137.73, 138.47, 144.96, 155.69, 156.40, 174.52. HRMS (ESI): m/z [M+Na]+ calcd for C22H25N3O4Na: 418.1743, found: 418.1745. X-ray analysis3.
4.1.6 1-Acetyl-3a,5-dihydroxy-N-(2-methoxyphenyl)-2,3,3a,8a-tetrahydropyrrolo[2,3-b]indole-8(1H)-carboxamide 14
The residue was purified by column chromatography (silica gel, 0–5% MeOH–Et2O) to give a white solid; yield: 53 mg (26%); mp 161–164 °C. 1H NMR (500 MHz, CD3OD): δ = 2.14 (s, 3 H, COCH3), 2.45 (m, 2 H, CH2), 3.08–3.13 (m, 1 H, CHH), 3.33–3.88 (m, 1 H, CHH), 3.88 (s, 3 H, OCH3), 5.81 (s, 1 H, H-8a), 6.80 (dd, 1 H, J1 = 2.5 Hz, J2 = 8.0 Hz), 6.86 (d, 1 H, J = 2.5 Hz), 6.90 (td, 1 H, J1 = 1.5 Hz, J2 = 8.0 Hz), 7.00 (dd, 1 H, J1 = 1.5 Hz, J2 = 8.0 Hz), 7.05 (td, 1 H, J1 = 1.5 Hz, J2 = 8.0 Hz), 7.43 (d, 1 H, J = 8.5 Hz), 7.76 (dd, 1 H, J1 = 1.5 Hz, J2 = 7.5 Hz). 13C NMR (125 MHz, CD3OD): δ = 22.15, 37.65, 48.51, 56.49, 84.45, 87.33, 110.91, 112.31, 118.00, 121.09, 121.62, 123.79, 125.46, 129.45, 135.00, 137.80, 152.63, 155.84, 157.08, 174.12. HRMS (ESI): m/z [M+Na]+ calcd for C20H21N3O5Na: 406.1379, found: 406.1378.
Acknowledgment
We thank Krystyna Wojtasiewicz for her valuable technical assistance.
1 The detailed structural parameters have been deposited with the Cambridge Crystallographic Data Centre under the number CCDC 910337.
2 The detailed structural parameters have been deposited with the Cambridge Crystallographic Data Centre under the number CCDC 910338.
3 The detailed structural parameters have been deposited with the Cambridge Crystallographic Data Centre under the number CCDC 910339.