

1 Introduction
The stereospecific functionalisation of chiral organolithiums [1] provides a valuable method for the synthesis of chiral tertiary thiols [2], tertiary alcohols [3] and α-tertiary amines [4–6]. Although stereospecific alkylation of chiral organolithiums is routine [1], stereospecific arylation or alkenylation to form more functionalised products requires specialised approaches [7–11]. Building on our discovery that metallated N-benzyl-N′-aryl ureas [12–19], carbamates [20–23] and thiocarbamates [24–26] undergo stereospecific aryl migration to the carbanionic centre, we previously reported that the stereospecific N to C vinyl migration of a variety of lithiated benzylic ureas 3, carbamates 4 and thiocarbamates 5 may be used to generate α-vinyl tertiary amines, alcohols and thiols from their parent compounds 1 after deprotection of the products 6–8 (Scheme 1a) [27].
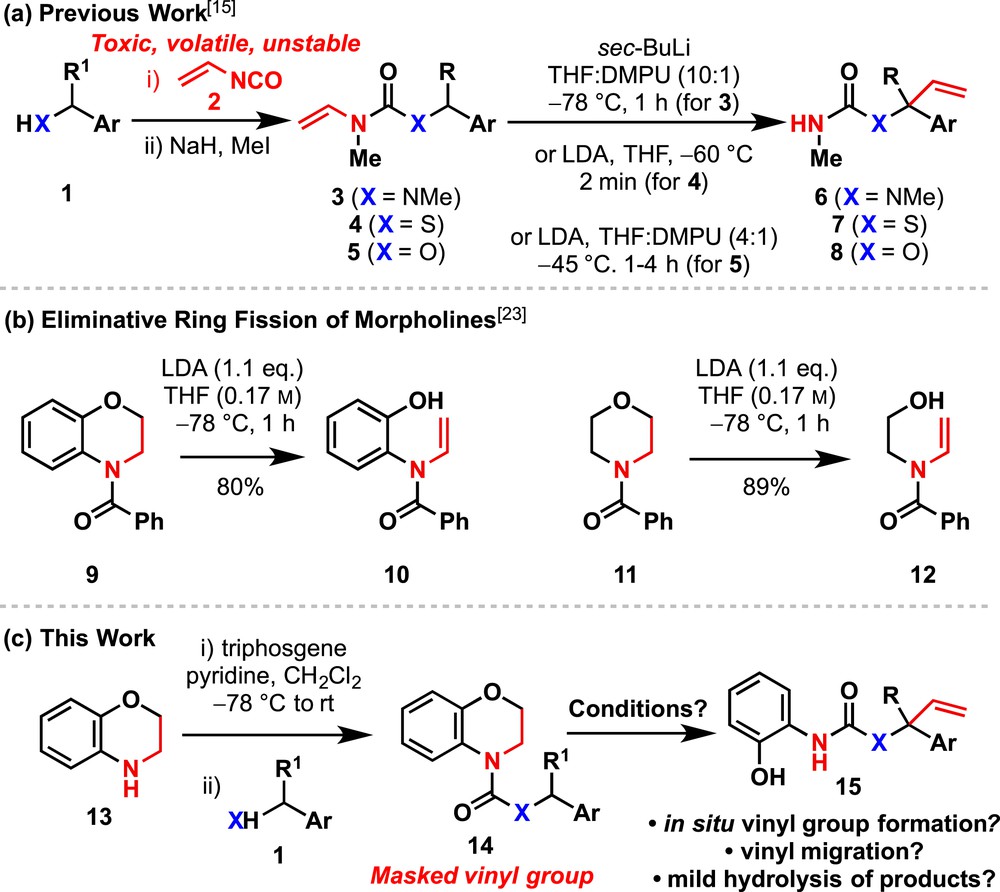
Formation and migration of N-vinyl substituents.
The N-vinyl substituted starting materials [14,17,27–30] for this reported vinylation had been synthesized by a two-step procedure that used vinyl isocyanate 2 to introduce the N-alkenyl substituent. Unfortunately, 2 is of limited commercial availability because of its toxicity and poor stability towards polymerisation. Although the in situ synthesis of 2 has been reported [31,32], isolation and storage of this volatile and reactive compound is problematic. In this article, we report a solution to the challenge of synthesizing and using N-vinyl ureas and carbamates that makes use of a benzomorpholine ring as a ‘masked N-vinyl group’, and we show how this strategy may be used in the organolithium-mediated synthesis of α-vinyl–substituted amines and alcohols.
Florio et al. reported that treatment of benzomorpholine 9 or morpholine 11 with lithium diisopropylamide (LDA) at −78 °C leads to enamides 10 and 12, respectively, by base-promoted reverse 6-endo-trig elimination of the phenoxide anion from a presumed intermediate amide-stabilised organolithium (Scheme 1b) [33]. We reasoned that benzylic ureas or carbamates 14 (X = NR or O) containing a benzomorpholine ring might likewise fragment upon treatment with LDA to generate an N-alkenyl urea [28] or carbamate, which could then undergo vinyl migration, ideally in a one-pot procedure (Scheme 1c). In this article, we report our success in developing this strategy for the C-vinylation of some carbanion nucleophiles.
2 Results and discussion
N-Carboxamidobenzomorpholines 16 and 18 were synthesized by standard conditions and treated with LDA in tetrahydrofuran (THF). With R = H, deprotonation of 16 at the benzylic position led to direct migration of the N-aryl ring of the benzomorpholine to the benzylic carbanion, resulting in a clean ring expansion to give 17 in good yield [19]. In contrast, treatment of 18, with R = Me, under the same conditions yielded a mixture of products 19 and 20 (Scheme 2a).
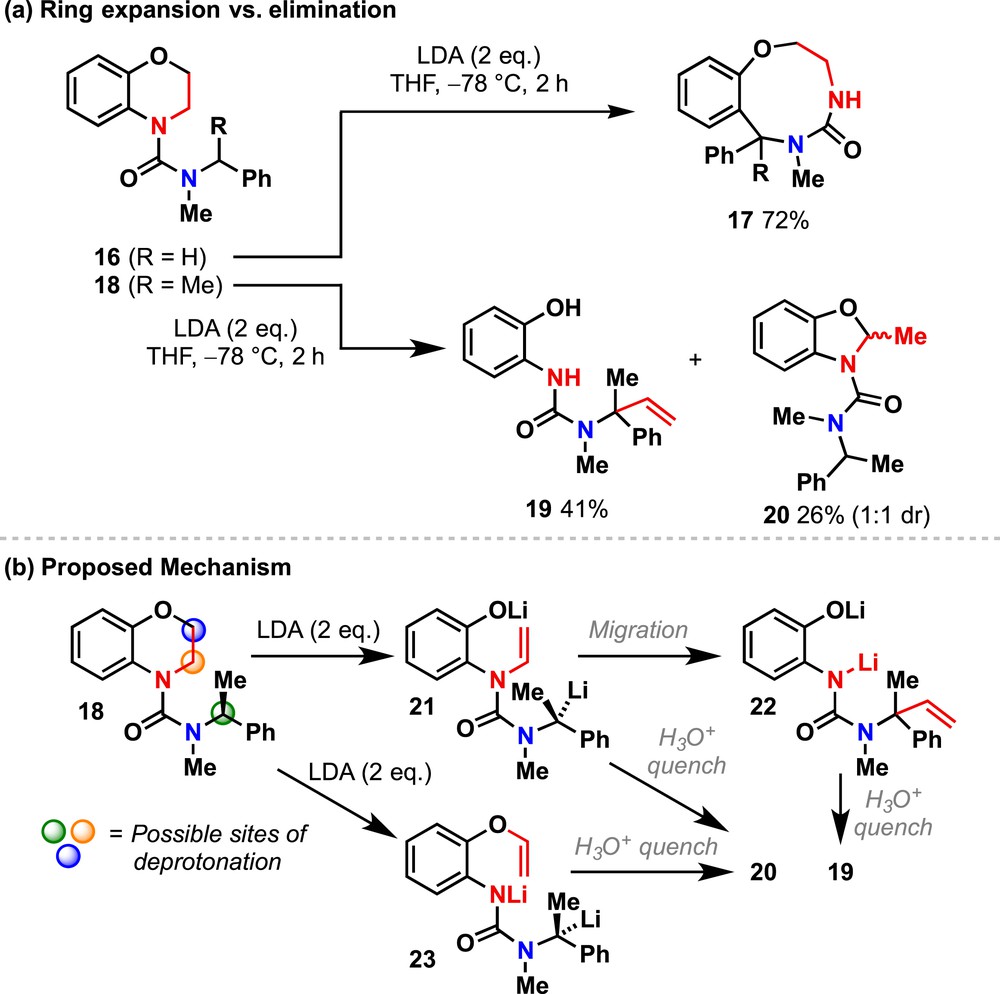
Eliminations and migrations in urea derivatives of benzomorpholine.
This differential reactivity can be rationalised by considering three alternative sites at which 16 or 18 may be lithiated (Scheme 2b): α to the morpholine nitrogen (orange), α to the morpholine oxygen (blue) or at the benzylic position (green). Assuming that the most acidic benzylic position (green) is deprotonated first, the organolithium resulting from 16 rapidly attacks the N-aryl substituent and ring expands to give 17. The increased steric bulk at the lithiated benzylic position of 18 presumably retards this ring expansion, allowing a second deprotonation to occur α to the morpholine nitrogen (orange). Elimination gives phenoxide 21, which then undergoes migration of the newly formed vinyl group to give 22, and hence 19.
The by-product 20 may be formed by two alternative pathways. Protonation of 21 before vinyl migration gives an N-acyl enamine, or deprotonation of α to oxygen (blue) and elimination would give a vinyl ether 23, either of which might undergo cyclisation under the conditions of the aqueous quench to give oxazolidine 20 [34].
Using enantiopure 18, a series of reactions was carried out to identify a set of conditions that suppressed the formation of 20. Switching to the more basic sec-BuLi (Table 1, entry 2) did not improve the yield of the vinyl-migrated product 13 and also gave poor conversion: 47% of 18 was recovered. The addition of 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU), a highly coordinating co-solvent [12,19], led to an improvement in the selectivity for vinyl migration product 19 (Table 1, entry 3), which was improved further by conducting the reaction on a larger scale (Table 1, entry 4). Analysis of 19 by supercritical fluid chromatography on a (S,S)-Whelk-01 Kromasil stationary phase furthermore confirmed that the reaction proceeded with complete enantiospecificity, presumably by stereochemically retentive (rather than invertive) attack [12,35].
Optimisation of the vinyl migration.
| Entry | SM | Base (equiv) | Additive (equiv) | 19a (%) | 20 (%) (dr)b |
| 1 | (S)-18 | LDA (2) | – | 41 | 26 (1:1) |
| 2 | (S)-18 | sec-BuLi (2) | DMPU (5) | 5c | 12 (1:1) |
| 3 | (S)-18 | LDA (3) | DMPU (5) | 53 | 5 (1:1) |
| 4 | (S)-18 | LDA (2.2) | DMPU (5) | 64d | >5 (1:1) |
| 5 | 24 | LDA (2) | – | –e | – |
| 6 | 25 | LDA (2.2) | DMPU (5) | 23f + 63g | – |
a Isolated yield from 0.2 mmol scale reaction.
b dr determined by 1H NMR of the crude reaction mixture.
c 47% remaining 18 recovered.
d Reaction carried out on 1 mmol scale.
e Starting 24 recovered unchanged.
f Yield of 26, the p-Cl analogue of 19.
g Yield of ring expansion/elimination product 27.
We also synthesized morpholine urea 24 for direct comparison with the reaction observed with 18, but attempted fragmentation/migration gave only recovered starting material, with no cleavage of the morpholine ring (entry 5). Fragmentation and rearrangement of the para-chlorophenyl–substituted urea 25 gave 23% of the vinyl migration product 26, along with 63% of the alkene 27 derived from ring expansion to a homologue of 17 followed by elimination. This result suggests that, like 16, the more reactive chlorophenyl-substituted organolithium derived from 25 undergoes ring expansion at a rate competitive with fragmentation to the vinyl urea.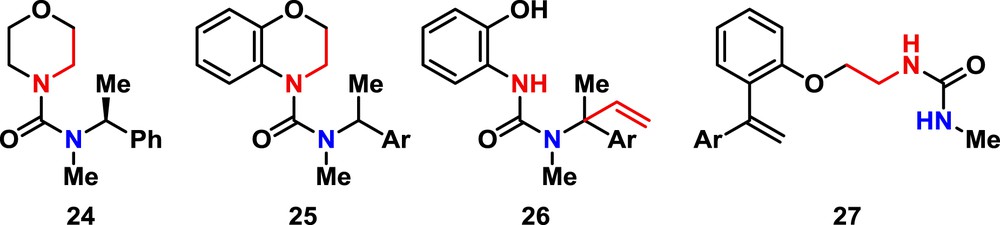
Having established conditions for the selective formation of 19, we next investigated the cleavage of the resulting urea to generate the tertiary amine. Attempts to hydrolyse 19 to its constituent α-tertiary amine under a wide range of basic (e.g., K2CO3/n-BuOH) and acidic (e.g., TFA, HCl) conditions met with limited success. However, exploiting the rapid methanolysis of related trisubstituted ureas reported by Booker-Milburn et al. [36] enabled us to liberate the desired α-tertiary amine (Scheme 3). Purification of the free amine by flash column chromatography followed by precipitation as the hydrochloride salt gave 28 in good yield. Application of these conditions to 26 also gave 29 in good yield.
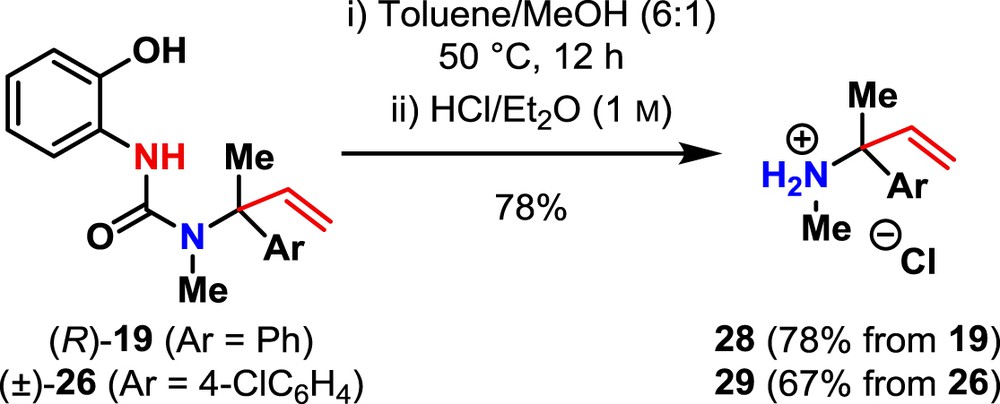
Methanolysis of ureas to give amine hydrochloride salts 28 and 29.
Comparable reactivity was shown by the carbamate 30, synthesized from benzomorpholine in two steps in good yield (see Section 4). Metallation of carbamate 30 with LDA under our optimized conditions gave the product of vinyl migration 32 in moderate yield (Scheme 4). Carbamate 32 was unstable and proved difficult to isolate, purify and characterize. With the aim of inducing direct fragmentation of 31 to the alcohol 33, the reaction was warmed to 70 °C overnight. Tertiary allylic alcohol 33 was isolated in good yield, meaning that the one-pot application of base and then heat allows the efficient vinylation of a carbamate α to oxygen. Alcohol 33 was nonetheless formed in essentially racemic form because of the relative configurational instability of dipole-stabilized benzylic organolithiums α to oxygen [1,20,21,27].
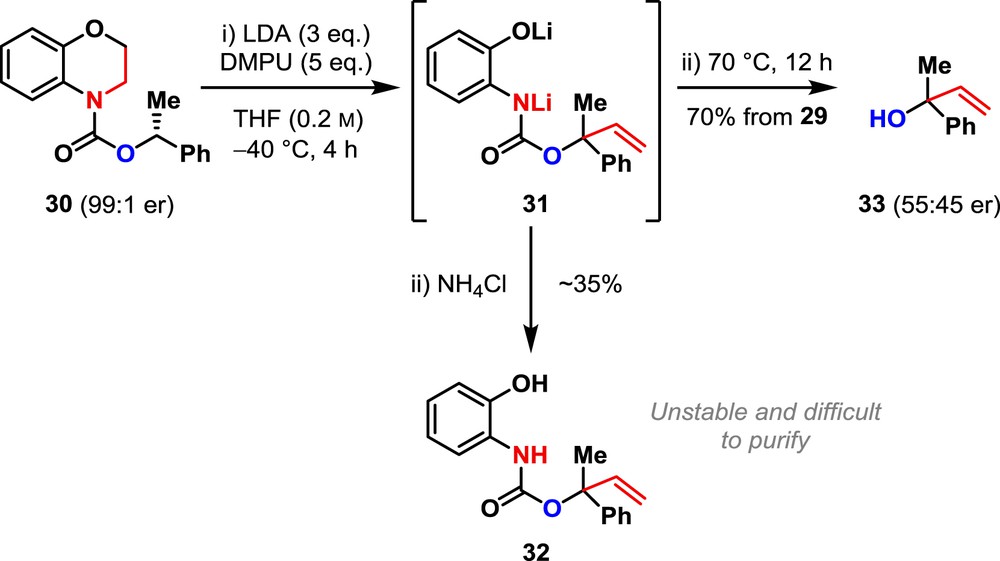
Carbamate vinylation and deprotection.
Ureas incorporating nitrile-stabilised anions undergo intramolecular migration chemistry [37], so α-cyano urea 34 was synthesized to investigate the applicability of the ‘masked vinylation’ strategy to non-benzylic carbanions. Treatment of 34 with LDA and DMPU under conditions similar to those used for the urea 18 and carbamate 30 gave an iminohydantoin 35 [37], which was hydrolysed under acidic conditions to the hydantoin 36 (Scheme 5).
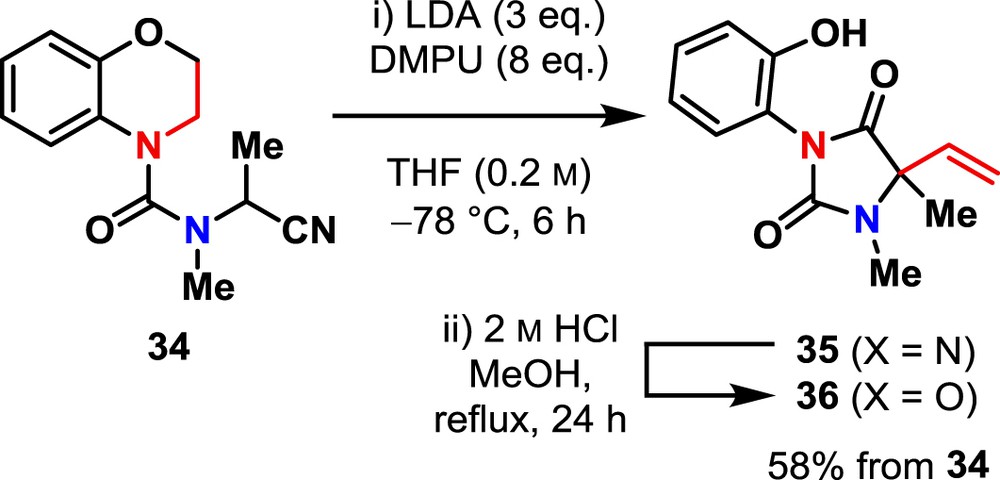
Vinylation of amino nitrile derivative 34.
3 Conclusions
We have demonstrated that benzomorpholine-derived ureas and carbamates behave as masked vinyl groups for the vinylation of a variety of carbon nucleophiles through a base-promoted cascade of elimination and vinyl migration. This approach overcomes difficulties in preparing simple N-vinyl ureas arising from the commercial unavailability of vinyl isocyanate and the associated difficulties in its use, relating to toxicity and storage.
4 Experimental section
4.1 General directions
Reactions requiring anhydrous conditions (where specified) were executed under dry nitrogen or argon atmospheres in glassware that was flame dried and allowed to cool under vacuum. Reaction mixtures were stirred magnetically. Air- and moisture-sensitive liquids and solutions were transferred via syringe or cannula into the reaction vessels through rubber septa. All reagents were purchased (unless specified) at highest commercial quality and used as received. Non-anhydrous solvents were purchased (unless specified) at the highest commercial quality and used as received. CH2Cl2, THF, dimethylformamide (DMF) and MeCN extra dry solvents over 3 Å molecular sieves were purchased from Acros and used as received. DMPU was distilled from CaH2 and stored under nitrogen in a Young's tube. NEt3 was distilled from CaH2 and stored under nitrogen in a Young's tube. 3,4-Dihydro-2H-benz-[b]-[1,4]oxazine was purchased from Alfa Aesar UK and used as received.
4.2 Equipment details
Thin-layer chromatography (TLC) was performed on aluminium-backed silica plates (0.2 mm, 60 F254), which were developed using standard visualizing agents: UV fluorescence (254 and 366 nm), phosphomolybdic acid/Δ, vanillin/Δ, potassium permanganate/Δ and Seebach/Δ. Flash chromatography was performed on an automated Biotage Isolera Spektra Four using gradient elutions on pre-packed silica gel Biotage SNAP Ultra/ZIP Sphere columns. Melting points were measured on a Kofler hot-stage melting point apparatus and are uncorrected. IR spectra were recorded on neat compounds using a Perkin Elmer (Spectrum One) FT-IR spectrometer (ATR sampling accessory). Only strong and selected absorbances (νmax expressed in cm−1) are reported. 1H NMR spectra were recorded on Jeol ECS (400 MHz) or Varian VNMR (400 or 500 MHz) instruments. Chemical shifts (δΗ) are quoted in parts per million (ppm) and referenced to the appropriate NMR solvent peak(s) and are assigned ArH, C, CH, CHH or CHH (diastereotopic protons), CH2, CH3. Two-dimensional (2D) NMR experiments COSY, HSQC and HMBC were used where necessary in assigning NMR spectra. Spin–spin coupling constants (J) are reported in Hertz (Hz). 13C NMR spectra were recorded on Jeol ECS (101 MHz) or Varian VNMR (101 MHz or 125 MHz) instruments. Chemical shifts (δC) are quoted in ppm and referenced to the appropriate solvent peak(s) and are assigned C, CH, CH2 and CH3 as determined using 2D NMR experiments HSQC and HMBC where necessary. Spin–spin coupling constants (J) are reported in Hz. High-resolution mass spectra were recorded on a Bruker Daltonics MicroTOF 2 mass spectrometer (ESI) with only molecular ions ([M]+ [M+H]+ and [M+Na]+) and major peaks reported. Optical rotations () were measured on a Bellingham and Stanley Ltd ADP220 polarimeter, where c is given in g/100 mL.
4.3 Starting material synthesis
4.3.1 (−)-(S)–N-Methyl-N-(1-phenylethyl)-2,3-dihydro-4H-benzo[b]-[1,4]oxazine-4-carboxamide (18)
Synthesis of 2,3-dihydro-4H-benzo[b]-[1,4]oxazine-4-carbonyl chloride: to a solution of triphosgene (3.4 g, 12 mmol, 0.46 equiv) in anhydrous CH2Cl2 (17 mL, 0.7 M) under a N2 atmosphere was added pyridine (2.0 mL, 25 mmol, 1.0 equiv) dropwise at −78 °C. A solution of 3,4-dihydro-2H-benz-[b]-[1,4]oxazine (3.5 g, 26 mmol, 1.0 equiv) in anhydrous CH2Cl2 (2 mL) was added dropwise at −78 °C before warming to room temperature. After completion, the reaction was quenched with HCl (1.0 M) and extracted three times with CH2Cl2. The combined organic layers were washed with saturated aqueous NaHCO3, dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum. The resulting residue was purified on a short pad of silica (eluting with 20% EtOAc/n-pentane) to give the carbamoyl chloride. Preparation of 18: to a dried Schlenk flask under a N2 atmosphere was added a solution of (S)–N-methyl-1-phenylethanamine (0.75 mL, 3.9 mmol, 1.3 equiv) and triethylamine (0.66 mL, 4.8 mmol, 1.6 equiv) in anhydrous acetonitrile (10 mL, 0.4 M). A solution of the carbamoyl chloride (0.79 g, 4.0 mmol, 1.0 equiv) in anhydrous acetonitrile (1 mL) was added dropwise to the mixture and stirred at room temperature until completion (followed by TLC). The reaction was quenched with saturated aqueous NaHCO3, extracted three times with CH2Cl2 and the combined organic layers were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum. The crude urea was purified by flash column chromatography (eluting with 7–60% Et2O/petrol on a ZIP Sphere 30 g column over 14 column volumes) to give the title compound 18; 1.07 g, 90% yield; light yellow solid; Rf 0.18 (30% Et2O/n-pentane); mp 79–80 °C (CH2Cl2/n-pentane); νmax (film)/cm−1 1642, 1601, 1584, 1496, 1431, 1391, 1338, 1306, 1282, 1217, 1178, 1061, 1050, 770; = −43 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 7.37–7.28 (m, 5H, ArH), 6.96–6.93 (m, 1H, ArH), 6.87–6.86 (m, 1H, ArH), 6.81–6.77 (m, 1H, ArH), 5.59 (q, J = 7.1 Hz, 1H, NCHPh), 4.35–4.31 (m, 2H, CH2), 3.72–3.68 (m, 2H, CH2), 2.60 (s, 3H, NCH3), 1.61 (d, J = 7.0 Hz, 3H, CH3); δC (101 MHz, CDCl3) 159.5 (CO), 145.0 (C), 140.7 (C), 128.7 (CH), 128.4 (C), 127.6 (CH), 127.6 (CH), 123.1 (CH), 120.7 (CH), 119.5 (CH), 117.3 (CH), 66.4 (CH2), 54.4 (CH), 44.0 (CH2), 31.0 (CH3), 16.2 (CH3); HRMS (ESI+) [M+H]+ C18H21N2O2+ requires 297.1598; found 297.1607. Chiral supercritical fluid chromatography ((S,S)-Whelk-01, 5/100 Kromasil, 25 cm × 4.6 mm ID), 125 bar CO2, 40 °C, 4 mL/min, 20% co-solvent (MeOH); tR 3.8 min (major), 5.2 min (minor) 99:1 er. Racemic sample was prepared by making a 1:1 mixture of (−)-18 and (+)-18, which was prepared in an analogous manner.
4.3.2 (−)-(S)–N-(1-(4-Chlorophenyl)ethyl)–N-methyl-2,3-dihydro-4H-benzo[b][1,4]oxazine-4-carboxamide (25)
See 18 for preparation of carbamoyl chloride. To a dried Schlenk flask under a N2 atmosphere was added a solution of (S)-1-(4-chlorophenyl)–N-methylethylamine (0.56 mL, 3.9 mmol, 1.3 equiv) and triethylamine (0.66 mL, 4.8 mmol, 1.6 equiv) in anhydrous MeCN (7.5 mL, 0.4 M). A solution of the carbamoyl chloride (0.59 g, 3.0 mmol, 1.0 equiv) in anhydrous acetonitrile (1 mL) was added dropwise to the mixture and stirred at room temperature until completion (followed by TLC). The reaction was quenched with saturated aqueous NaHCO3, extracted three times with CH2Cl2 and the combined organic layers were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum. The crude urea was purified by flash column chromatography (eluting with 7–60% Et2O/petrol on a ZIP Sphere 30 g column over 14 column volumes) to give the urea. Methylation: to a flame dried Schlenk flask under a N2 atmosphere was added sodium hydride (0.21 g, 5.1 mmol, 2 equiv, 60% suspension on mineral oil) suspended in DMF (10 mL, 0.25 M). To this suspension was added a solution of the urea (∼2.6 mmol) from the previous step in DMF (1 mL) dropwise at 0 °C. Methyl iodide (0.47 mL, 7.6 mmol, 3 equiv) was added dropwise and the reaction was allowed to warm to room temperature for 24 h. The reaction mixture was quenched with saturated aqueous NH4Cl and extracted three times with EtOAc. The combined organic phases were washed with saturated aqueous LiCl to remove the DMF, dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum. The resulting residue was purified by flash column chromatography (eluting with 7–60% Et2O/petrol on a ZIP Sphere 30 g column over 15 column volumes) to give the title compound 25; 0.63 g, 66% yield over two steps; white solid; Rf 0.15 (30% Et2O/n-pentane); mp 117–119 °C (CH2Cl2/n-pentane); νmax (film)/cm−1 2922, 2853, 1636, 1498, 1389, 1342, 1244, 1179, 1057, 769; = −46 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 7.33 (s, 4H, ArH), 6.94–6.76 (m, 4H, ArH), 5.56 (q, J = 7.0 Hz, 1H, NCHPh), 4.39–4.28 (m, 2H, CH2), 3.76–3.63 (m, 2H, CH2), 2.58 (s, 3H, NCH3), 1.59 (d, J = 7.1 Hz, 3H, CH3); δC (101 MHz, CDCl3) 159.4 (CO), 145.0 (C), 139.3 (C), 133.5 (C), 129.0 (CH), 128.8 (CH), 128.2 (C), 123.2 (CH), 120.7 (CH), 119.4 (CH), 117.4 (CH), 66.4 (CH2), 53.8 (CH), 43.9 (CH2), 31.1 (NCH3), 16.2 (CH3); HRMS (ESI+) [M+H]+ C18H20ClN2O2+ requires 331.1208; found 331.1201.
4.3.3 (−)-(R)-1-Phenylethyl-2,3-dihydro-4H-benzo[b][1,4]oxazine-4-carboxylate (30)
See 18 for preparation of carbamoyl chloride. In a dried Schlenk flask under a N2 atmosphere, sodium hydride (0.36 g, 9.0 mmol, 3.0 equiv, 60% suspension on mineral oil) was suspended in anhydrous CH2Cl2 (12 mL, 0.25 M) and cooled to 0 °C before (R)-(+)-phenylethanol (1.1 mL, 9.0 mmol, 3.0 equiv) was added. A solution of the carbamoyl chloride (0.59 g, 3.0 mmol, 1.0 equiv) in anhydrous CH2Cl2 (1 mL) was added dropwise to the mixture. The ice bath was removed and the mixture was heated to 40 °C overnight. The reaction was quenched with saturated aqueous NH4Cl and extracted three times with CH2Cl2. The combined organic phases were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum. The product was purified by flash column chromatography (eluting with 2–25%, EtOAc/n-hexane + 1% NEt3 on an SNAP Ultra 25 g column over 10 column volumes) to yield the title compound 30; 0.56 g, 66% yield, colourless oil; Rf 0.25 (25% Et2O/n-pentane); νmax (film)/cm−1 2979, 2934, 1699, 1494, 1373, 1259, 1224, 1210, 1145, 1056, 1028, 748; = −13 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 7.85 (br s, 1H, ArH), 7.43–7.27 (m, 5H, ArH), 7.03–6.96 (m, 1H, ArH), 6.93–6.85 (m, 2H, ArH), 5.94 (q, J = 6.6 Hz, 1H, CH), 4.25 (t, J = 4.7 Hz, 2H, CH2), 3.95–3.92 (m, 2H, CH2), 1.63 (d, J = 6.6 Hz, 3H, CH3); δC (101 MHz, CDCl3) 153.0 (CO), 146.1 (C), 141.8 (C), 128.7 (CH), 128.1 (CH), 126.1 (CH), 125.9 (C) 124.8 (CH), 123.4 (CH), 120.5 (CH), 117.2 (CH), 74.8 (CH), 65.6 (CH2), 42.5 (CH2), 22.8 (CH3); HRMS (ESI+) [M+Na]+ C17H17NO3Na+ requires 306.1101; found 306.1107.
4.3.4 (±)–N-(1-Cyanoethyl)–N-methyl-2,3-dihydro-4H-benzo[b]-[1,4]oxazine-4-carboxamide (34)
Step 1: methylamine (42 mmol, 3.0 equiv, 8.0 M in EtOH) was added to a solution of lactonitrile (14 mmol, 1.0 equiv) in EtOH (6 mL, 2.5 M) at 0 °C in the presence of an excess of MgSO4. The reaction mixture was allowed to reach room temperature and stirred overnight. The mixture was filtered and the excess MeNH2 and solvent were removed from the filtrate carefully under vacuum to afford the crude nitrile amine. Step 2: in a round-bottom flask under a N2 atmosphere, triphosgene (0.40 equiv) was dissolved in anhydrous CH2Cl2 (1.0 M) and cooled to −78 °C. 2,6-Lutidine (1.2 equiv) was added dropwise to the solution, and the reaction mixture was stirred for 10 min before a solution of the nitrile amine (1.0 equiv) was added dropwise. The reaction mixture was allowed to reach room temperature and was stirred for 2 h. Upon completion, the reaction was quenched with HCl (1.0 M aq) and extracted three times with CH2Cl2. The combined organic layers were washed with saturated aqueous NaHCO3 and brine and dried over MgSO4. Filtration and concentration of the filtrate under vacuum gave the crude carbamoyl chloride. Step 3: in a round-bottom flask under a N2 atmosphere, the carbamoyl chloride (1.0 equiv) was dissolved in anhydrous MeCN (0.4 M) and 2,6-lutidine (1.1 equiv) was added. A solution of 3,4-dihydro-2H-benz-[1,4]oxazine (1.1 equiv) in MeCN (0.4 M) was added dropwise to the mixture. The mixture was warmed to 60 °C and stirred for 24 h. Upon completion, the solvent was removed under vacuum. The remaining oil was dissolved in EtOAc, washed with HCl (1.0 M, aq), with saturated aqueous NaHCO3 and brine and dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum. Purification by flash column chromatography (eluting with 10–80% Et2O/petrol on an SNAP Ultra 10 g column over 15 column volumes) gave the title compound 34; 2.4 g, 70% over three steps: white solid; Rf 0.30 (50% EtOAc/n-hexane); mp 95–96 °C (CH2Cl2/n-pentane); νmax (film)/cm−1 2953, 1651, 1585, 1496, 1377, 1359, 1311, 1278, 1249, 1057, 750; δH (400 MHz, CDCl3) 7.02–6.83 (m, 4H, ArH), 5.19 (q, J = 7.2 Hz, 1H, NCHCN), 4.40–4.28 (m, 2H, CH2), 3.80 (ddd, J = 13.3, 5.4, 3.2 Hz, 1H, CH2), 3.66 (ddd, J = 13.3, 6.7, 3.4 Hz, 1H, CH2), 2.89 (s, 3H, NCH3), 1.59 (d, J = 7.3 Hz, 3H, CH3); δC (101 MHz, CDCl3) 158.3 (CO), 145.2 (C), 126.9 (C), 124.4 (CH), 120.9 (CH), 119.9 (CH), 118.5 (CN), 117.7 (CH), 66.5 (CH2), 44.3 (CH3), 43.5 (CH2), 33.1 (CH3), 17.2 (CH3); HRMS (ESI+) [M+Na]+ C13H15N3O2Na+ requires 268.1056; found 268.1069.
4.4 Vinyl migration products
4.4.1 (+)-(R)-3-(2-Hydroxyphenyl)-1-methyl-1-(2-phenylbut-3-en-2-yl)urea (19)
In a dried Schlenk flask under a N2 atmosphere, urea 18 (0.30 g, 1 mmol, 1.0 equiv) was dissolved in anhydrous THF (6.7 mL, 0.15 M) and DMPU (0.6 mL, 5 mmol, 5.0 equiv) was added at room temperature. The mixture was cooled to −78 °C and LDA (1.1 mL, 2.2 mmol, 2.2 equiv, 2.0 M in THF/heptane/ethylbenzene) was added dropwise and the mixture was stirred for 4 h at −78 °C. The reaction was quenched with saturated aqueous NH4Cl and extracted three times with CH2Cl2. The combined organic layers were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum to give a crude residue. Purification of this residue by flash column chromatography (eluting with 7–60% Et2O/petrol on an SNAP Ultra 10 g column over 13 column volumes) gave the title compound 19; 0.19 g, 64% yield, light yellow solid; Rf 0.20 (30% Et2O/n-pentane); mp 94–96 °C (CH2Cl2/n-pentane); νmax (film)/cm−1 3392, 3059, 1631, 1586, 1538, 1448, 1359, 1242, 754, 699; = +24 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 9.77 (s, 1H, NH), 7.52–7.36 (m, 5H, ArH), 6.98–6.89 (m, 2H, ArH), 6.58 (ddd, J = 8.5, 7.1, 1.8 Hz, 1H, ArH), 6.39 (dd, J = 17.3, 10.6 Hz, 1H, CHHH), 6.26 (br s, 1H, OH), 5.74 (dd, J = 7.9, 1.5 Hz, 1H, ArH), 5.35 (d, J = 10.6 Hz, 1H, CHHH), 5.27 (d, J = 17.3 Hz, 1H, CHHH), 3.20 (s, 3H, NCH3), 1.83 (s, 3H, CH3); δC (101 MHz, CDCl3) 158.0 (CO), 149.5 (CH), 144.9 (C), 140.4 (CHCH2), 129.9 (CH), 128.4 (CH), 126.5 (C), 126.2 (CH), 125.8 (CH), 122.0 (CH), 119.9 (CH), 119.8 (CH), 115.3 (CHCH2), 65.3 (C), 32.8 (NCH3), 26.9 (CH3); HRMS (ESI+) [M+Na]+ C18H20N2O2Na+ requires 319.1417; found 319.1417. Chiral supercritical fluid chromatography ((S,S)-Whelk-01, 5/100 Kromasil, 25 cm × 4.6 mm ID), 125 bar CO2, 40 °C, 4 mL/min, 20% co-solvent (MeOH); tR 12.3 min (major), 13.3 min (minor) >99:1 er. Racemic sample was prepared by making a 1:1 mixture of (+)-19 and (−)-19, which was prepared in an analogous manner.
4.4.2 (±)-1-(2-(4-Chlorophenyl)but-3-en-2-yl)-3-(2-hydroxyphenyl)-1-methylurea (26)
In a dried Schlenk flask under a N2 atmosphere, urea 25 (0.17 g, 0.50 mmol, 1.0 equiv) was dissolved in anhydrous THF (3.3 mL, 0.15 M) and DMPU (0.30 mL, 2.5 mmol, 5.0 equiv) was added at room temperature. The mixture was cooled to −78 °C and LDA (0.60 mL, 1.1 mmol, 2.2 equiv, 2.0 M in THF/heptane/ethylbenzene) was added dropwise and the mixture was stirred for 4 h at −78 °C. The reaction was quenched with saturated aqueous NH4Cl and extracted three times with CH2Cl2. The combined organic layers were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum to give a crude residue. Purification of this residue by flash column chromatography (eluting with 5–100% EtOAc/petrol on an SNAP Ultra 10 g column over 16 column volumes) gave the title compound 26; 38 mg, 23% yield; colourless oil; Rf 0.71 (60% EtOAc/n-pentane); mp 138–140 °C (CH2Cl2/n-pentane); νmax (film)/cm−1 2923, 1759, 1634, 1594, 1520, 1496, 1450, 1342, 1094, 1011, 827, 746; δH (400 MHz, CDCl3) 9.48 (br s, 1H, NH), 7.42–7.36 (m, 4H, ArH), 7.03–6.96 (m, 1H, ArH), 6.93 (dd, J = 8.1, 1.6 Hz, 1H, ArH), 6.67 (ddd, J = 7.9, 7.1, 1.6 Hz, 1H, ArH), 6.36 (dd, J = 17.3, 10.6 Hz, CHCHH) 6.34 (br s, 1H, OH), 6.09 (dd, J = 7.9, 1.6 Hz, 1H, CH), 5.36 (d, J = 10.6 Hz, 1H, CHCHH), 5.25 (d, J = 17.3 Hz, 1H, CHCHH), 3.16 (s, 3H, NCH3), 1.81 (s, 3H, CH3); δC (101 MHz, CDCl3) 157.9 (CO), 149.4 (C), 143.5 (C), 140.6 (CHCH2), 140.5 (C), 134.0 (C), 129.7 (CH), 127.1 (CH), 126.4 (CH), 126.3 (CH), 122.0 (CH), 120.1 (CH), 119.9 (CH), 115.6 (CHCH2), 65.1 (C), 33.1 (NCH3), 26.7 (CH3); HRMS (ESI+) [M+H]+ C18H20ClN2O2+ requires 331.1208; found 331.1207.
4.4.3 (±)-2-Phenylbut-3-en-2-ol (33)
Carbamate 30 (0.11 g, 0.40 mmol, 1.0 equiv) was added to a dried Schlenk flask under a N2 atmosphere and dissolved in THF (2 mL, 0.2 M). DMPU (0.24 mL, 2.0 mmol, 5.0 equiv) was added to the flask at room temperature. The mixture was cooled to −40 °C and LDA (0.60 mL, 1.2 mmol, 3.0 equiv, 2.0 M in THF/heptane/ethylbenzene) was added dropwise and the mixture was stirred for 4 h. The mixture was warmed to 70 °C and stirred overnight. The reaction was quenched with saturated aqueous NH4Cl and extracted three times with CH2Cl2. The combined organic phases were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum. The product was purified by flash column chromatography (eluting with 2–25% EtOAc/n-hexane on an SNAP Ultra 10 g column over 14 column volumes) to give the title compound 33; 41 mg, 70% yield; colourless oil; Rf 0.72 (30% Et2O/n-pentane); νmax (film)/cm−1 3398, 3060, 2908, 1492, 1446, 1177, 1064, 1028, 994, 922, 765, 699; δH (400 MHz, CDCl3) 7.50–7.45 (m, 2H, ArH), 7.38–7.32 (m, 2H, ArH), 7.29–7.23 (m, 1H, ArH), 6.18 (dd, J = 17.3, 10.6 Hz, 1H, CHCHH), 5.30 (dd, J = 17.3, 1.1 Hz, 1H, CHCHH), 5.15 (dd, J = 10.6, 1.1 Hz, 1H, CHCHH), 1.92 (br s, 1H, OH), 1.66 (s, 3H, CH3); δC (101 MHz, CDCl3) 146.5 (C), 145.0 (CHCH2), 128.4 (CH), 127.1 (CH), 125.3 (CH), 112.5 (CHCH2), 74.9 (C), 29.5 (CH3). HRMS (ESI+) [M+Na]+ C10H12NONa+ requires 171.0780; found 171.0778; chiral HPLC (Chiralcel OJ column 25 cm × 4.6 mm ID), 98:2 n-hexane/iPrOH, 1 mL/min, 25 °C, tR 22.6 min (major), 33.1 min (minor) er 55:45. Spectroscopic data for 33 match those reported in the literature [38].
4.4.4 (±)-3-(2-Hydroxyphenyl)-1,5-dimethyl-5-vinylimidazolid-ine-2,4-dione (35)
In a dried Schlenk flask under a N2 atmosphere, urea 34 (600 mg, 2.45 mmol, 1.0 equiv) was dissolved in anhydrous THF (25 mL, 0.1 M) and DMPU (2.4 mL, 19.57 mmol, 8.0 equiv) was added at room temperature. The mixture was cooled to −78 °C and LDA (3.7 mL, 7.34 mmol, 3 equiv, 2.0 M in THF/heptane/ethylbenzene) was added dropwise and the mixture was stirred for 6 h at −78 °C. The reaction was quenched with saturated aqueous NH4Cl and extracted three times with EtOAc. The combined organic layers were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum to give a crude residue. Purification of this residue by flash column chromatography (eluting with 25–100% EtOAc/petrol on an SNAP Ultra 25 g column over four column volumes followed by 100% EtOAc over four column volumes) gave the title compound 35; 0.35 g, 58% yield; light yellow solid; Rf 0.2 (EtOAc); mp 75–78 °C; νmax (film)/cm−1 3280, 3400–2500 (br), 2936, 1743, 1656; δH (400 MHz, CDCl3) 7.25–7.12 (m, 2H, ArH), 7.02–6.82 (m, 2H, ArH), 5.84 (dd, J = 17.3, 10.5 Hz, 1H, CHCHH), 5.52–5.32 (m, 2H, CHCH2), 2.87 (s, 3H, NCH3), 1.61 (s, 3H, CH3); δC (101 MHz, CDCl3) 167.3 (CNH), 155.3 (CO), 152.8 (C), 135.8 (C), 130.1 (CH), 128.6 (CH), 120.5 (CH), 119.2 (CH), 118.9 (CHCH2), 64.7 (C), 25.6 (NCH3), 20.7 (CH3); HRMS (ESI+) [M+H]+ C13H16N3O2+ requires 246.1237; found 246.1237. Then an aqueous HCl 2 M solution (2 mL, 4 mmol, 7.5 equiv) was added to the iminohydantoin (130 mg, 0.53 mmol, 1.0 equiv) in methanol (2 mL, 0.26 M) and the reaction mixture was heated to reflux for 24 h. The reaction was quenched by adding saturated aqueous NaHCO3 and extracted three times with EtOAc. The combined organic layers were dried over MgSO4, filtered and the resulting filtrate was concentrated under vacuum to give a crude residue. Purification of this residue by flash column chromatography (eluting with 10–80% EtOAc/petrol on an SNAP Ultra 10 g column over eight column volumes followed by 80% EtOAc over two column volumes) gave the hydantoin 36; 130 mg; 99% yield; white solid; Rf 0.40 (80% EtOAc/n-hexane); mp 142–143 °C (CH2Cl2/n-pentane; νmax (film)/cm−1 3301, 2984, 1172, 1697; δH (400 MHz, CDCl3) 7.29–7.19 (m, 2H, ArH), 7.01–6.95 (m, 2H, ArH), 6.92 (br s, 1H, OH), 5.85 (dd, J = 17.3, 10.5 Hz, 1H, CHCHH), 5.51–5.39 (m, 2H, CHCH2), 2.97 (s, 3H, NCH3), 1.64 (s, 3H, CH3); δC (101 MHz, CDCl3) 173.6 (NCO), 155.4 (NCON), 151.0 (C), 133.7 (CHCH2), 130.0 (CH), 127.5 (CH), 121.0 (CH), 120.3 (C), 119.6 (CH), 119.4 (CHCH2), 65.9 (C), 25.6 (NCH3), 19.5 (CH3); HRMS (ESI)+ [M+H]+ C13H15N2O3+ requires 247.1077; found 247.1069.
4.5 Methanolysis procedure
4.5.1 (−)-(R)-N-Methyl-2-phenylbut-3-en-2-amine hydrochloride (28)
To a solution of urea 19 (24 mg, 81 μmol, 1 equiv) in anhydrous toluene (0.69 mL, 0.1 M), in a dried Schlenk flask under a N2 atmosphere, was added anhydrous MeOH (0.11 mL, 0.1 M) and the mixture was heated to 50 °C and stirred overnight. The solvents were removed under vacuum, and the product was purified by flash column chromatography (eluting with 10–80% Et2O/petrol on an SNAP Ultra 10 g column over 13 column volumes). The combined fractions were concentrated until a small amount of solvent remained. HCl in Et2O (1 mL, 1.0 M) was added to this solution to precipitate a white solid. The solution was decanted and the yellow solid was triturated with Et2O a further three times. The yellow solid was dried under high vacuum to give the title compound 28 (NB: this compound is hygroscopic and therefore we were unable to obtain a satisfactory melting point); 12.5 mg, 78% yield, white solid; Rf 0.11 (70% EtOAc/n-pentane); νmax (film)/cm−1 3213, 1760, 1734, 1479, 1253, 748, 739; = −35 (c 0.2, MeOH); δH (500 MHz, DMSO-d6) 7.54–7.51 (m, 4H, ArH), 7.50–7.45 (m, 1H, ArH), 6.25 (dd, J = 17.5, 11.0 Hz, 1H, CHCHH), 5.64 (d, J = 11.0 Hz, 1H, CHCHH), 5.49 (d, J = 17.5 Hz, 1H, CHCHH), 2.54 (s, 3H, NCH3), 1.86 (s, 3H, CH3); δC (126 MHz, DMSO-d6) 129.6 (C), 129.2 (CHCH2), 120.93 (CH), 120.90 (CH), 118.1 (CH), 110.0 (CHCH2), 56.2 (C), 18.7 (NCH3), 12.3 (CH3); HRMS (ESI+) [M]+ C11H16N+ requires 162.1277; found 162.1285.
4.5.2 (±)-2-(4-Chlorophenyl)–N-methylbut-3-en-2-amine (29)
To a solution of urea 26 (35 mg, 0.11 mmol, 1 equiv) in anhydrous toluene (0.86 mL, 0.1 M), in a dried Schlenk flask under a N2 atmosphere, was added anhydrous MeOH (0.15 mL, 0.1 M) and the mixture was heated to 50 °C and stirred overnight. The solvents were removed under vacuum, and the product was purified by flash column chromatography (eluting with 12–100% EtOAc/petrol on an SNAP Ultra 10 g column over 12 column volumes) to give the title compound 29; 13 mg; 67% yield; colourless oil; Rf 0.21 (50% Et2O/n-pentane); = +50 (c 0.18, CHCl3); δH (500 MHz, CDCl3) 7.40–7.37 (m, 2H, ArH), 7.30–7.27 (m, 2H, ArH), 5.95 (dd, J = 17.4, 10.7 Hz, 1H, CHCHH), 5.22 (dd, J = 10.7, 1.1 Hz, 1H, CHCHH), 5.17 (dd, J = 17.4, 1.1 Hz, 1H, CHCHH), 2.24 (s, 3H, NCH3), 1.47 (s, 3H, CH3); δC (126 MHz, CDCl3) 144.2 (C), 132.5 (C), 128.4 (CH), 128.3 (CHCH2), 128.2 (CH), 113.6 (CHCH2), 60.6 (C), 29.6 (NCH3), 25.3 (CH3); HRMS (ESI+) [M+H]+ C11H15ClN+ requires 196.0888; found 196.0886.
4.6 Side products
4.6.1 N,2-Dimethyl-N-(–1-phenylethyl)benzo[d]oxazole-3(2H)-carboxamide (20)
This compound is formed as a side-product during the optimization of the vinyl migration (see Table 1, entry 2). Purification by flash column chromatography (eluting with 5–50% EtOAc/petrol on a ZIP Sphere 10 g column over 18 column volumes) gave the title compound 20 as a 1:1 mixture of diastereomers; 7 mg, 12% yield; colourless oil; Rf 0.70 (20% EtOAc/n-pentane); δH (400 MHz, CDCl3) 1:1 mixture of diastereomers 7.47–7.28 (m, 10H, ArH), 6.86–6.69 (m, 7H, ArH), 6.58–6.55 (m, 1H, ArH), 6.26 (2 × q, J = 5.8, 5.1 Hz, 2H, 2 × NCHPh), 5.63 (q, J = 7.1 Hz, 1H, OCHN), 5.45 (q, J = 7.0 Hz, 1H, OCHN), 2.71 (s, 3H, NCH3), 2.68 (s, 3H, NCH3), 1.68–1.63 (m, 12H, 2 × OCHNCH3 and 2 × CH3); δC (126 MHz, CDCl3) 157.9 (CO), 150.34 (C), 150.28 (C), 140.6 (C), 140.4 (C), 132.7 (C), 132.5 (C), 128.7 (CH), 128.6 (CH), 127.8 (CH), 127.7 (CH), 127.6 (CH), 122.32 (CH), 122.28 (CH), 121.04 (CH), 121.03 (CH), 111.5 (CH), 111.2 (CH), 109.1 (CH), 109.0 (CH), 94.04 (OCHN), 94.03 (OCHN), 54.3 (NCHPh), 53.5 (NCHPh), 30.9 (NCH3), 30.5 (NCH3), 20.32 (CH3), 20.26 (CH3), 16.9 (CH3), 15.4 (CH3) (1 × CH not observed because of overlapping signals); HRMS (ESI+) [M+H]+ C18H21N2O2+ requires 297.1598; found 297.1594.
4.6.2 1-(2-(2-(1-(4-Chlorophenyl)vinyl)phenoxy)ethyl)-3-methyl urea (27)
Formed during the vinylation of 25 (we were unable to fully purify this compound from the suspected ring expansion product, therefore characterization data reflects this); 122 mg, 74% yield; colourless oil; Rf 0.29 (60% EtOAc/n-pentane); δH (400 MHz, CDCl3) 7.40–7.31 (m, 7H, ArH), 7.06 (t, J = 7.4 Hz, ArH), 6.84 (d, J = 8.2 Hz, ArH), 5.66 (d, J = 1.1 Hz, CCHH), 5.40 (d, J = 1.1 Hz, CCHH), 3.85 (t, J = 4.7 Hz, CH2), 3.34 (t, J = 4.7 Hz, CH2), 2.74 (s, 3H, NCH3); HRMS (ESI+) [M+Na]+ C18H19ClN2O2+ requires 353.1027; found 353.1024.
4.6.3 N-Acetyl-N-methyl-2,3-dihydro-4H-benzo[b][1,4]oxazine-4-carboxamide
Formed during the optimization of the nitrile vinylation chemistry when using s-BuLi as the base; 15 mg; 22% yield; yellow liquid; νmax (film)/cm−1 2935, 2879, 1674, 1495, 1365, 1246, 1058, 752; δH (500 MHz, CDCl3) 7.16 (br dd, J = 8.1, 1.5 Hz, 1H, ArH), 7.08–7.04 (m, 1H, ArH), 6.93 (dd J = 8.1, 1.5 Hz, 1H, ArH), 6.90–6.86 (m, 1H, ArH), 4.36 (t, J = 4.8 Hz, 2H, CH2), 3.91 (br s, 2H, CH2), 3.02 (s, 3H, NCH3), 2.24 (s, 3H CH3); δC (126 MHz, CDCl3) 171.6 (CO), 156.0 (NCON), 146.2 (C), 126.5 (CH), 125.0 (C), 121.7 (CH), 121.2 (CH), 117.9 (CH), 66.0 (CH2), 43.0 (CH2), 33.3 (NCH3), 23.5 (CH3); HRMS (ESI+) [M+Na]+ C12H14N2O3Na+ requires 257.0897; found 257.0898.
Acknowledgements
This work was supported by the ERASMUS+ Programme, the Groningen University Fund and the Hilmar Johannes Backer Foundation, EPSRC (grant EP/L018527), and the University of Bristol.
Appendix A Supplementary data
Copies of 1H NMR and 13C NMR spectra and HPLC traces can be found in the supplementary information document related to this article.
The following is the supplementary data related to this article: