

1 Introduction
With the development of national defense and economy, more and more high energy density compounds (HEDCs) have been extensively studied as ingredients for explosives, fuels and propellant formulations [1–7]. Molecules, which possess the superior explosive performance, have been receiving highly heated attention because they may be substituted for traditional energetic compounds as HEDCs [8–12]. At present, a commonly used method of obtaining HEDCs is to add highly energetic groups as substitutes to a compound or polymer chain in order to increase the energetic character and improve oxygen balance, such as nitro, nitrate, nitramine groups and so on [13,14]. In support of computer technology and theoretical chemistry, computer modeling and quantum simulation have been extensively used to facilitate “molecular design”. Nowadays, an increasing number of scientists throw themselves into “molecular design” to look for good HEDCs. Most research works are focused on high-nitrogen heterocyclic compounds and some important results have been achieved [15,16].
As everyone knows, heterocyclic nitramines constitute a class of organic energetic compounds and have played important roles in the civil and military fields for a long time. Typical examples of these well-known explosives are RDX (1,3,5-trinitro-1,3,5-triazinane) [17,18], HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane) [19,20] and TNAZ (1,3,3-tirnitoazetidine). Recently, another type of organic nitamines compound, polydifluoroaminopurines, entered our line of sight. The basic structures and the atom numbering schemes used for the polydifluoroaminopurines are presented in Fig. 1. Purine received extremely widespread attention in biology because it is a mother frame for many important biologic compounds: guanine, xanthine adenine, hypoxanthine, theobromine and many others [21–23]. In addition, purine has a nitrogen content of more than 46%, and it is also fascinating because of four hydrogen atoms at the ring that can be replaced with functional groups. It is well known that the addition of energy-rich substituents, such as –NO2, –N3, –NO3, and –NNO2, create exceptionally powerful explosives, propellants, and fuels with excellent explosive properties. In the present work, we have conducted a systematic study on the basis of the purine structure modification in order to get satisfactory HEDCs. Compounds containing two fluorine atoms bonded to nitrogen, i.e., a difluoroamino (NF2) group, have been extensively studied as ingredients for propellants and explosives [24–26]. When the difluoroamino groups are attached to the purine skeleton, ploydifluoroaminopurines are formed. In this article, 15 derivatives of purine are designed through substituting the hydrogen atoms in purine using difluoroamino groups, and the geometry, thermodynamic characters, and detonation performance are calculated in detail. These results provide useful information for the molecular design of novel high energetic density materials.
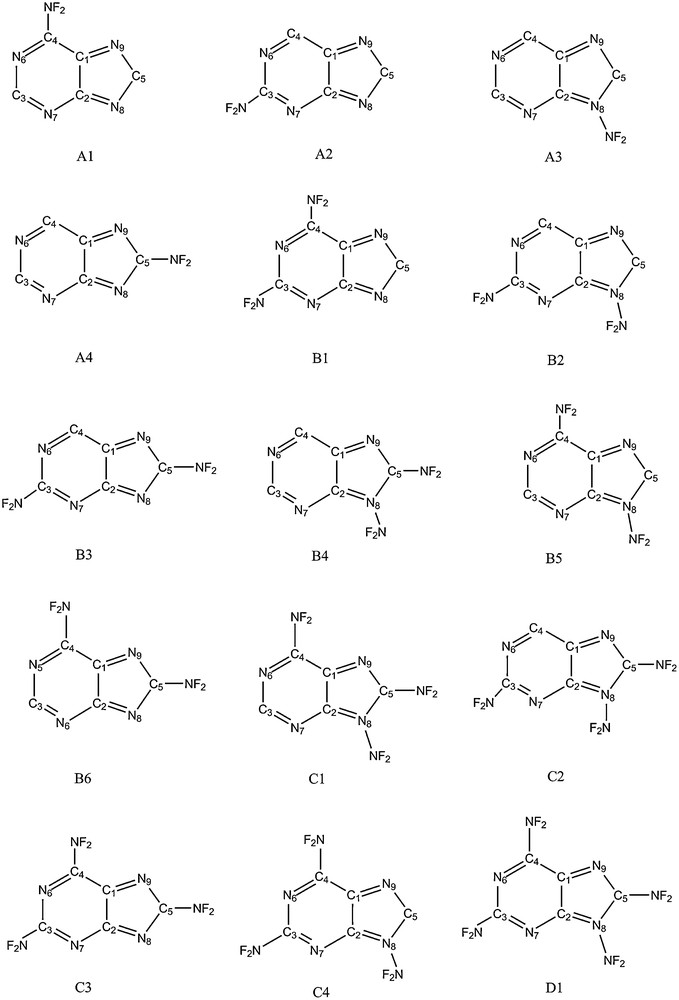
Structures and atom numbering schemes used for the polydifluoroaminopurines.
The remainder of this paper is organized as follows. A description of our computational method is given in Section 2. The results and discussion are presented in Section 3, followed by a summary of our conclusions in Section 4.
2 Computational methods
The geometry of the molecular structures was fully optimized without symmetry restriction using DFT-B3LYP, B3P86 methods with the 6-311G** basis set, in the Gaussian 03 software package [27]. Each optimized structure was checked to ensure that there was a local energy minimum on the potential energy surface by vibrational analysis. Based on these structures, Wiberg bond orders were calculated by using nature bond orbital (NBO) analyses.
In this paper, the predictions of HOFs adopt the hybrid DFT-B3LYP, B3P86 methods with 6-311G** basis set via designed isodesmic reactions [28–31]. The method of isodesmic reations has been employed very successfully to calculate the standard gas-phase heat of formation (ΔfH0Gas). Isodesmic reations are designed to decrease the calculation errors, in which the numbers of all kinds of bonds are kept invariable. These isodesmic reactions that were used to calculate the HOFs of the polydifluoroaminopurines at 298 K are as follows:
| C5H4–xN4(NF2)x + XCH4C5H4N4 + XCH3NF2(X = 1, 2, 3, 4) | (1) |
For reaction, the heats of formation at 298 K by Eq. (3) [32] can be deduced from Eq. (2):
| (2) |
| (3) |
The experimental thermodynamic parameters of C5H4N4 and CH4 are taken from the references [33,34]. An accurate value ΔHf for CH3NF2 was obtained from the references. It was obtained from the replacement reaction CH3NH2 + F2→CH3NF2 + H2 using the G2 theory [35]. The data mentioned above are listed in Table 1. ΣE (0 K, p) corresponds to the energies for the sum of products at 0 K, ΣE (0 K, r) is the sum of the energies of the reactant at 0 K. ΔZPE is the sum of the zero-point correction energies of products and reactants. ΔHT is the thermal correction value, Δ(PV) equal ΔnRT for ideal gas, and it will be zero because Δn is zero in an isodesmic reaction. All values are calculated by us and the final results are listed in Table 2.
Calculated total energy (E0, a.u), zero-point energy (ZPE, a.u), values of thermal correction (HT, a.u), and heats of formation (HOF, kJ/mol) of the reference compounds.
| Compound | B3LYP/6-311G** | B3P86/6-311G** | |||||
| E0 | ZEP | HT | E0 | ZEP | HT | HOF | |
| CH3NF2 | −294.29833 | 0.04676 | 0.00518 | −294.89503 | 0.09563 | 0.00515 | −115.23 |
| C5H4N4 | −412.04628 | 0.09494 | 0.00664 | −413.15575 | 0.04708 | 0.00661 | 18.00 |
| CH4 | −40.53375 | 0.04459 | 0.00381 | −40.71398 | 0.04475 | 0.00382 | −74.40 |
Total energies (E0, a.u), zero-point energies (ZPE, a.u), thermal correction values (HT, a.u), heat of formation (HOF, kJ/mol) of the derivatives of purine.
| Compound | B3LYP/6-311G** | B3P86/6-311G** | ||||||
| E | EZPE | HT | HOF | E | EZPE | HT | HOF | |
| A1 | −665.80411 | 0.09354 | 0.00972 | −40.93 | −667.33032 | 0.09448 | 0.00873 | −43.92 |
| A2 | −665.80719 | 0.09350 | 0.00962 | −49.00 | −667.33353 | 0.09444 | 0.00957 | −49.89 |
| A3 | −665.77312 | 0.09294 | 0.00956 | 43.24 | −667.29889 | 0.09387 | 0.00949 | 43.77 |
| A4 | −665.80133 | 0.09354 | 0.00971 | −29.22 | −667.32756 | 0.09446 | 0.00965 | −29.88 |
| B1 | −919.56535 | 0.09240 | 0.01260 | −76.90 | −921.50952 | 0.09360 | 0.01250 | −81.14 |
| B2 | −919.53248 | 0.09161 | 0.01253 | 5.83 | −921.47570 | 0.09279 | 0.01243 | 4.02 |
| B3 | −919.56171 | 0.09209 | 0.01268 | −70.70 | −921.50497 | 0.09326 | 0.01258 | −72.36 |
| B4 | −919.52148 | 0.09153 | 0.01265 | 38.27 | −921.46430 | 0.09271 | 0.01255 | 37.53 |
| B5 | −919.52846 | 0.09149 | 0.01269 | 16.36 | −921.47106 | 0.09265 | 0.01166 | 13.68 |
| B6 | −919.55867 | 0.09223 | 0.01270 | −61.86 | −921.50182 | 0.09339 | 0.01261 | −63.49 |
| C1 | −1173.27534 | 0.09017 | 0.01573 | 15.60 | −1175.63507 | 0.09160 | 0.01560 | 13.89 |
| C2 | −1173.27597 | 0.08993 | 0.01572 | 12.55 | −1175.63556 | 0.09136 | 0.01558 | 11.19 |
| C3 | −1173.31459 | 0.09066 | 0.01582 | −86.77 | −1175.67818 | 0.09229 | 0.01556 | −98.41 |
| C4 | −1173.28802 | 0.09029 | 0.01562 | −17.33 | −1175.64870 | 0.09175 | 0.01547 | −21.52 |
| D1 | −1427.02969 | 0.08851 | 0.01884 | −7.05 | −1429.80626 | 0.09016 | 0.01869 | −9.59 |
Their solid phase enthalpies of formation [ΔHf(gas)] can be obtained from the gas-phase enthalpies of formation using the Hess law of constant heat summation:
| (4) |
Here, ΔHsub is the sublimation enthalpy evaluated using the following equation suggested by Politzer et al. [36]:
| (5) |
The strength of bonding, which could be evaluated by bond dissociation energy (BDE), is fundamental to understand the chemical processes [37]. The energy required for bond homolysis at 298 K and 1 atm corresponds to the enthalpy of reaction A–B(g) → A(g) + B(g), which is the bond dissociation enthalpy of the molecule A–B by definition [38]. For many organic molecules, the terms “bond dissociation energy” and “bond dissociation enthalpy” often appear interchangeably in the literature [39]. Therefore, at 0 K, the homolytic BDE can be given in terms of Eq. (6):
| BDE0(A–B) = E0(A·) + E0(B·) | (6) |
The bond dissociation energy with zero-point energy (ZPE) correction can be calculated by Eq. (7):
| BDE(A–B)ZPE = BDE0(A–B) + ΔZPE | (7) |
For each compound of interest, the explosive reaction was identified by applying the “most exothermic principle”; for some C–H–N–O–F explosives, the class of HEDCs is calculated using an “HF–CF4–H2O–CO2 arbitrary” [40], according to which the order of precedence in the formation of detonation products is as follows:
- • hydrogen, as available, first reacts with fluorine to form HF;
- • remaining fluorine, if any, goes to CF4;
- • remaining hydrogen, if any, next interacts with oxygen to form H2O;
- • remaining oxygen, if any, oxidizes carbon directly to CO2.
This corresponds to a condition where the equilibria
| (8) |
| (9) |
In detail, the theoretical density of each compound was predicted from the molecular volume divided by molecule weight. The volume was obtained by finding the arithmetic average of 100 single-point molar volumes, defined as the volume of the 0.001 electrons/Bohr3 electro density envelope and computed by Monte Carlo integration [42]. Q was evaluated by the HOF difference between products and explosives according to the principle of exothermic reactions. The corresponding D and P values can be evaluated on the basis of the ρ and Q values. But the procedure used to estimate densities could lead to significant errors. An electrostatic interaction correction for improved crystal density prediction also had been studied by Politzer et al. [43,44]. The method is showed by eqs. (10–14).
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
Here, υ is balance parameter, V(r) is electrostatic potential, V(ri) is the value of V(r) at any point ri on the surface,
3 Results and discussion
3.1 Heats of formation
Fig. 1 shows the numbering scheme for 15 polydifluoroaminopurine isomers. Table 1 lists the total energies, zero-point energies and the values of thermal correction at the B3LYP/6-311G** and B3P86/6-311G** levels for three reference compounds being enlisted in the isodesmic reaction. The experimental thermodynamic parameters of C5H4N4 and CH4 are taken from the references [33,34]. An accurate value ΔHffor CH3NF2 was obtained from the references. It was obtained from the replacement reaction CH3NH2 + F2→CH3NF2 + H2 using the G2 theory [35]. Table 2 summarizes the total energies, zero-point energies and the value of HOFs obtained through using Eq. (3). Previous studies showed that the theoretically predicted values of HOF were in good agreement with experiments by choosing appropriate reference compounds in the isodesmic reaction. An efficient strategy of reducing errors of HOFs is to keep bonds unbroken as much as possible. This approach has been proved to be reliable [31,45]. On this basis, data in Table 2 are supposed to be credible, although the experimental values of HOFs are unavailable for comparison.
We calculated the HOFs at B3LYP/6-311G** and B3P86/6-311G** levels to evaluate the method dependence. The results show that the HOFs for the polydifluoroaminopurine derivatives at the B3P86/6-311G** level are systematically smaller than those at the B3LYP/6-311G** level. There are very good linear relationships between the HOFs from B3LYP and B3P86 methods with 6-311G**: HOFB3P86 = 1.0373HOFB3LYP – 1.7813 with R = 0.9986. Also noteworthy is the HOFs of polydifluoroaminopuine isomers with the same number of –NF2 groups are affected by the position of difluoroamine groups. As for four monodifluoroaminoadamantane isomers, the HOFs of A1, A2, and A4 are close to each other, while the HOF of the A3 is higher than the other three derivatives. The HOFs of six didifluoroaminepurines isomers showed that the HOF values of B2, B4, B5 are bigger than B1, B3, B6. Similar results can be found in the three substation groups. From which we can see that when –NF2 attaches to the N atom of derivatives, the HOFs values is bigger than when –NF2 attaches to the C atom of derivatives. It is suggested that title compounds are more stable when the difluoroamino groups are attached to the C atom of purine than to the N atom.
3.2 Electronic structure and molecular stability
Table 3 lists the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies and the energy gaps (ΔELUMO–HOMO), at the B3LYP/6-311G** and B3P86/6-311G** Level. Results show that the EHOMO values and ELUMO values at the B3P86/6-311G** level are systematically smaller than those at the B3LYP/6-311G** level. The variations of ΔELUMO–HOMO are very similar at the two levels. Many studies have indicated that the HOMO–LUMO energy gap is an indicator of chemical stability [46,47]: the smaller the energy gap, the lower the chemical stability. From Table 3, it can be seen that the ΔELUMO–HOMO values decrease as the number of difluoroamino groups increases. In addition, all of the polydifluoroaminopurines have lower ΔELUMO–HOMO values than the unsubstituted purine. It can be deduced that the stability generally decreases as the substituent number increases. Among A1–A4, the ΔELUMO–HOMO value of A4 is the smallest, which is 0.18945 a.u.; those of A1, A2 and A3 are large, i.e. 0.19526 a.u., 0.20367 a.u. and 0.20069 a.u., respectively. Therefore, it indicates that A1, A2 and A3 are slightly more stable than A4. For series C, the stabilities of C1, C2, and C3 are higher than that of C4. As for B1–B6 compounds, the ΔELUMO–HOMO value of B3 is 0.19643 a.u., which is larger than that of other compounds. The stability here refers to the chemical or photochemical processes with electron transfer or electron leap [48,49]. It should be pointed out that the sensitivity was estimated using ΔELUMO–HOMO, which gives only a rough estimate.
Calculated HOMO and LUMO energies (in a.u) as well as the energy gaps (ΔELUMO–HOMO) of the title compounds, calculated at the B3LYP/6-311G** and B3P86/6-311G** levels.
| Compound | B3LYP/6-311G** | B3P86/6-311G** | ||||
| ELUMO | EHOMO | ΔELUMO–HOMO | ELUMO | EHOMO | ΔELUMO–HOMO | |
| C5H4N4 | −0.05496 | −0.25743 | 0.20247 | −0.07819 | −0.28122 | 0.20303 |
| A1 | −0.08775 | −0.28301 | 0.19526 | −0.10932 | −0.30467 | 0.19535 |
| A2 | −0.08037 | −0.28404 | 0.20367 | −0.10207 | −0.30604 | 0.20397 |
| A3 | −0.07639 | −0.27708 | 0.20069 | −0.09827 | −0.29821 | 0.19994 |
| A4 | −0.08950 | −0.27895 | 0.18945 | −0.11057 | −0.29898 | 0.18841 |
| B1 | −0.09418 | −0.27895 | 0.18477 | −0.11463 | −0.29945 | 0.18482 |
| B2 | −0.09395 | −0.28726 | 0.19331 | −0.11518 | −0.30801 | 0.19283 |
| B3 | −0.10017 | −0.29660 | 0.19643 | −0.12062 | −0.31862 | 0.19800 |
| B4 | −0.10114 | −0.28945 | 0.18831 | −0.1212 | −0.31098 | 0.18978 |
| B5 | −0.10549 | −0.29867 | 0.19318 | −0.12694 | −0.31998 | 0.19304 |
| B6 | −0.10549 | −0.29580 | 0.19031 | −0.12604 | −0.31777 | 0.19173 |
| C1 | −0.12307 | −0.31066 | 0.18759 | −0.14344 | −0.33148 | 0.18804 |
| C2 | −0.12584 | −0.31505 | 0.18921 | −0.14643 | −0.33578 | 0.18935 |
| C3 | −0.12330 | −0.31198 | 0.18868 | −0.13477 | −0.32032 | 0.18555 |
| C4 | −0.02767 | −0.18808 | 0.16041 | −0.13086 | −0.31478 | 0.18392 |
| TATB | −0.10280 | −0.26480 | 0.16210 |
Bond order is a measure of the overall bond strength between two atoms. A high value of the bond order indicates a covalent bond, while a low value shows an ionic nature. The ring–NF2 of the purine derivations presents the smallest bond order by NBO analyses in Table 4. Therefore, the ring–NF2 bonds are the weakest and are easier to break than other bonds according to the principle of the smallest bond order (PSBO) [50]. Moreover, numerous studies show that the energy barriers for the skeleton C–C or C–N ruptures of ring compounds are much larger than those of the ring –NF2 ruptures [51,52]. The rupture of the ring –NF2 bond is affected by the strong electronegativity of the F atom of NF2 group. It can be predicted that the ring –NF2 bond is an initiator during the thermolysis process. To elucidate the pyrolysis mechanism and thermal stability of title compounds, we calculated the BDE for the possible initial steps in the pyrolysis route. The BDE is a key parameter for estimating the thermodynamic stability, and it can provide useful information for understanding the stability of a compound. Generally speaking, the stronger are the weakest bonds, the more stable are the energetic materials. Therefore, the BDE is directly related to the stability of energetic compounds. Table 5 presents the bond order, BDE and BDEZPE of purine derivatives along with those of commonly used explosives RDX and HMX. However, we should also note that such trigger bonds are not the only possible route to decomposition for some HEDCs [53].
Calculated Wiberg bond index (WBI) of part bonds of the purine derivatives at the B3LYP/6-311G** level.
| Compound | B3LYP/6-311G** | B3P86/6-311G** | ||
| PC-NF2 | PN-NF2 | PC-NF2 | PN-NF2 | |
| A1 | 0.9647 | 0.9642 | ||
| A2 | 0.9497 | 0.9490 | ||
| A3 | 1.0864 | 1.0814 | ||
| A4 | 0.9963 | 0.9951 | ||
| B1 | 0.9825 | 0.9867 | ||
| B2 | 0.9854 | 1.0782 | 0.9894 | 1.0732 |
| B3 | 0.9496 | 0.9492 | ||
| B4 | 1.0107 | 1.0675 | 1.0121 | 1.0623 |
| B5 | 0.9649 | 1.0774 | 0.9644 | 1.0731 |
| B6 | 0.9655 | 0.9652 | ||
| C1 | 0.9656 | 1.0594 | 0.9654 | 1.0555 |
| C2 | 0.9488 | 1.0551 | 0.9483 | 1.0506 |
| C3 | 0.9520 | 0.9513 | ||
| C4 | 1.1647 | 0.9824 | 0.9961 | 1.0668 |
| D1 | 0.9540 | 1.0501 | 0.9534 | 1.0467 |
Calculated bond dissociation energies (BDE, kJ/mol) for the rupture of the weakest bond, calculated at the UB3LYP/6-311 G** level.
| Compound | BDE | BDEZPE | Trigger bonder | Bonder order |
| A1 | 252.64 | 238.65 | C4–NF2 | 0.9647 |
| A2 | 272.00 | 257.40 | C3–NF2 | 0.9497 |
| A3 | 179.35 | 160.35 | N8–NF2 | 1.0864 |
| A4 | 282.17 | 267.50 | C5–NF2 | 0.9963 |
| B1 | 271.85 | 257.06 | C3–NF2 | 0.9825 |
| B2 | 270.94 | 255.94 | C3–NF2 | 0.9854 |
| B3 | 268.81 | 254.23 | C3–NF2 | 0.9496 |
| B4 | 271.47 | 256.90 | C5–NF2 | 1.0107 |
| B5 | 247.96 | 234.08 | C4–NF2 | 0.9649 |
| B6 | 249.77 | 235.58 | C4–NF2 | 0.9655 |
| C1 | 245.60 | 231.56 | C4–NF2 | 0.9656 |
| C2 | 263.79 | 249.39 | C3–NF2 | 0.9488 |
| C3 | 264.53 | 250.25 | C3–NF2 | 0.9520 |
| C4 | 165.00 | 132.74 | N8–NF2 | 0.9824 |
| D1 | 147.90 | 131.80 | C3–NF2 | 0.9540 |
| RDX | 166.19 | 145.62 | – | – |
| HMX | 178.77 | 160.41 | – | – |
As shown in the Table 5, it is found that all the calculated BDE and BDEZPE are larger than 120 kJ/mol. Especially, the BDE and BDEZPE of purine derivatives except A3, C3 and C4 are more than 200 kJ/mol, which indicated that all the polydifluoroaminopurines have good thermal stabilities. Based on the BDE and bond order values of the title compounds, we found that BDEs are consistent with bond order values. From the BDE and BDEZPE values, it is found that those without zero-point energies correction are larger than those including zero-point energies correction. However, the order of the dissociation energy is not affected by the zero-point energies. Compared with the commonly used explosives RDX and HMX, the dissociation energies of polydifluoroaminopurines are larger than those of RDX (152.09 kJ/mol) and HMX (166.48 kJ/mol), except for A3, C4 and D1, which means that these compounds suffice stability request of explosives and they are promisingly well-behaved HEDCs. It is found that polydifluoroaminopurines have higher BDE for the weakest for initial steps in the thermal decompositions; it may be inferred that these derivatives are more insensitive to thermal impact.
3.3 Detonation properties
Detonation velocity and detonation pressure are two important performance parameters for an energetic material. The Kamlet–Jacobs equations have been proved to be reliable for predicting the explosive properties of polydifluoroaminopurines. The Kamlet–Jacobs equations show that ρ is a key factor to influence D and P. However, it should be pointed out that calculations by equations (6) and (7) reproduced the total body of experimental detonation property measurements for ideal C–H–N–O explosives [41,54]. Commonly used classic explosives were not covered with halogen elements when the Kamlet–Jacobs equations were demonstrated. But even so, most people calculate the detonation performance for C–H–N–O–F explosives by means of K–J equations, and the results seem to be reliable [26,55–58]. In this article, Table 6 presents the calculated ρ, Q, D, and P of the polydifluoroaminopurines. For comparison, the detonation performances of four known explosives RDX, HMX, are listed in Table 6.
Predicted detonation properties of the purine derivatives together with RDX, HMXa.
| Compound | ρ (g/cm3) | Q (cal/g) | D (km/s) | P (GPa) |
| A1 | 1.82 | 733.22 | 6.44 | 18.53 |
| A2 | 1.85 | 721.94 | 6.48 | 18.90 |
| A3 | 1.75 | 850.86 | 6.50 | 18.48 |
| A4 | 1.76 | 749.58 | 6.31 | 17.45 |
| B1 | 1.93 | 1037.82 | 7.52 | 26.11 |
| B2 | 1.94 | 1126.89 | 7.73 | 27.70 |
| B3 | 1.97 | 1044.49 | 7.66 | 27.40 |
| B4 | 1.89 | 1161.81 | 7.64 | 26.71 |
| B5 | 1.95 | 1138.22 | 7.77 | 28.12 |
| B6 | 1.96 | 1054.01 | 7.63 | 27.13 |
| C1 | 2.01 | 1341.08 | 8.42 | 33.54 |
| C2 | 2.05 | 1338.41 | 8.52 | 34.63 |
| C3 | 2.05 | 1251.46 | 8.38 | 33.60 |
| C4 | 2.13 | 1312.26 | 8.71 | 36.96 |
| D1 | 2.08 | 1463.93 | 8.90 | 38.16 |
| RDXb | 1.78(1.82) | 1591.03 | 8.87(8.75) | 34.67(34.00) |
| HMXb | 1.88(1.91) | 1633.90 | 9.28(9.10) | 39.19(39.00) |
As is evident in Table 6, it is found that ρ values become large as the number of dinitroamino groups is increased from A1 to C6, but when the number of substituents is four, the ρ values become slightly smaller. That is because the range of D1 molecular volume increased more than the degree of molecular mass. For the 15 purine derivatives, ρ is in the range 1.75–2.13 g/cm3, D is in the range 6.31–8.90 km/s, and P is the range 17.45–38.16 GPa, respectively. As the numbers of substituents increase, the Q, D, and P values of the polydifluoroaminopurines enhance. It is observed from Table 6 that purine derivatives except monodifluoroaminopurines have higher ρ values than RDX (1.82 g/cm3) and HMX (1.91 g/cm3). Compared with the conventional explosives, the explosive performances of C4 and D1 have higher D and P values than RDX. It is a pity that none of them exhibit better explosive performances than HMX. So, these results support the claim that introducing difluoroamino groups usually enhances its detonation performance. We suggest that the replacement of the hydrogen atoms of purine molecule with difluoroamino groups would make the resulting molecules some new potential HEDCs.
However, Short et al. put forward a calibrated equation according to the Chapman–Jouguet theory in the early 1980s [41]. It is plausible to seek a method of rationalizing all cylinder test results for ideal explosives. The method pointed up that a relation could serve as a valuable tool in comparing the ideal explosives, and of C–N–O, H–N–O, and C–H–N–O–F explosives relative to C–H–N–O explosives. In the literature, Short et al. stated in details that the equation for predicting wall velocities in the cylinder test:
| (15) |
| (16) |
Cylinder wall veloities in the cylinder test data used to calibrate equation.
| Wall velocites mm/μs at R–R0 (cm) | ||||||||||||
| Compound (ρ, ϕ) | 0.6 | 0.8 | 1.0 | 1.2 | 1.4 | 1.6 | 1.8 | 2.0 | 2.2 | 2.4 | 2.6 | |
| A1 (1.82 g/cm3, ϕ = 3.680) | 1.17 | 1.21 | 1.23 | 1.25 | 1.27 | 1.29 | 1.30 | 1.31 | 1.33 | 1.34 | 1.35 | |
| A2 (1.85 g/cm3, ϕ = 3.652) | 1.18 | 1.22 | 1.24 | 1.26 | 1.28 | 1.30 | 1.31 | 1.32 | 1.33 | 1.35 | 1.35 | |
| A3 (1.75 g/cm3, ϕ = 3.934) | 1.18 | 1.21 | 1.24 | 1.26 | 1.28 | 1.29 | 1.31 | 1.32 | 1.34 | 1.35 | 1.36 | |
| A4 (1.76 g/cm3, ϕ = 3.704) | 1.14 | 1.17 | 1.20 | 1.22 | 1.24 | 1.26 | 1.27 | 1.28 | 1.30 | 1.31 | 1.32 | |
| B1 (1.93 g/cm3, ϕ = 4.585) | 1.39 | 1.42 | 1.45 | 1.48 | 1.50 | 1.51 | 1.53 | 1.54 | 1.56 | 1.57 | 1.58 | |
| B2 (1.94 g/cm3, ϕ = 4.775) | 1.43 | 1.47 | 1.50 | 1.52 | 1.54 | 1.56 | 1.57 | 1.59 | 1.60 | 1.61 | 1.62 | |
| B3 (1.97 g/cm3, ϕ = 4.605) | 1.42 | 1.46 | 1.48 | 1.51 | 1.53 | 1.54 | 1.56 | 1.57 | 1.59 | 1.60 | 1.61 | |
| B4 (1.89 g/cm3, ϕ = 4.839) | 1.41 | 1.45 | 1.47 | 1.50 | 1.52 | 1.54 | 1.55 | 1.57 | 1.58 | 1.59 | 1.61 | |
| B5 (1.95 g/cm3, ϕ = 4.798) | 1.44 | 1.48 | 1.51 | 1.53 | 1.55 | 1.57 | 1.58 | 1.60 | 1.61 | 1.62 | 1.63 | |
| B6 (1.96 g/cm3, ϕ = 4.625) | 1.42 | 1.45 | 1.48 | 1.50 | 1.52 | 1.54 | 1.55 | 1.57 | 1.58 | 1.59 | 1.60 | |
| C1 (2.01 g/cm3, ϕ = 5.366) | 1.57 | 1.61 | 1.64 | 1.67 | 1.69 | 1.71 | 1.72 | 1.74 | 1.75 | 1.76 | 1.77 | |
| C2 (2.05 g/cm3, ϕ = 5.362) | 1.60 | 1.63 | 1.66 | 1.69 | 1.71 | 1.73 | 1.74 | 1.76 | 1.77 | 1.78 | 1.79 | |
| C3 (2.05 g/cm3, ϕ = 5.189) | 1.57 | 1.61 | 1.64 | 1.66 | 1.68 | 1.70 | 1.71 | 1.73 | 1.74 | 1.75 | 1.76 | |
| C4 (2.13 g/cm3, ϕ = 5.308) | 1.64 | 1.68 | 1.71 | 1.73 | 1.75 | 1.77 | 1.78 | 1.80 | 1.81 | 1.82 | 1.83 | |
| D1 (2.08 g/cm3, ϕ = 5.720) | 1.68 | 1.72 | 1.74 | 1.77 | 1.79 | 1.81 | 1.83 | 1.84 | 1.85 | 1.87 | 1.88 | |
| HMX | ||||||||||||
| (1.891 g/cm3, ϕ = 6.772) | 1.69 | 1.73 | 1.77 | 1.81 | 1.84 | 1.85 | 1.86 | 1.87 | 1.88 | 1.90 | 1.91 | |
| 1.69 | 1.73 | 1.77 | 1.80 | 1.82 | 1.84 | 1.86 | 1.88 | 1.90 | 1.91 | 1.92 |
a
From Table 7, it can be seen that the wall velocity
4 Conclusions
From the above calculations and analyses above, the following conclusions can be drawn:
- • the HOFs of the title compound increase almost constantly with each difluoroamino group attached to the purine skeleton. And different substituted positions and the number of the substituent can affect the polydifluoroaminopurines HOFs. From which, we can see that when –NF2 attaches to the N atom of derivatives, the heat of formation is bigger than when –NF2 attaches to the C atom of derivatives;
- • the bond dissociation energies values of all title compounds are over 120 kJ/mol. It indicates that all the polydifluoroaminopurines have good thermal stabilities. In addition, this result also shows that to judge the thermal stability of purine derivatives is not possible by the bond order simply;
- • the calculated molecular densities, detonation velocities, and detonation pressures of the polydifluoroaminopurines indicate that difluoroamino groups play a very important role in enhancing their density and detonation performances. As it is well known, a superior performance HEDCs candidate not only has excellent detonation properties but also could exist stably. In addition, we also calculate the cylinder wall velocities by means of the simplified calculational method. It is worth noting that these results of wall velocity are very close to the explosive velocity (D) using K–J equation.
To sum up, it may be concluded that C3 and C4 have a chance as potential candidates of HEDMs, which have less sensitivity and higher performance, good detonation performance (D and P) and thermal stability (BDE) over RDX. These two compounds present good performance and are worthy of synthesis and further investigation. Our results should also provide some useful information for the molecular design of novel HEDCs.
Acknowledgments
This work is supported by the Natural Science Foundation of Shanxi Province (No.2010021009-2 and 2010011012-1), the Natural Science Foundation of China (No.20871077, No.21031003), the Research Project Supported by Shanxi Scholarship Council of China (No.201063) and the Natural Science Foundation of Shanxi Normal University.
Vous devez vous connecter pour continuer.
S'authentifier