

1 Introduction
Fluid lipid bilayer is the landmark structure of all biological membranes [1,2]. Lipids segregate within the membrane into different domains depending on their alkyl chain, head group and degree of saturation [3,4]. Saturated lipid molecules are unique for their ability to self-assemble into crystalline ordered domains because of the linear geometry of the alkane chains and the van der Waals interactions between methylene groups [5]. Their self-assembly ability has already resulted in numerous examples of artificial monolayer and bilayer films [6]. However, little has been reported on expanded multilayers, which upon successive addition of saturated lipid bilayers could stack to form non-fluid three-dimensional (3D) crystalline materials [7].
Non-lipid saturated amphiphiles appear to have been more successful to afford 3D lamellar films [8–11]. A very promising methodology relies on organosilane hybrid precursors of the structure: H3C(CH2)nSiX3 [12–17] and X3Si(CH2)nSiX3 [18,19] (X = alkoxy, Cl) that self-assemble into inorganic–organic multilayer structures. Hybrid materials with organic–inorganic character represent a very dynamic field of research [20,21]. Their novel properties and multifunctional nature has opened up many opportunities of applications in very different domains [22]. In particular, these lamellar hybrids represent attractive materials in polymer nanocomposites [22,23], catalysis [24], optoelectronics [12,25,26] and photolithography [27,28]. In contrast to the first class of metallic hybrid mesostructures forming spontaneously, the self-organization of alkylsilanes is triggered by concomitant hydrolysis and polycondensation of the reactive group X, referred to as the sol–gel process. Acid-catalyzed hydrolysis replaces the –SiX functions by –SiOH silanol functions, thus, driving the in situ generation of amphiphilic n-alkyltrisilanol species. Subsequent polycondensation reactions yield a cross-linked siloxane layers separating bilayered stacks of alkyl chains. In general, it is very difficult to achieve the long-range order of crystalline films with monosilylated precursors [29], and a more popular approach relies on symmetric bridged precursors [18,30–36]. The result is a very small number of studies reporting the lamellar structuring of n-alkylsilsesquioxane films, in this way, mimicking the organization of saturated lipids [12–19].
In 2011, we introduced an alternative UV-driven sol–gel process performed without solvent and water, enabling the formation of crystalline multilayer n-alkylsilsesquioxane films from trichloro- [37] or trimethoxy-based [38] monosilylated n-alkylsilanes. In our procedure, a photoacid generator (C12H25)2Φ2I+SbF6–) was solubilised into the precursor. Upon UV irradiation, the liberated H+SbF6– Brønsted superacids caused both the fast sol–gel photopolymerization and the self-assembly process. The important benefits of using a solvent-free pathway are to circumvent the long hydrocarbon chain insolubility and the solvation effect perturbing the self-assembly process. The resulting 3D lamellar structures with bilayered hydrocarbon chains exhibited a strong resemblance with crystalline lipid domains. A very intriguing feature, which serves as a starting point to this study, has been the possibility to tailor the alkyl packing arrangement, depending on the chemical nature of the anchoring group (X). As depicted in Fig. 1, we performed the photoacid-catalyzed sol–gel process of two n-octadecyl monosilylated precursors, which differ in the nature of the –SiX hydrolyzable function –C18H37SiCl3 (C18TCS) and C18H37Si(OCH3)3 (C18TMS): the trichlorosilane derivative yielded a stacking of non-interpenetrated bilayers while the trimethoxysilane precursor favored an equivalent interdigitated structure, with alkyl chains of a first layer intercalated in the space between two chains of an opposing layer.

Effect of the hydrolyzable group (X = OCH3 or Cl) on the packing geometry of the lamellar n-octadecylsilsesquioxane mesostructure.
Our characterization effort was mainly focused on a combination of standard solid-state nuclear magnetic resonance (NMR) techniques: 1H, 29Si, 13C, 2D WISE. The chemical structure of the siloxane and hydrocarbon sheets, the alkyl conformational order and thermal stability are all important issues that were systematically investigated and compared in the two mesostructures. Another goal of this study was to examine whether the bilayer arrangement has an effect on the alkyl chain dynamics. It seems intuitively plausible that an interdigitated mesostructure by constraining the mobility of the chain ends may reduce the extent of chain motion compared to a non-interpenetrated system. The dependency between dipolar couplings and the respective mobility and separation of the nuclei involved makes solid-state NMR a well-suited technique to assess the local dynamical properties of these polymeric structures [39,40].
2 Experimental
2.1 Chemicals
n-Octadecyl trimethoxysilane (C18TMS, 97% wt) and n-octadecyl trichlorosilane (C18TCS, 97% wt) were purchased from ABCR. Benzophenone was used as a photosensitizer and the PAG UV 1241 (bis-dodecyl diphenyliodonium hexafluoroantimonate salt) were provided by Aldrich and Deuteron, respectively. All of the chemicals were used as received without further purification.
2.2 Synthesis of multilayer hybrid films based on C18TMS and C18TCS
In a typical procedure, photoacid generator UV1241 (2% wt) and benzophenone (2% wt) were dissolved in the C18TMS or C18TCS precursors to form a photolatent solution in the absence of UV light. The resulting formulation was then deposited on a glass substrate or silicon wafers (SAXS analysis) previously treated by a 20% wt NaOH solution using an Elcometer 4340 automatic film applicator equipped with a wire wound bar to produce a 5-μm-thick liquid film. Irradiation was performed at room temperature under a UV conveyor with a belt speed of 10 m/min using a UV lamp (H lamp, Fusion) with a light intensity of 1.46 J/cm2 per pass. The C18TMS and C18TCS hybrid films were subjected to five successive passes under the conveyor to yield solid poly(n-alkylsilsesquioxane) films labelled as C18PSTCS and C18PSTMS, respectively. During UV irradiation, the room's humidity rate was maintained between 30 and 35% as assessed with a hygrometer.
2.3 Characterization
X-ray diffraction patterns were obtained on a PANalytical X’pert Pro diffractometer with fixed slits using Cu Kα radiation (λ = 1.5418 Å) and θ–2θ mounting. Before analysis, films on silicon wafers were directly deposed on a stainless steel sample holder. Data were collected between 0.5 and 10° 2θ degrees with a scanning step of 0.01° s−1. 13C (I = 1/2) magic angle spinning (MAS) and 1H–13C cross polarization magic angle spinning (CP–MAS) NMR spectra were recorded with a Bruker double-channel 4-mm probe with a spinning frequency of 12 kHz on a Bruker Avance II 400 spectrometer operating at B0 = 9.4 T (Larmor frequency ν0 (13C) = 100.63 MHz and ν0 (1H) = 400.17 MHz). 13C single-pulse MAS NMR experiments were performed with a π/4 pulse duration of 2.5 μs and a 60 s recycling delay, these recording conditions ensuring the quantitative determination of the proportions of the different carbon species. Typically, 1000 scans were recorded. 1H–13C CPMAS NMR experiments were acquired using a ramp for Hartmann–Hahn matching with 8 s recycling delay and a contact time of 1 ms. The radiofrequency field strength used for 1H decoupling was set at 66 kHz. For the variable-temperature CP-MAS experiments, the sample temperature was controlled to within ± 0.2 °C by a Bruker variable-temperature unit (BVT3000). Typically, 132 scans were recorded. 1H–13C HETCOR/WISE experiments were performed under the conditions of MAS. A 1H 90° pulse of 5.1 μs was followed by a proton evolution period t1 consisting of 256 increments of 20 μs. After each t1 period, cross polarization (contact time = 1 ms) followed by carbon detection with continuous wave proton decoupling of 64 kHz gives a carbon spectrum that is modulated as a function of t1 by free induction decay of the associated protons. The spectral widths were 28 and 200 kHz in F1 and F2 dimensions, respectively. Typically, 256 scans were taken for each slice, with a repetition time of 8 s.The processed data sets contained 1024 points in the F2 (13C) dimension and 256 points in the F1 (1H) dimension. 1H (I = 1/2) MAS NMR experiments were performed at room temperature on a Bruker Avance II 400 spectrometer operating at B0 = 9.4 T (Larmor frequency ν0 = 400.17 MHz). Single-pulse experiments (SPE) were carried out with a double-channel 2.5-mm Bruker MAS probe, a spinning frequency of 30 kHz and a π/2 pulse duration of 3.5 μs. 1H spin lattice relaxation times (T1) were measured with the inversion-recovery pulse sequence for all samples. Typically, 64 scans were recorded. 29Si (I = 1/2) 1H–29Si CP–MAS NMR spectra were recorded at room temperature, using a ramp for Hartmann–Hahn matching, 8 s recycling delay and a contact time of 4 ms, on a Bruker Avance II 300 spectrometer operating at B0 = 7.1 T (Larmor frequency ν0 (29Si) = 59.62 MHz and ν0 (1H) = 300.13 MHz) with a Bruker double-channel 7-mm probe with a spinning frequency of 4 kHz. The radiofrequency field strength used for 1H decoupling was set at 55 kHz. Typically, 1000 scans were recorded. Chemical shifts reported thereafter are relative to tetramethylsilane for 29Si, 13C and 1H. Deconvolutions of the spectra were peformed using Dmfit software [41]. Thermally induced weight-loss curves were obtained through Q500 thermogravimetric analyzer (TA instruments) with a nitrogen flow of 20 mL/min; the temperature range was over 25–1000 °C at a heating rate of 5 °C/min.
3 Results
3.1 Small-angle X-Ray diffraction
Fig. 2 displays the X-ray diffraction patterns of the C18PSTCS and C18PSTMS hybrid films, derived respectively from C18TCS and C18TMS sol–gel photopolymerization. Both patterns are consistent with a multilayer mesostructure composed of alkyl chains bilayers separated by siloxane sheets. The presence of several peaks at the intermediate scattering angles suggests a long-range periodicity [42], although a relative broad 001 peak is observed in both structures (full width at half maximum of 2θ value of 0.11° and 0.20° for C18PSTCS and C18PSTMS, respectively). The major difference between the two films prepared from alkoxy- and chloro- precursors appears in the position of the low-angle 001 peak, which is indicative of a distinct packing arrangement of the hydrocarbon chains forming the interior bilayer. Compared to the theoretical length of a fully extended C18H37SiOx unit (l = 2.62 nm), the d-spacing value of 5.08 nm (∼2 × l) found with C18PSTCS can be related to a conventional bilayer organization with a head-to-head arrangement. In the case of C18PSTMS, the distance of 2.73 nm, close to the theoretical chain length suggests distinctively fully interdigated bilayers with alternating up-and-down octadecyl chains. Well-defined strips are clearly observed through the TEM images of both samples (Fig. 2), which are consistent with a lamellar morphology. The inter-lamellar distances of 5.1 nm and 2.6 nm found with C18PSTCS and C18PSTMS, respectively (utilizing Fourier transform for image processing), are quite close to those obtained by X-ray diffraction data. Differences in self-aggregation kinetics or in the size of the leaving group (OMe vs Cl) could be at the origin of the different chain packing. The wide-angle diffraction data shown in Supplementary data, Fig. S1 provides additional insight into the chain–chain spacing. The distances of 4.16 Å and 4.12 Å for C18PSTCS and C18PSTMS are close to those observed in dense solid phases of alkyl chains [13]. While the general mesoscopic organization of the hybrid films can be established through XRD and TEM analyses, information about composition, mobility and molecular interactions that form the basis of the self-assembled lamellar mesostructure was studied by a combination of solid-state NMR methods.

(Top) XRD patterns of C18PSTCS and C18PSTMS hybrid lamellar films. A trend for interdigitation is evidenced in C18PSTMS sample while C18PSTCS is characterized by non-interpenetrated bilayers. (Bottom) TEM images showing the multilayered mesostructures.
3.2 Solid-state NMR
3.2.1 29Si CP–MAS NMR
The partial condensation of the inorganic silica interlayer is clearly evidenced using 29Si CP–MAS NMR. As shown in Fig. 3, the spectra of the irradiated samples C18PSTMS and C18PSTCS are dominated by T2 ((HO)(R)Si(OSi)2, –58 ppm) and T3 ((R)Si(OSi)3, –68 ppm) siloxane species. Additional FT–IR analysis (not presented) revealed for both samples, a complete hydrolysis after the photo-irradiation process. Compared to C18PSTCS, the minor contribution of trisiloxane units (T3) in C18PSTMS indicates less condensed siloxane sheets, a result supported also by the presence of T1 ((HO)2(R)Si(OSi), –50 ppm) subspecies in this latter sample. The higher reactivity of the chlorosilyl functions compared to alkoxysilyl groups, presumably, accounts for the greater degree of condensation reached by C18PSTCS [43]. On the other hand, the hydrolysis of the chlorosilyl moieties produces in situ HCl, which is able to catalyze condensation reactions and might also contribute to the formation of denser films. Quantitative single-pulse experiments (SPE) (not provided) revealed a tendency similar to that of the C18PSTMS film, exhibiting a slightly less condensed structure (T3: 52.3%, T2: 47.7%) than the C18PSTCS film (T3: 54.7%, T2: 45.3%). As expected, the difference in condensation is less pronounced than in the CP–MAS experiment, thus, suggesting that this parameter is unlikely to be behind the variation in chain packing (Fig. 3).

1H–29Si CP–MAS NMR spectra of the UV-cured C18PSTCS and C18PSTMS films.
3.2.2 13C CP– and SPE–MAS NMR
The n-octadecyl chain conformation and its thermal stability have been assessed by 13C solid-state NMR for both precursors. Of particular interest in alkane-based compounds is indeed the internal methylene carbon region (C3 to C16) between 30 and 34 ppm, as shown in the CP–MAS spectra in Fig. 4. The other resonances at lower chemical shifts are assigned to tethering (C1, C2) and terminal (C17, C18) carbons [13]. The chemical shift of the inner methylene carbons is known to be sensitive to alkane chain conformation: the upfield (30.5 ppm) and downfield (33.2 ppm) signals are attributed to gauche and trans conformers, respectively [44,45]. Therefore, it can be concluded that at ambient temperature, the C18PSTMS film exhibits a higher conformational order than its chlorosilane analogue, even though the resonance of the trans signal remains prevalent in both films. Variable-temperature 13C CP–MAS NMR have been implemented to probe the thermal stability of the octadecyl bilayer. Fig. 4 shows that upon heating, the trans component at 33 ppm diminishes and the 30 ppm signal associated with gauche defects increases. The disordering effect of temperature is much greater with C18PSTCS, with a higher proportion of chains becoming completely disordered at 50 °C, suggesting the lower thermal stability of this latter film. Accordingly, the disordering of the chains appears as a reversible process for the interdigitated structure, since the 13C spectra of C18PSTMS are observed to return to their original state upon cooling. By contrast, C18PSTCS keeps a memory of the heat-induced disorder with a higher number of gauche defects upon returning to ambient temperature, further supporting the higher thermal stability of C18PSTMS.

1H–13C CP-MAS NMR spectra of C18PSTCS and C18PSTMS at various temperatures showing the thermal stability of the two lamellar mesostructures.
In addition to 13C chemical shifts, containing information about molecular structure and conformation, the shape of the resonances must be discussed in detail because they may give insight into the motional processes of the hydrocarbon chain. By creating spatial proximity between alkyl chains, interdigitation can possibly change the dynamic character of the chain-end terminal methyl (C18) and that of the adjacent methylene (C17). To assess the difference in molecular mobility between the two multilayer films, SPE instead of cross polarization (CP) experiments spectra should be preferably performed. In CP spectra, 1H→13C magnetization transfer relies on the strength of the heteronuclear dipolar couplings, which decreases with fast motions. Consequently, the more mobile carbons located at the chain end, such as C17 or C18, are significantly under-represented or barely detectable in a CP experiment. In contrast, SPE experiments should make the observations of these terminal carbons easier (as sharp spectral features) but require recycling delays of 60 s and then long experimental times for collecting a spectrum with a good signal-to-noise ratio [46]. The 13C SPE–MAS NMR spectra of two self-assembled hybrid films are provided in Fig. 5.
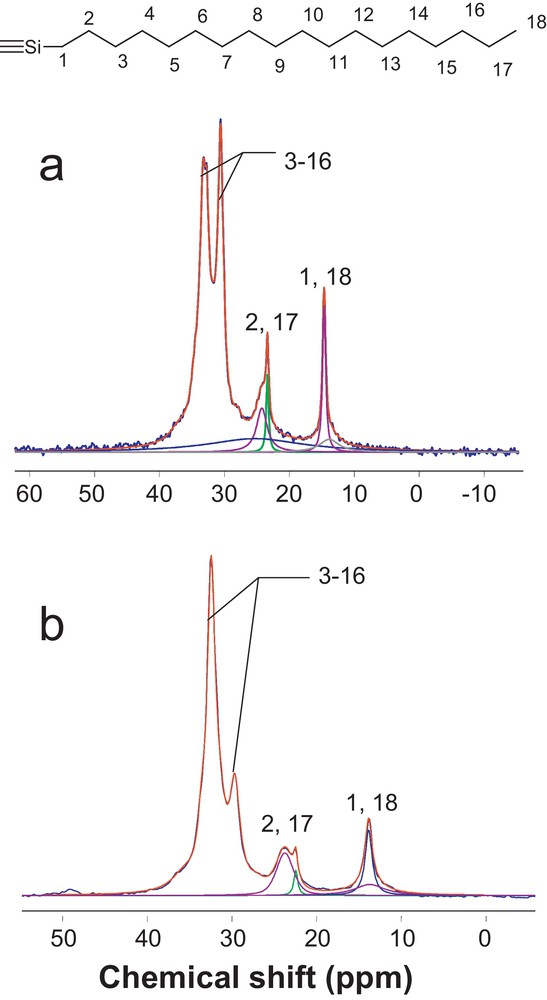
13C SPE–MAS NMR spectra of the C18PSTCS (a) and C18PSTMS (b) films. Color available on line.
As expected, the disordered gauche methylenes experiencing weak dipolar couplings are more represented in the SPE spectra.
In addition, narrow lines from mobile carbons now arise in the high-field range of the SPE spectra, whereas they are not seen in the CP spectra (Fig. 4). Unfortunately, the α-methylene carbon (C17) adjacent to the terminal CH3 arises in the ∼24 ppm region, in which C2 makes also a contribution. In both SPE spectra, the double maxima resonance can be decomposed into a broad and a sharp signal, whose proportions are very different. The sharp spectral feature is assumed to be essentially representative of mobile C17, while the broad resonance is a mixed contribution from more rigid carbons (C17 and C2) in the close vicinity of the Si atom. No quantitative interpretation is attempted here because the deconvolution is not unique, and consequently unreliable. The proposed idea is that this sharp line shape (intensity, width) could be used as a qualitative marker of C17 carbons’ mobility. Clearly, the contribution of this narrow line is smaller and broader in C18PSTMS compared to C18PSTCS. Consequently, such a difference may be the sign of a higher mobility restriction of the α-methylenes carbons imposed by the interdigitation of the alkyl chains in C18PSTMS.
Likewise, the carbon C18 and tethering carbon C1 are not well resolved in both SPE spectra and appear in the 14 ppm region as the sum of a broad and a sharp component with different relative areas. Again, we assume that the narrow line arises mainly from the most mobile terminal methyl groups (C18), whereas the broad resonance originates from carbon C1 attached to the rigid siloxane network and more rigid C18 carbons. In the SPE spectra, it is clear that the interpenetrated C18PSTMS film is characterized by a decreased contribution of the sharp line, which appears also narrower in comparison with C18PSTCS. Such qualitative difference might be indicative of a lower fraction in mobile terminal methyl groups in C18PSTMS, which is in accordance with the previous interpretation on C17.
Even though the latter results are consistent with chain end mobility restrictions resulting from interdigitation, both samples also differ in conformational order. It is not unlikely, from our point of view, that the higher proportion of trans segments found in C18PSTMS may also limit the motions of the α-methylenes and the terminal methyl groups. In contrast, we think that the weak difference in the degree of condensation of the siloxane interlayer is less likely to affect the mobility of the chain ends.
3.2.3 1H-MAS NMR
In the 1H-MAS NMR spectra displayed in Fig. 6, the double maximum signal is readily assigned to CH3 (∼0.8 ppm) and CH2 (∼1.2 ppm) protons. In contrast to the 13C spectrum, it is not possible to distinguish the α-methylene. However, the signal at 1.2 ppm, representative of all the methylene carbons, is again decomposable into a sharp and a broad component, whose relative contribution will depend on the rigidity of their environment. As expected, the methylenes in the interdigitated octadecyl chains (C18PSTMS) are characterized by an increased fraction of the broad feature (1.2 ppm, 59%) compared to C18PSTCS (1.3 ppm, 52%), originating from the most tightly dipolarly coupled protons, lacking of mobility. In agreement with the 13C SPE–MAS data, a lower mobility of the terminal methyl in C18PSTMS is evidenced by a significantly broader resonance at 0.8 ppm with Δω1/2 = 75 kHz (45 kHz with C18TCS). The rest of the proton assignment is discussed in the caption of Fig. 6.

1H MAS NMR spectra of C18PSTCS (a) and C18PSTMS (b). To complete the proton assignment, we note the weakly intense signals at 1.9 ppm (0.4%) and 1.6 ppm (3.2%) in C18PSTCS and C18PSTMS, respectively, assigned to isolated non-hydrogen-bonded silanols. Two other spectral features attributed to hydrogen-bonded hydroxyls (silanol, physisorbed water) are detected in C18PSTMS at 3.5 ppm (1.8%) and 5.3 ppm (0.7%) – where a higher chemical shift reflects stronger interactions – instead of a single one in C18PSTCS at 5.3 ppm (1%). The overall higher population in silanols found in C18PSTMS agrees with its lower degree of condensation, already evidenced by 29Si solid-state NMR. The last small spectral features at 7.0 ppm (C18PSTCS) and 7.1 ppm (C18PSTMS) were ascribed to the aromatic protons from the photoacid generator ((C12H25)2Φ2I+PF6–) and the photosensitizer (Φ2CO). Color available on line.
3.2.4 2D 1H–13C WISE NMR
Qualitative information about hydrocarbon chain molecular motion can be obtained by combining proton line width measurements with 13C detection in a 2D NMR experiment commonly referred to as WIde-line SEparation (WISE) [47]. Indeed, it is a general principle that the proton line width characterizes the strength of the dipolar coupling among protons, which is strongly correlated with their local dynamics. For example, crystalline or rigid segments are known to increase the homonuclear dipolar interactions, leading to broad proton line width. Conversely, flexible or disordered regions are manifested by a substantial sharpening of the proton resonance, since the dipolar coupling is almost averaged out by fast molecular motions. The WISE technique has been introduced initially in the characterization of polymer to address dynamic heterogeneities caused by the coexistence of rigid and flexible moieties in the same macromolecule [48,49]. More recently, 2D WISE spectra were used to assess the chain mobility in self-assembled monolayers (SAM) [50,51].
The 2D WISE NMR spectra have been recorded for C18PSTCS and C18PSTMS. In the 13C NMR spectra displayed in Fig. 7, both films show different components at 33 and 30 ppm in the 13C NMR spectra related to trans and gauche conformers respectively. Since the 13C chemical shift is sensitive to conformation, slices taken of the 1H dimension in a 2D WISE spectrum allow the separation of the broad proton resonances for ordered and disordered regions, which would completely overlap in a 1D 1H NMR spectrum. These slices in the 1H dimension for the methylene carbons at 33 ppm and 30 ppm are given in Fig. 7. (The full 2D WISE NMR spectra are shown in Supplementary data, Fig. S2). As expected from the larger dipolar couplings of conformationally ordered chains, the 1H slices of the 33 ppm methylene signal display a broader line (1.1 kHz) than that of the gauche resonance at 30 ppm (0.3 kHz). However, both films have essentially the same dipolar slices at 33 and 30 ppm. Such a remarkable result implies that it is not possible to detect mobility differences of the inner methylenes (C3–C16), whether trans or gauche, depending on their packing geometry. Presumably, the cohesive interactions developed in an interdigitated structure do not provide any additional mobility restriction, which could induce a detectable increase in the dipolar coupling. Nevertheless, one must bear in mind that only a part of the 13C nuclei can be observed with a 2D-WISE experiment based on CP data. With regards to the terminal methyl, the 14 ppm peak contains contributions from carbons 1 and 18, and cannot be used to draw conclusions on methyl dynamics.

13C CP MAS recorded at room temperature and 1H wide-line spectra of C18PSTCS and C18PSTMS. The 1H wide-line spectra shown correspond to the 1H slices of gauche and trans components at 30 and 33 ppm, respectively in the 13C dimension in the 2D wide-line separation experiments (WISE).
3.3 Thermal gravimetric analysis
In Fig. 8, the TGA thermograms of C18PSTMS and C18PSTCS show respectively two and three discernible weight-loss regions. The small transition initiated at ∼105 °C, found in both films, can be related to the loss of physisorbed water and the progressive post-condensation of residual silanols, liberating new water molecules [13]. The slowly decreasing slope suggests that silanol condensation reactions are effective over a wide range of temperatures, from 105 to 300 °C approximately. For this first small transition, the larger weight-loss noted in C18PSTCS is not consistent, a priori, with its more condensed siloxane network. This suggests the hypothesis of a lower temperature degradation of the disordered octadecyl chains, which are significantly represented in this latter sample. Consequently, the bimodal population in gauche and trans conformers in C18PSTCS could account for the existence of two distinct large transitions at 280 and 350 °C, marking the degradation of the alkyl chain. The progressive and low-temperature transition may be assigned to gauche defects displaying a lower thermal stability and a range of possible conformations. The higher temperature transition is sharper, which is in agreement with ordered trans alkyl chains with a single conformation. Accordingly, the degradation of high trans conformers populations in C18PSTMS leads to a single sharp transition at a similar temperature of 350 °C. The final weight-losses of C18PSTCS (22%) and C18PSTMS (18%) observed at high temperature are consistent with the theoretical values of the Si–O–Si moiety, around 17%.

Thermogravimetric analysis of C18PSTCS (—) and C18PSTMS (----).
4 Conclusion
We have taken advantage of an original sol–gel photopolymerization process to generate two multilayer poly(n-octadecylsiloxane) films characterized by a different arrangement of the alkyl bilayer. A head-to-head geometry was favoured by the self-assembly of C18TCS whereas a head-to-tail configuration was preferentially formed with C18TMS. On the basis of various characterization techniques (SAXS, MAS NMR methods), we proved that C18PSTMS differed in the interdigitation of the alkyl bilayers, a higher trans population, and a slightly lower siloxane condensation, in comparison with C18PSTCS. The 13C SPE–MAS NMR spectra established a lower mobility for the α-methylene carbons (C17) and the methyl chain ends (C18) in this interpenetrated structure, a result confirmed by 1H MAS NMR in the case of the terminal methyl group. As illustrated in Fig. 9, C17 and C18 are parts of the alkyl chain whose local environment is most likely to be affected by the packing arrangement. In an interdigitated structure, these headgroups are surrounded by stiff methylene groups (tethering C1 carbon) and the cross-linked siloxane of the opposite layer. This is in sharp contrast with their local environment in a non-interpenetrated mesostructure that is much more mobile. We suggest that mobility difference is to be expected for reasons of (a) interdigitation, but also (b) conformational ordering, and to a less extent, (c) the overall density of the siloxane interlayer. Obviously, we cannot separate all of the above contributions and claim that the interdigitated environment is a single source of a stronger immobilization of the hydrocarbon chains. Future studies including n-alkylsilane alkoxides functionalized at their chain end by CF3 or PO3H2 groups should provide new insights into the structural and dynamic processes of these supramolecular materials [52].

Local and mesoscopic structures of the two n-alkylsilsesquioxane films differing in the packing geometry of the alkyl tail. The two configurations show the effect of the alkyl chain interdigitation on the local environment of the CH3 and α–CH2 groups.