

1 Introduction
Since the beginning of the 1990s, there is a growing interest in the synthesis of new materials with original textures within the nanometer scale range, and promoted by the self-assembly of amphiphile molecules [1–8]. The physico-chemical nature as well as the formation kinetics and thermodynamics of the interfaces between self-assembled amphiphiles (surfactants, block copolymers) and oxide based networks (silica, transition metal oxides, hybrid materials) play a key role in the final properties of these materials [9–20]. Therefore, a deep characterisation and modelling of these interfaces is needed. Different spectroscopies have been used for that purpose. For example, the first ESR studies gave insights in the local mobility of probes covalently bond to surfactants, [12,13] whilst fluorescence of diluted dyes provided information about micelle surfaces and ordering [14,21]. Several NMR studies have been undertaken for different topics (Scheme 1). For example, various groups explored the mechanisms of mesophase formation, the charge matching effects during synthesis and the spatial location of chemical functions or confined molecules [9,22–34]. In the course of our solid-state NMR research on materials’ syntheses, we focus on the characterisation of the interfaces in hybrid and/or textured materials [35–43].
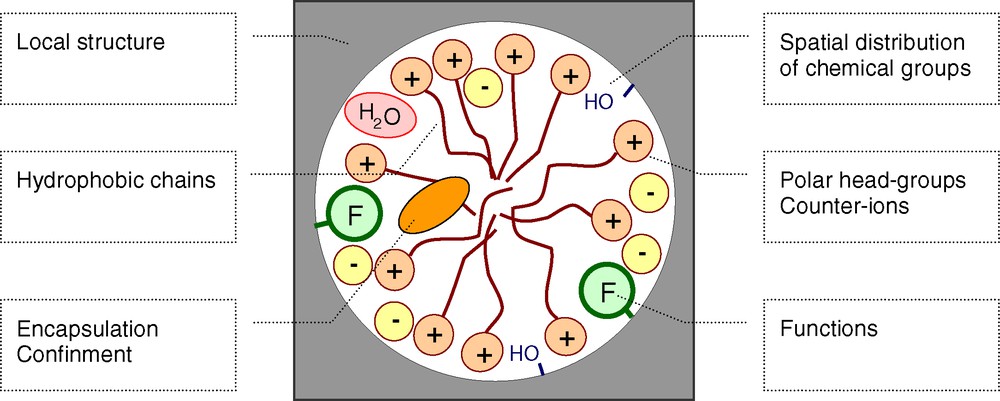
Examples of topics studied by NMR for materials textured by self-assembled amphiphiles.
Our first interest was driven by the new opportunities offered by 1H solid-state NMR developments. Increased proton resolution can often be achieved in the 1H spectra of the synthesised materials by using simple fast magic angle spinning (MAS) – today up to 70 kHz in frequency - and a high static magnetic field, helped in that by the residual mobility of the organic groups. This increased resolution allows taking advantage of the richness of the direct dipolar (D) or indirect (J) homonuclear (1H-1H) and heteronuclear (1H-X) spin-spin couplings. We will present some examples of a 1H multi-scale characterisation in the case of block copolymers, hybrid mesotructured membranes and mesoporous silica microspheres.
More recently, we have performed 14N and 81Br solid-state NMR experiments for the study of the charged entities (ions, polar head groups) of ionic surfactants like the well-known hexadecyltrimethylammonium bromide (HTAB). These charged entities play a major role in building the interfaces through their electrostatic interactions. We will present first a more academic study on the crystal structures of n-alkyltrimethylammonium bromide salts, followed by preliminary applications to the characterisation of interfaces in mesoporous materials made of cationic surfactants.
2 1H NMR characterisation at different length scales
2.1 Towards simplified approaches
Because of its high gyromagnetic ratio associated to its high natural abundance, 1H nucleus is a very sensitive probe in solid-state NMR which can provide valuable information about the nature of the chemical groups present in biological matter, polymers, hybrid organic-inorganic materials, etc. However, these intrinsic spin-properties of 1H nucleus and the high content of hydrogen in the organic molecules yield to both specific drawbacks and advantages in solid-state NMR (and especially for rigid solids). In particular, the presence of very strong 1H homonuclear dipolar couplings gives rise to large homogeneous broadenings of the 1H resonances, which are not efficiently removed by MAS at a moderate spinning frequency (∼15 kHz). Due to these line-broadening effects combined with the relatively small proton chemical shift range (typically from 0 to 15 ppm), 1H solid-state NMR spectra obtained with these experimental conditions greatly suffer from a lack of spectral resolution. However, the 1H homonuclear dipolar couplings also bear valuable structural information since they allow directly probing proton-proton inter-atomic distances and, hence, the spatial proximities between similar or different functional groups. Nowadays, the merging of new techniques and methods facilitates the use of 1H solid-state NMR, which can in turn allow a more precise characterisation of the materials at different length scales (0.1 to about 100 nm) (Scheme 2).

1H solid-state NMR characterisation at different length scales.
Typically, the dominating homonuclear dipolar 1H-1H couplings can be decreased by acting on:
- • the number of spins through an isotopic dilution (e.g. by deuteration);
- • the spin part of the dipolar Hamiltonian through homonuclear decoupling schemes;
- • the geometric part of the dipolar Hamiltonian through MAS.
In parallel, the precise selection of a 1H-X pair is now possible using the direct dipolar (DHX) or indirect (JHX) spin-spin couplings existing between protons and other heteronuclei. In the case of DHX couplings, major advances have been the cross-polarisation 1H → X under proton homonuclear decoupling like in the CPLG sequence [52,53] and the use of proton detected methods – like the already known REDOR [54,55] – which benefits from the increased spectral resolution. It becomes thus possible to properly select spatially close 1H-X pairs, and even to obtain their distances through the measurements of DHX couplings. In case of JHX couplings, fast MAS and/or efficient proton decoupling have allowed the use of pulse sequences often restricted to liquid-state NMR applications [56–58]. Again, 1H or X detection can be chosen, and measurements of JHX couplings are possible.
The strong 1H-1H homonuclear dipolar couplings allow exploring longer distances. In the range 0.1–1 nm, excitation of multiple-quantum coherences between coupled 1H spins can be used for that purpose typically in a Double Quantum (DQ) – Single Quantum (SQ) correlation experiment, recorded using the Back to Back (BaBa) pulse sequence, for example [59,60]. In the higher range 1–100 nm, proton spin diffusion (transfer of polarisation through “flip-flop” transitions) can be used to perform long-range magnetisation exchange experiments [61,62].
These three axes (increase of proton resolution, precise selection of H-X pairs, exploration of distances) constitute the basis of a multi-scale characterisation approach for the study of hybrid and/or textured materials [35–37,58]. Nowadays, the progress in solid-state NMR are such that this approach can be readily implemented in most of the recent spectrometers using different appropriated methods. It also benefits from the deep knowledge acquired in the study of polymers [63].
2.2 Application to the study of interfaces
The characterisation of polymers or hybrid materials possessing organic-organic or organic-inorganic interfaces between domains separated at the nanometer scale really benefits from a 1H multi-scale characterisation approach. Some years ago, we have shown how it applies to the study of a crystalline zinc phosphonate layered compound [35,37]. It was particularly significant to show how we can obtain a signature of the organic-inorganic interface by selecting precisely a 1H-31P pair, and how far from this interface are the different organic groups located between the organometallic sheets. In addition, it was possible to identify the presence of a minoritary phase-separated impurity using a triple CP 31P{1H}31P sequence [64] implying a 1H spin diffusion step.
2.3 Block copolymers
More recently, we have studied a family of new block copolymers under fast MAS (33 kHz) and high magnetic field (17.6 T) conditions. The structure of one of the investigated block copolymers, which is obtained by ring opening metathesis polymerisation (ROMP), is presented in Fig. 1a. This polymer consists of a hydrophobic block containing butyl ester functionalities and a second hydrophilic block containing carboxylic acid groups. In this context, ROMP is a versatile method as it allows us to prepare a variety of amphiphilic block copolymers by choosing the functional groups, by varying the block size and by preparing di, tri, tetra- (and more) block copolymers [65]. Therefore these amphiphilic block copolymers possess self-assembly properties as shown recently [66,67]. They are also potential candidates for the texturation of hybrid organic-inorganic materials, and we have started to analyse their interaction with Al precursors.
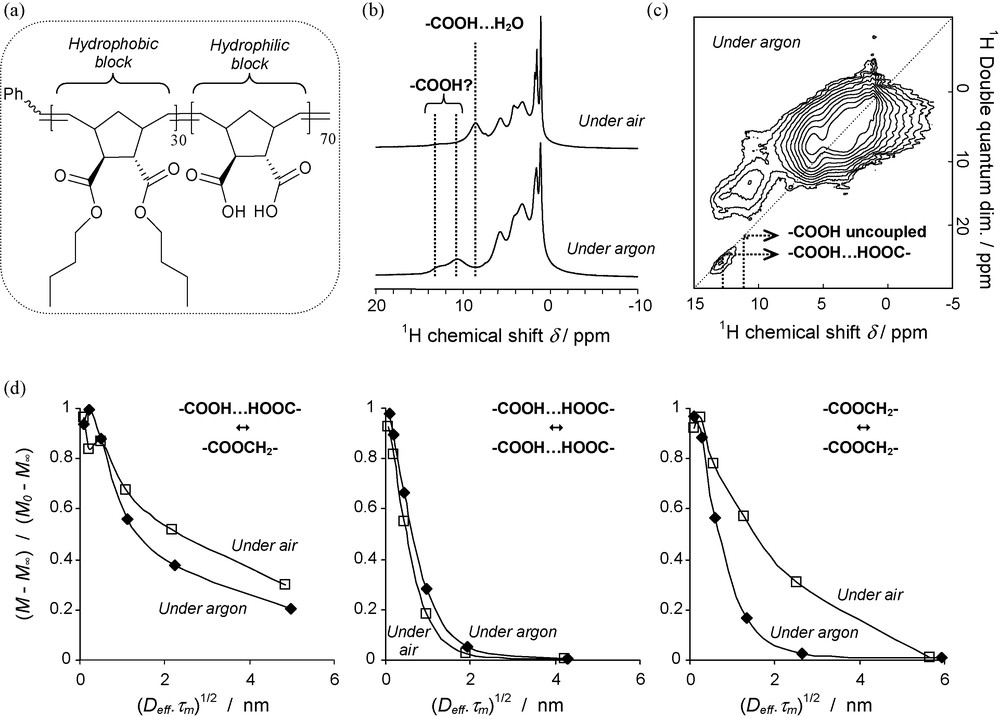
1H NMR multi-scale characterisation of block copolymers; a: ROMP copolymer studied. The pending groups of determine the amphiphile properties: butyl ester functions for the hydrophobic block, carboxylic functions for the hydrophilic block; b: 1H simple pulse spectra of copolymer undried (air) and dried (argon) recorded at fast MAS (νMAS = 33 kHz) and high magnetic field (ν0 = 750 MHz); c: 2D 1H-1H DQ-SQ BaBa correlation spectrum of dried copolymer; d: 1H spin diffusion curves from 2D 1H-1H exchange experiments recorded for copolymer dried (♦) and undried (□).
1H spectra of the copolymer of Fig. 1.a are presented in Fig. 1b (νMAS = 33 kHz, ν0 = 750 MHz). The spectrum recorded under air without any drying procedure (top) differs from that recorded under argon on a dried copolymer (bottom). The main differences concern the three peaks observed at chemical shifts δ above 9 ppm. These peaks are related to the protons of the –COOH carboxylic function with different environments. For the undried sample, most of the –COOH functions are interacting with water molecules, leading to a decrease of their chemical shift δ. For the dried sample, the assignment of the two peaks solely from chemical shift databases is not straightforward. In such case, a 1H-1H DQ–SQ correlation experiment gives directly the answer: the presence of an autocorrelation cross-peak is the signature of coupled –COOH functions, while its absence is the signature of uncoupled –COOH functions (Fig. 1c). Therefore, 1D and 2D relatively simple experiments recorded at fast MAS and high magnetic field allow analysing the environment of –COOH functions. When adding Al precursors (nitrate or alkoxide) for the formation of hybrid materials, we observed the disappearance of such –COOH signals, accounting for the possible complexation of Al3+ cations by –COO– groups.
We also studied the spatial distributions of the ester and carboxylic functions using 2D 1H-1H magnetisation exchange experiments. For the self-assembled copolymers, the signals of these functions are the signature of the hydrophobic and hydrophilic domains, respectively. We present in Fig. 1.d the proton spin diffusion curves obtained after spectrum modelling. According to previous references, [68] the magnetisation has been normalised to (M – M∞)/(M0 – M∞), where M0 is the initial magnetisation and M∞ the magnetisation at equilibrium. In order to account for the differences in diffusivity constants, the abscissa has the dimension of a length and corresponds to (Deff.τm)1/2, where Deff is an effective diffusivity estimated by (Deff)1/2 = (DA.DB) 1/2/2.(DA1/2 + DB1/2) [63] using individual diffusivities (e.g. DA, DB) obtained from T2 values following previous empirical correlations [69].
For dried and undried samples, the spin diffusion curves of the exchange between ester and carboxylic proton magnetisations across the interface between hydrophobic and hydrophilic domains are presented at the left side of Fig. 1d. Both curves indicate the presence of domains which size reaches some nanometers. However, we observe that the spin diffusion, and hence the spatial organisation, depends on the dryness. Shorter distances between domains appear to exist in the dried sample. But surprisingly, no clear difference between samples is observed for the exchange between carboxylic protons (curves in the middle). On the contrary, the curves related to the magnetisation exchange between protons of the ester functions are significantly different (right side). It appears therefore that the interactions and chemical environments of the carboxylic functions inside the hydrophilic blocks have an effect on the spatial organisation of the neighbouring hydrophobic blocks. Such an effect can be important for the further use and processing of the copolymers alone or as templates.
2.4 Self-assembled copolymers in hybrid siloxane membranes
Block copolymers can indeed be used for the preparation of ordered mesoporous materials [70]. We have studied hybrid organic-inorganic siloxane networks templated by self-assembled tri-block copolymer PEO-PPO-PEO (106-70-106), where PEO are poly-ethylene oxide hydrophilic blocks, and PPO poly-propylene oxide hydrophobic blocks (Fig. 2a). Monolithic hybrid membranes, with a high level of mesostructured organisation can be easily prepared [71]. The siloxane network is made of Q units SiO4/2 associated with T units RSiO3/2 or D units R2SiO2/2, where R stands here for a methyl group (Fig. 2a). By choosing either T or D units and by varying their relative amount, the hydrophobicity of the siloxane-copolymer interface (and hence of the siloxane surface after template removal) can be tuned.

1H NMR multi-scale characterisation of hybrid mesostructured membranes; a: chemical groups encountered in the textured hybrid siloxane membranes; b: Typical 1H single spectra obtained for series D and T at fast MAS and high magnetic field (νMAS = 30 kHz, ν0 = 750 MHz); c: 1D selective exchange pulse sequence and related 1H spectra obtained at selected mixing times (τm = 0, 10, 60, 200, 700 ms) for series T; d: 1H spin diffusion curves (series T) and schematised evolutions; e: example of exchange from methyl groups in PPO domains to methyl groups bound to the siloxane network for the series D (♦) and T (□).
1H spectra recorded at fast MAS (30 kHz) and high magnetic field (17.6 T) showed enough resolution (Fig. 2b), helped in that by a non-negligible internal mobility. Spectrum assignment has been made using spectral databases [72] and 1H-13C INEPT correlation spectra run at fast MAS, an appropriate experiment for this kind of samples [58]. In order to collect information about the spatial organisation of the different chemical groups, we have undertaken a proton spin diffusion study [73]. A specific pulse sequence has been used for this purpose (Fig. 2c). This sequence consists in a spin echo (90° – τ/2 – 180° – τ/2) involving a 180° selective Gaussian pulse followed by a longitudinal magnetization exchange block (90° – mixing time τm – 90°). The selective pulse has been centred on the signal of the methyl groups of the propylene oxide (PO) units. This signal marks the presence of a hydrophobic domain at the core of the copolymer aggregates. When increasing τm, the other signals appear as expected in a typical magnetisation exchange experiment (Fig. 2c). The advantages of this method compared to standard 2D magnetisation exchange experiments are the reduction of the overall experimental time and the easier modelling of the 1D spectra. In turn, this allows obtaining more complete spin diffusion curves (typically 30 spectra can be recorded in 12 h) as shown in Fig. 2d. Two types of curves can be distinguished as schematised at the bottom of Fig. 2d. The type I corresponds to curves that strongly decrease from the beginning and pass through a minimum at negative values of the normalised magnetisation (M - M∞)/(M0 - M∞). They are related here to the spin diffusion from CH3– in PO to –CH– or –CH2– in PO. The magnetization from CH3– in PO is here first transferred to the protons of the same domain, and afterwards to the other groups present in the sample. Similar kind of curves have been already observed or modelled for other mesoporous materials, [40] for lipid membranes, [74,75] and for organic crystals [76]. The type II corresponds to curves that do not vary at small mixing times and then decrease almost linearly as a function of τm1/2 before reaching equilibrium. They are related to the diffusion towards protons that are located outside the PPO domains. The presence of extended interfaces between different domains (PPO, PEO, siloxane network) can also contribute to the observed initial delay [68] or at least to a slower spin diffusion at small τm values [77,78]. The initial slope after the delay is indicative of the spatial distribution of the protonated groups from the PPO domain but depends on various parameters such as the dimensionality of the domains, the volume fractions of each domain and the diffusivity constants [63,68]. Here, volume fractions and mesophases are identical, and the variations in diffusivity can be integrated by rescaling the abscissa from (τm)1/2 to (Deff.τm)1/2 as mentioned above. By studying in detail each curve, we have been able to depict schematically the spatial organisation and the differences between samples. For example, the spin diffusion between the methyl groups in PO and the methyl groups of T or D units of two series of samples is compared in Fig. 2e. It appears that the methyl groups of the D units are much closer to the PPO domains than the methyl groups of the T units. This is consistent with other results indicating that the interface between the siloxane network and the copolymer differs for the two samples: the presence of the less polar D units favours the formation of more disordered interfaces and smaller inter-aggregates distances. It appears also that the introduction of D or T units can be an efficient way to avoid the inclusion of PEO segments inside the siloxane network [79].
2.5 Spray-dried mesoporous microspheres
Micelle aggregates of ionic surfactants like HTAB are also ideal templates to prepare ordered mesoporous textures. They were indeed used in the first works published on the field [1,80,81]. They are also used for the preparation of mesoporous microspheres through spray-drying and aerosol processes [82,83]. Some of the advantages of these syntheses are the incorporation of all the non-volatile matter in the final materials, the easy and scalable processing, and the limitation of the number of steps. However, both texture and morphology are defined by solvent evaporation and solidification during the short drying step (4 seconds and below), and are therefore interdependent properties. We have been working on these spray-drying routes, and we have particularly studied the relationships between the morphology, the texture and the siloxane-surfactant interfaces presented in Fig. 3a [38–40,84,85].

1H NMR multi-scale characterisation of mesoporous spray-dried microspheres; a: morpology, texture and interfaces defined at different length scales; b: XRD characteristic distances plotted as a function of the molar proportion of alkoxy groups bound to the siloxane network nROSi/nSi as obtained from 1D 1H spectra and modelling (νMAS = 30 kHz, ν0 = 750 MHz). 2D 1H-1H exchange experiments demonstrated the proximity between surfactant polar head groups and alkoxy groups; c: spatial proximity between the trimethylammonium polar head group of the surfactant and the methyl groups bound to the siloxane network proven by 1H-1H DQ-SQ BaBa correlation.
Hydrolysis and condensation reactions in solution prior to spray drying allow preparing siloxane oligomers from molecular siloxane precursors. The oligomers will afterwards interact together and with the surfactant micelles in order to form the siloxane network and the final texture. From SEM, TEM and SAXS measurements on the powdered samples, we have evidenced a correlation between the observed texture (phase, degree of ordering), the morphology (isolated or agglomerated spheres) and the ageing time of the sol which governs the extent of hydrolysis and condensation in solution [38]. Using additional data from SAXS measurements on sols, we have shown that siloxane oligomers with a radius of gyration in the 0.5–1.0 nm range allow producing well separated spheres with a significant degree of textural ordering. This size range is close to the wall thickness estimated for several mesoporous materials made with HTAB [86]. From intermolecular interaction considerations, it seems that this size range corresponds also to the optimal distances between surfactant aggregates in mesophases made of HTAB and silica [87]. However, we observed a gradual decrease in the characteristic X-rays correlation distance and the degree of textural ordering as a function of the proportion of alkoxy groups bound to the siloxane network and coming from an incomplete hydrolysis in solution (Fig. 3b). From 1H-1H magnetisation exchange experiments recorded at fast MAS (30 kHz) and high magnetic field (17.6 T), we have proven that these alkoxy groups are spatially close to trimethylammonium surfactant head-groups. The presence of these alkoxy groups might disturb the aggregation properties of the surfactants as noticed for surfactants micelles in contact with small alcohol or amine molecules [88,89]. Therefore, both the size and the nature of the pending groups of the siloxane oligomers are important parameters to be controlled in order to optimise the properties of the spray-dried materials. This observation is also important when the incorporation of organic groups bound to the siloxane oligomers is envisaged as discussed below.
The functionalisation of the siloxane surfaces in mesoporous materials is a key issue from which the potential applications may depend. Two methods exist:
- • a post-synthesis functionalisation using grafting procedures;
- • a direct funtionalisation using co-condensation reactions between Q and T or D siloxane units [90].
In the case of one-pot spray-drying methods applied to the production of mesoporous microspheres, the latter method can be highly useful. We have been working on this direction,[39,40,84] and we have defined a robust protocol consisting in the preliminary formation of siloxane oligomers of known characteristics containing only Q units, followed by the condensation on these oligomers of T units bearing the organic functions [84]. With this one-pot but two-steps protocol, spray-dried mesoporous microspheres with functionalised surfaces have been obtained for organic functions of different kinds (–CH3, –C6H5, –[CH2]3–CN, –(CH2)3–SH…). 1H-1H magnetisation exchange experiments and 1H-1H DQ-SQ correlation experiments (Fig. 3c) have both highlighted the spatial proximity between the organic function introduced and the trimethylammonium polar head-groups (before surfactant removal), [40,84] proving that these functions are effectively distributed on the final siloxane surface.
3 14N and 81Br as new NMR probes for the interfaces between oxide network and cationic surfactants
Whilst there have been several solid-state NMR studies of ordered mesoporous silica-based materials using 1H, 13C and 29Si nuclei as local probes, there were almost no structural studies using 14N or 79/81Br nuclei although they are potential local probes of the head-groups and counter-anions of cationic surfactants. Therefore, we have investigated the potentiality of NMR experiments involving 14N and 81Br nuclei. For that purpose, we first revisited the crystalline forms of tetraalkylammonium bromide surfactants. In parallel, we started the study of mesoporous materials (Scheme 3).
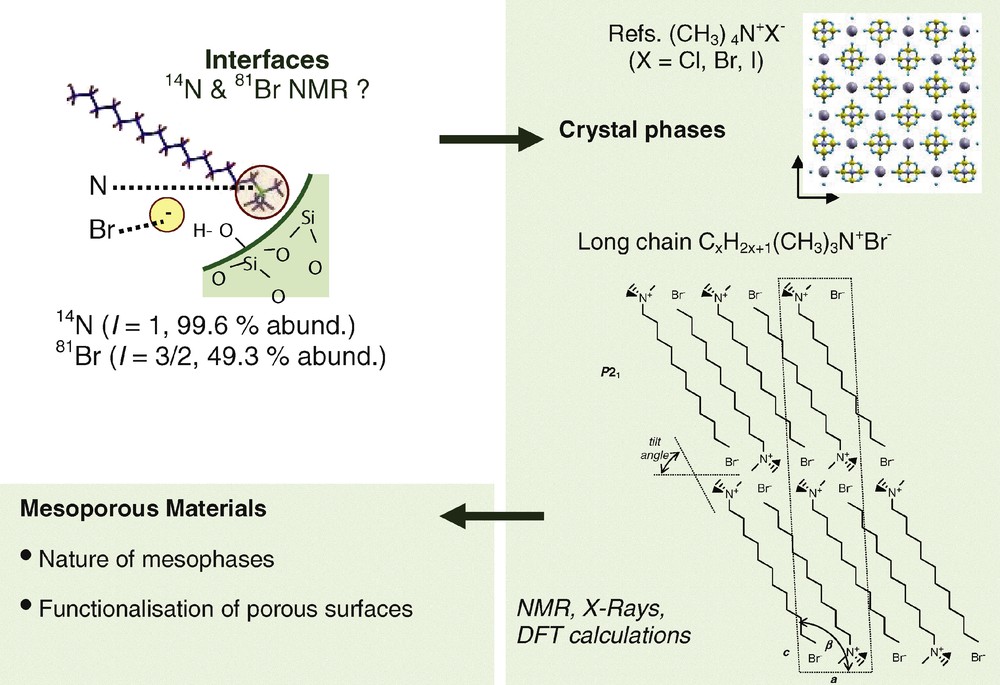
14N and 81Br NMR studies of cationic surfactants in crystals and mesoporous materials.
3.1 14N and 81Br data on crystalline cationic surfactants
In contrast to several nitrogen containing organic groups, tetraalkylammonium compounds give rise to moderate 14N quadrupolar coupling constants CQ (in the 100 kHz range), which allow a direct observation of 14N resonances in conventional MAS spectra [91,92]. We have undertaken a systematic study of n-alkyltrimethylammonium bromide crystals of different chain length (from 12 to 16 carbons) and compared their spectra to that of smaller tetraalkylammonium salts [93]. Experimental and modelled 14N spectra of tetramethylammonium bromide (TMAB) and HTAB are presented in Fig. 4a. Contrasted values of CQ and asymmetry parameter ηQ were obtained for TMAB (26.2 kHz and 0.04 resp.) and HTAB (97.8 kHz and 0.84 resp.). These differences in quadrupolar interaction parameters mainly come from differences in molecular and crystal structures. Fig. 4b shows the atomic arrangement around nitrogen atoms. Using experimental results and theoretical calculations, we have found a correlation between CQ and the deviation of C-N-C angles from Td symmetry (Fig. 4c). In addition, the spatial distribution of Br- counter anions is found to modify the Electric Field Gradient (EFG) tensor [Vij] when going from more symmetric (TMAB) to less symmetric (HTAB) structures.

14N solid-state NMR study of tetraalkylammonium bromide crystals; a: experimental spectra, modeling and difference spectra for TMAB and HTAB; b: schematic presentation of the local site geometry of nitrogen atoms from the optimised structures of TMAB and HTAB; c: variations of the 14N quadrupole coupling constant CQ as a function of the angle difference (θTd - θmin) for calculated CQ (♦) and experimental CQ (□).
To our knowledge, no previous work was published on 81Br (or 79Br) NMR studies of TMAB crystals. Here, CQ values are more important (5–10 MHz range) but direct observation is still possible at high magnetic field (17.6 T) [93]. Fig. 5a presents the 81Br spectra and TMAB and HTAB crystals recorded at different MAS frequency and the related modelled spectra. From these spectra, the quadrupolar interaction parameters but also the chemical shift anisotropy (CSA) parameters can be determined. For HTAB, we found CQ values higher (8.03 MHz) than those for TMAB (6.03 MHz). In TMAB and HTAB crystal structures (Fig. 5b), Br− anions have similar environments excepting the absence of two ammonium positively charged groups and a related decrease in symmetry for HTAB. This creates again differences in the EFG tensors as observed from theoretical calculations. Moreover, we found an empirical correlation between CQ values of these and other n-alkyltrimethylammonium bromide crystals, and the sum of N-Br distances at the power minus three (Fig. 5.c). This correlation exemplifies how very small variations in the spatial distribution of charges can have significant effects in the 81Br quadrupolar interaction parameters.

81Br solid-state NMR study of tetraalkylammonium bromide crystals, a: experimental spectra, modeling and difference spectra for TMAB and HTAB span at different MAS frequencies; b: Schematic presentation of the local site geometry of bromine atoms from the optimised structures of TMAB and HTAB; c: variations of the 81Br quadrupole coupling constant CQ as a function of the sum Σ ri-3 where ri are recalculated values for the r(N-Br) distances.
3.2 Application to mesoporous materials
This dependence of 81Br quadrupolar interaction parameters on the spatial distribution of charges is an explanation for our failure in directly observing 81Br resonances in conventional MAS spectra in the case of mesoporous materials containing Br− counter anions (e.g. samples obtained by an acidic sol-gel route). Indeed it is now well-accepted that models describing in simple terms the molecular interactions at the interfaces between the oxide network and the surfactant assemblies might be revised, and that these interfaces are far from being ordered, in particular for materials made under acidic conditions [28,29,33]. Therefore, the spatial distribution of charges will be more disordered than in surfactant crystals. The inability to directly observe 81Br would likely be due to that. However, it is possible to use indirect 1H-detection of the 81Br resonances like in 1H{81Br} TRAPDOR experiments [94]. We have undertaken such experiments on spray-dried mesoporous microspheres obtained from acidic sols and with phenyl functions bound to the siloxane network following a previous synthesis procedure [84]. Our results, presented in Fig. 6b, show that Br− anions are distributed close to the trimethylammonium head-groups but also close to the surfactant n-alkyl chains and to the phenyl groups bound to the siloxane network. This again suggests a disordered siloxane-surfactant interface. Indirectly, it also proves that the phenyl groups are indeed spatially located at this interface, and therefore at the siloxane surface after surfactant removal. The 1H{14N} TRAPDOR curves presented in Fig. 6c give similar information but from the point of view of the positively charged trimethylammonium groups. We observe that the curve of the phenyl groups is between that of the polar head-groups (direct H-C-N bonding scheme) and that of the n-alkyl chain (H-(C)x-N bonding scheme). Herein, phenyl groups are spatially close to the head-groups.
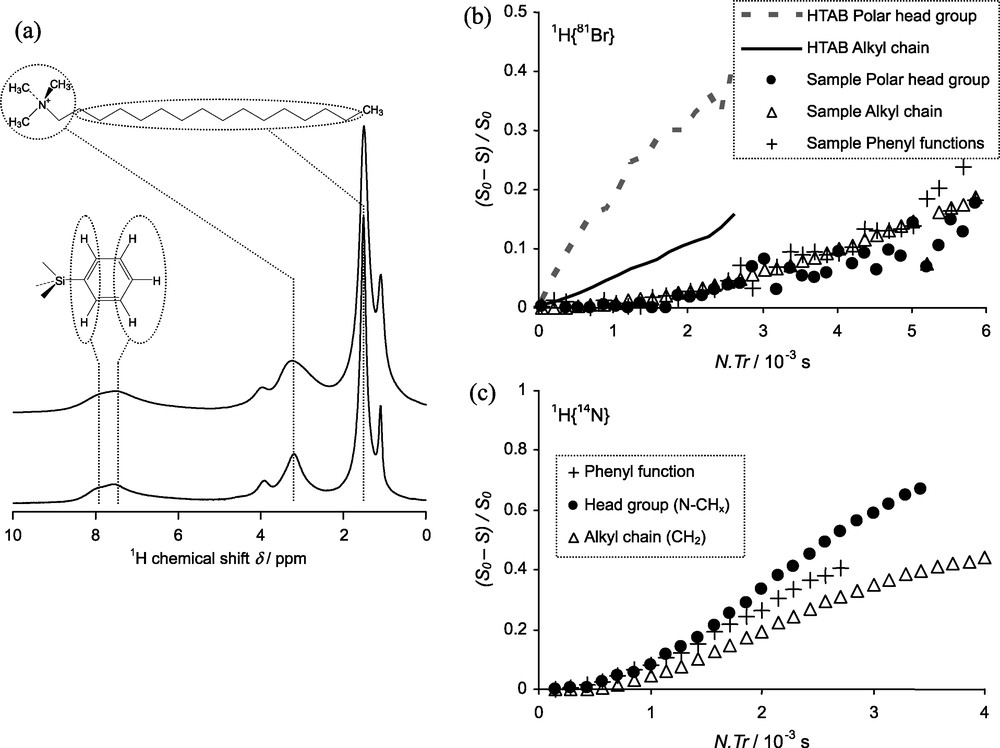
Study of surfactant-siloxane interfaces using proton detected 1H{81Br} and 1H{14N} NMR experiments; a: 1H single pulse spectra of mesoporous microspheres with phenyl groups bound at the siloxane surface: νMAS = 14 kHz (top) or 30 kHz (bottom) (ν0 = 750 MHz); b: 1H{81Br} TRAPDOR curves obtained for HTAB (νMAS = 29 kHz) and the mesoporous functionalised microspheres (νMAS = 30 kHz); c: 1H{14N} TRAPDOR curves obtained for the mesoporous functionalised microspheres (νMAS = 14 kHz).
Direct observation of 14N resonances for mesoporous materials is however possible. For example, in the case of spray-dried mesoporous microspheres with mercaptopropyl groups at the siloxane-surfactant interface [40], we have observed interesting variations in 14N spectra. When a hydrothermal treatment in basic conditions (NH3) is applied to these materials, there is an important increase in textural ordering as observed in Fig. 7a by the appearance of longer range diffraction peaks corresponding to the p6 mm phase. This effect comes from the restructuration of the siloxane-surfactant interface after local dissolution and precipitation processes promoted by the strong electrostatic interactions between negatively charged siloxane species and positively charged surfactant. It has been already exploited for other systems [95]. As observed in Fig. 7b, this effect gives rise to a significant increase of the 14N signal, which can be related to the increase in textural ordering. However, these and other observed spinning sidebands patterns often differ from that of initial surfactants (e.g. HTAB in Fig. 7b). Combined effects of spatial distribution of charges and internal mobility are suspected. And we are currently analysing a series of spectra, which tend to show an important and sample-dependent contribution of the internal mobility of the trimethylammonium head-groups.
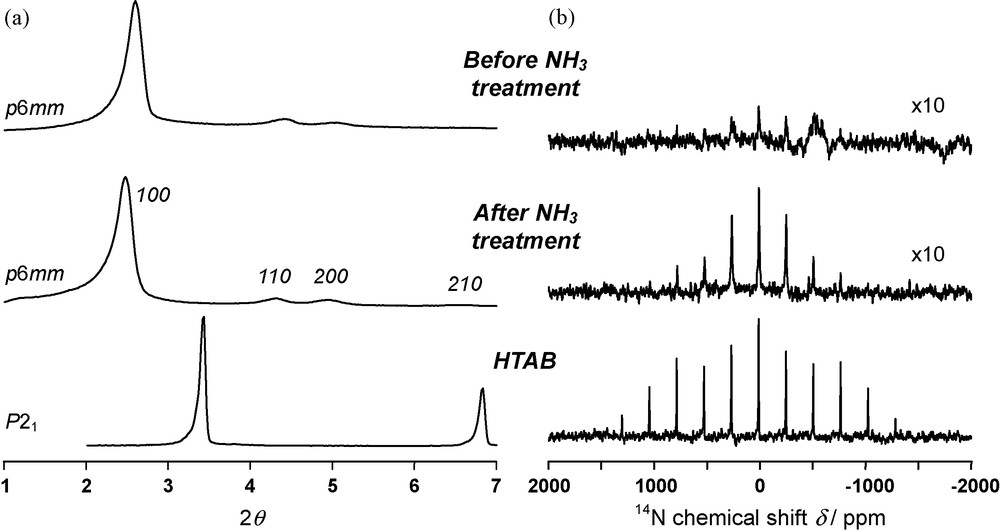
Textural improvement of mesoporous functionalised (-SH) microspheres through a NH3 hydrothermal treatment; a: XRD patterns: sample before treatment (top), sample after treatment (middle), HTAB for comparison (bottom); b: 14N quadrupolar echo spectra: sample before treatment (top), sample after treatment (middle), HTAB for comparison (bottom). Intensities are normalised by the number of scans.
4 Conclusions
Through several examples we have shown how informative 1H, 14N and 81Br solid-state NMR can be for the study of interfaces between self-assembled amphiphile molecules and oxide-based networks. Recent methodological and technological achievements have made the use of proton NMR more popular. The growing access to increased MAS frequencies and static magnetic fields will indeed facilitate the characterisation of a wider range of materials. Nowadays, a complete characterisation at different length scales of the textured materials is achievable by non-specialised spectroscopists. Herein information about the local environment and the spatial distribution of different protonated chemical groups is readily accessible. This information allows a better understanding of the self-assembly processes, at the basis of the final materials’ properties. Incidentally it appears often that the studied interfaces are less ordered than what was imagined.
Direct observation of 14N and 81Br resonances in conventional MAS spectra of cationic surfactant crystals is also affordable but its transposition to the characterisation of materials textured by these surfactants remains difficult. However, first results obtained with 14N NMR are encouraging, and might complete the study of the order and dynamics in mesoporous materials from the local point of view of the polar head-groups. Alternatively proton detected 1H{14N} and 1H{81Br} NMR experiments give interesting and additional information on the spatial distribution of chemical groups at the interfaces. The continuous technological and methodological improvements in terms of sensitivity and resolution will continue to beneficiate all these studies.
Acknowledgements
CNRS, région Centre and région Languedoc-Roussillon are acknowledged for funding the NMR spectrometers. CNRS, Austrian Agency for International Cooperation in Education and Research, EU, EGIDE and INTAS are acknowledged for supporting international collaborations.