

1 Introduction
A current challenge is the development of efficient strategies for the design of switchable non-linear optical (NLO) materials. The ability to switch “ON/OFF” the NLO activity of a molecule is of relevance for the development of molecular photonic devices those properties can be switched by modifying one of the component parts [1]. To achieve an efficient switching effect, the molecule must be stable in the two ON and OFF states and the response time must be relatively fast. As most molecules with large quadratic hyperpolarizability values comprise π-systems end-capped with donor and acceptor moieties, various strategies have been proposed and categorized into three types (Fig. 1) [1a]: types (I) and (II) include the alteration of either the electron-donor or the electron-acceptor capacity of the end groups by using external stimuli, such as redox and protonation/deprotonation. Type (III) includes the alteration of the π-bridge using an external trigger, such as light, and in this category photochromic compounds seem to be promising candidates for the design of photoswitchable NLO materials [2].
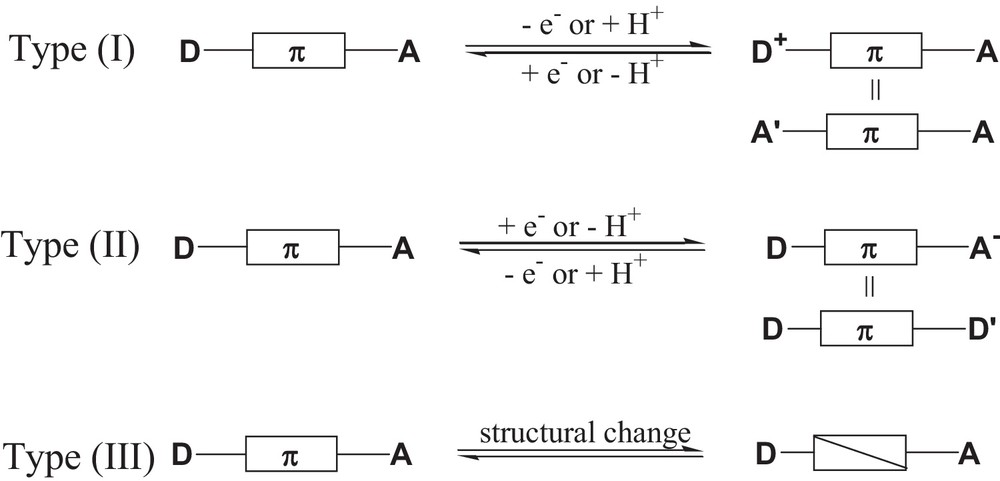
Schematic representations of strategies for switching NLO responses.
Photochromism, which refers to the reversible colour change of a compound with light irradiation, is attracting much attention for the construction of molecular devices, and there has been a growing interest in the synthesis, properties and applications of organic photochromic materials during the last decade [1b,3]. Among them, dithienylethene (DTE) derivatives are the most promising because of their good fatigue resistance, remarkable thermal stability of both isomers and rapid response time [3a]. Typically, DTE undergo reversible interconversion between a non-conjugated open form and a π-conjugated closed form when irradiated in the UV and visible spectral ranges, respectively (Scheme 1), and changes in the π-conjugated chain of DTE derivatives can be successfully used to control donor-acceptor interactions. Recently, the combination of transition metals and ligands featuring diarylethene units has received much attention and opened up new perspectives for the design of metal-based photoswitchable molecules [4]. For example, versatile diarylethene-containing polyimine ligands and their transition metal complexes have been synthesized and their photochromic behaviour has been widely exploited for the photomodulation of luminescence and electronic properties [5–10].
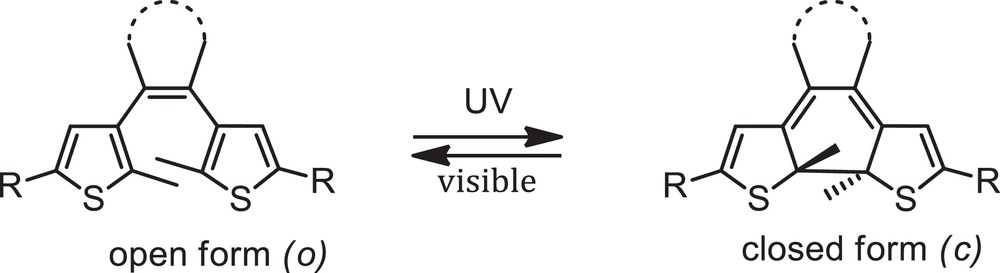
Photochromic interconversion of a dithienylethene (DTE) unit.
Our research group has been involved for the past 15 years in the NLO properties of bipyridyl metal complexes [11]. We have previously shown that compounds, such as donor-substituted bipyridines are excellent building blocks for the construction of either dipolar compounds [12] or non-dipolar (octupolar) metal complexes of D3 [13] and D2d [14] symmetry (Fig. 2). We have shown that the NLO response is mainly dictated by the intense intra-ligand charge-transfer (ILCT) transition from the donor group to the acceptor pyridine ring, and the role of the metal fragment is that of an inductive acceptor. These works have also underlined the role of the metallic core not only as a template but also for its participation in the quadratic non-linear optical responses, and suggested that other factors, such as the geometry of the complexes and the low-energy MLCT transitions, which may also contribute to the global NLO activity.
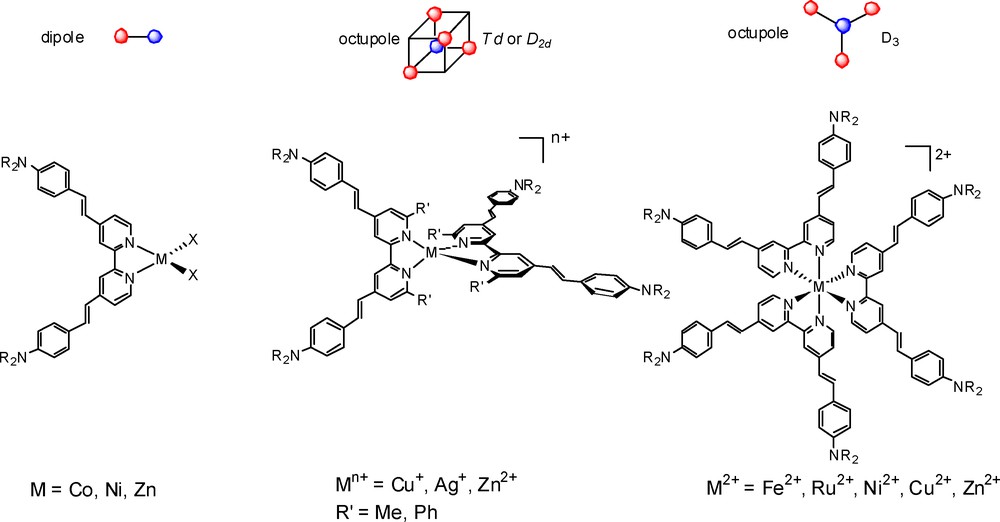
Representative examples of bipyridyl metal NLO chromophores.
The scope of this account is to discuss some of the recent work made in the area of photochromic organometallic and coordination compounds, which have been used for the photomodulation of the quadratic non-linear optical (NLO) properties, as well as of the photoregulation of the emission properties of the resulting systems. For this purpose, we have designed new chromophores combining DTE-based bipyridines with different metallic fragments, giving rise to multi-photochromic metal complexes containing from two to six DTE units, and studied the photocontrol of both NLO and luminescence properties.
2 Design and photochromic properties of bis-DTE 2,2′-bipyridine derivatives
2.1 Synthesis
The synthesis of bipyridine derivatives La-d (R = H; D = H, OMe, NMe2, NBu2) [15,16] and L′c (R = Me; D = NMe2) [17] was achieved by functionalization of the 4,4′ positions of 4,4′-dimethyl-2,2′-bipyridine and 4,4′,6,6’-tetramethyl-2,2’-bipyridine, respectively (Scheme 2). Dilithiation with LDA in THF at –78 °C, followed by reaction with chlorotrimethylsilane and subsequent chlorination with hexachloroethane afforded the resulting bis-chloromethyl derivatives in good yields Finally, the treatment of diethylphosphonatomethyl-2,2′-bipyridines, readily obtained through an Arbuzov reaction, with aldehydes a–d under normal Wadsworth–Emmons conditions afforded the target bipyridine derivatives La-d(o,o) and L′c(o,o) in their open forms in 50–60% yield after purification by recrystallization.
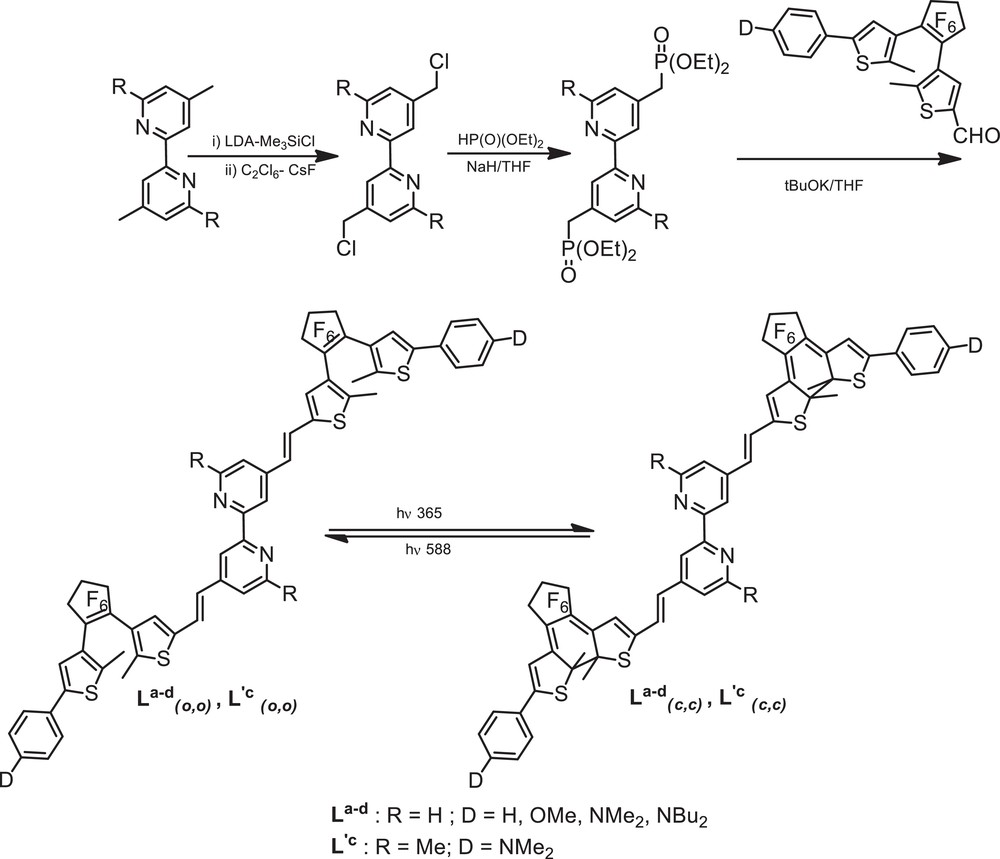
Synthesis of the photochromic bipyridine ligands and their photocyclization properties.
All compounds L and L′(o,o) show good transparencies in the visible region whatever the nature of the end D group: the UV–visible spectra in dichloromethane show an intense absorption at 340–350 nm which is tentatively assigned to (IL) π → π* transitions of the bipyridyl moieties with some mixing of the DTE units.
2.2 Photochromic properties
For all bipyridyl derivatives, the photocyclization process was confirmed by 1H NMR experiments: the colorless (or pale yellow) solution of La-d(o,o) and L′c(o,o) in CD2Cl2 turns to blue-green, upon UV irradiation at 365 nm. Their 1H NMR spectra show the characteristic upfield shift by ca. 0.5–0.6 ppm of the two thiophene protons, meanwhile, the methyl signals are shifted downfield by ca. 0.2 ppm. Integration of the methyl groups indicates a nearly quantitative conversion to the fully ring-closed isomers. The photocyclization and photocycloreversion processes were also followed by absorption spectroscopy [15–17] (Fig. 3). Irradiation of CH2Cl2 solutions at 365 nm resulted in the increase of a new broad band located between 620 and 670 nm (Table 1), which is attributed to the intra-ligand (ILc) π → π* transition of the closed form of the DTE units, the red-shift arising from the formation of a π-conjugated system. The influence of the electron-donor end group D is reflected by the bathochromic shift of the absorption band within the series NBu2 > NMe2 > OMe > H. For example, replacement of the H end group with the strong donating NBu2 group induces an additional 56-nm red shift of the ILc band (Table 1). Excitation in the absorption band of the closed forms leads to the quantitative regeneration of the open isomers, showing the reversibility of the process.
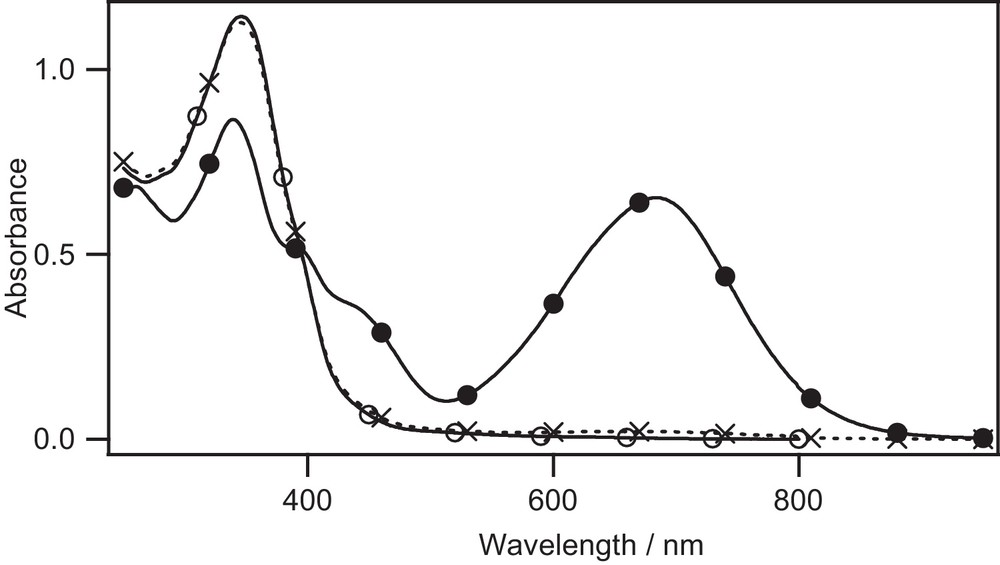
UV–vis absorption change of Lc in CHCl3 (1.5 × 105 M) upon irradiation. open form, PSS after irradiation at 365 nm, PSS after backward irradiation at 588 nm.
Main electronic absorption data for bipyridyl ligands in the open and closed (PSS) forms, % conversion at the PSS and photochromic quantum yields.
| Compound | λabs/nma open | λabs/nma,b closed (PSS) | % Convc | Φ Ring-closureb (o,o) → (o,c)/(o,c) → (c,c) |
| La | 343 | 623 | > 95 | 0.83/0.16 |
| Lb | 345 | 629 | > 95 | 0.65/0.15 |
| Lc | 348 | 669 | > 95 | 0.58/0.043 |
| Ld | 346 | 679 | > 95 | 0.12/0.012 |
| L′c | 340 | 668 | > 95 | – |
a At 298 K in CH2Cl2.
b Irradiation at λ = 365 nm.
c Determined by 1H NMR spectroscopy.
In order to gain further insight into the mechanistic closing process operating for the biphotochromic compound of these series, we have investigated quantitatively the photo-induced formation of the (c,c) species in solution under light irradiation [18]. We have measured the photochromic quantum yields by means of absorption spectroscopy and photokinetic models. All the spectroscopic experiments were conducted in CH2Cl2 solutions under continuous irradiation at λ = 365 nm (resp. λ = 588 or 650 nm) to promote the cyclization (resp. retrocyclization) reactions. This study revealed two different sets of quantum yields, the first one related to the (o,o)→(o,c) reaction being systematically larger by one order of magnitude than the second one describing the second ring-closure (o,c)→(c,c) (Fig. 4, Table 1). It thus becomes clear that although both photochromic reactions (o,o)→(o,c) and (o,c)→(c,c) can actually take place, the efficiency of the reaction is strongly reduced for the second step, and we believe that this can be the consequence of a strong cross-talk between a given open-DTE unit in the excited state and its neighbour. Concerning the ring opening, the quantum yields were found identical for both retrocyclization processes, which tend to confirm the weak electronic coupling between the closed-DTE units through the twisted bipyridyl central bridge. Their values were found to be lower than 10-2, namely, much smaller than those related to cyclization as usually reported for non-constraint DTE units. Systematic analyses of the ligands show a monotonous decrease of the quantum yields with increasing electron-donating ability of the end group D, namely H < OMe < NMe2 < NBu2.
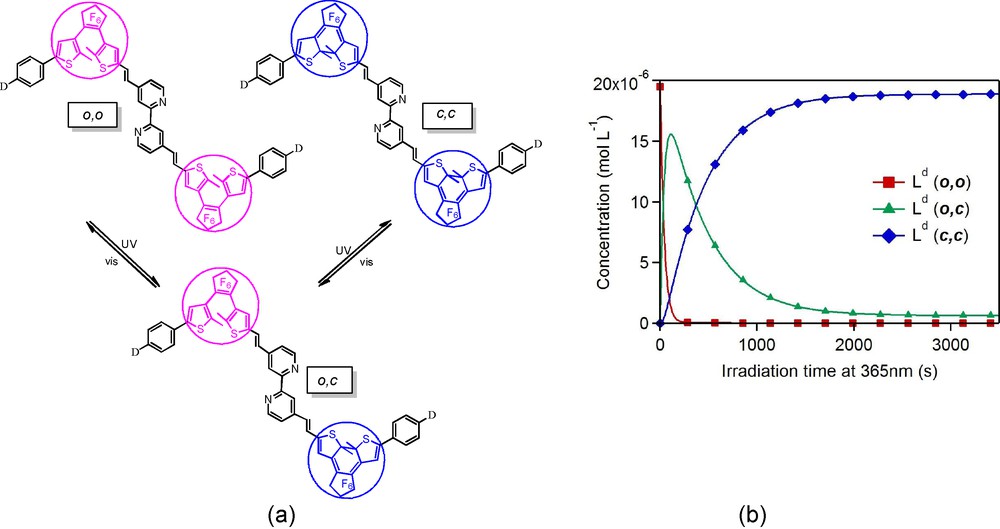
(a) Stepwise ring-closure and ring opening reactions of the photochromic bipyridines. (b) Time-evolution of the concentration of the three interconverting isomers of Ld.
3 Photochromic, NLO and luminescence properties of metal complexes featuring one bis-DTE 2,2′-bipyridine ligand
3.1 Synthesis
Fig. 5 depicts the structures of the Zn(II), Re(I), Ru(II) and Ir(III) complexes featuring a. bis-DTE bipyridine ligand. The Zn(II) complexes 1 a–d were readily obtained after room temperature treatment of La-d(o,o) with one equivalent of zinc diacetate dihydrate or zinc dichloride in dichloromethane [15,16]. Reaction of Lc(o,o) with Re(CO)5Br under refluxing anhydrous toluene afforded fac-Lc(o,o)Re(CO)3Br 2c as brown crystals. The ruthenium complex [(Lc(o,o))(dmbipy)2Ru](PF6)2 3c (dmbipy = 4,4′-dimethyl-2,2′-bipyridine) was obtained from the corresponding cis-(dmbipy)2RuCl2 upon treatment first with 2 equiv of silver triflate in methanol, then, the reaction with ligand Lc(o,o) in dichloromethane at room temperature, followed by anion-exchange from triflate with hexafluorophosphate [16]. The cationic Ir(III) complex 4c was readily synthesized upon treatment of the corresponding μ-chloro dimer [Ir(N^C-ppy-Me)2(μ-Cl)]2 with Lc(o,o) in the presence of AgPF6 [19]. All complexes were fully characterized by means of 1H and 13C NMR and UV–visible spectroscopy.
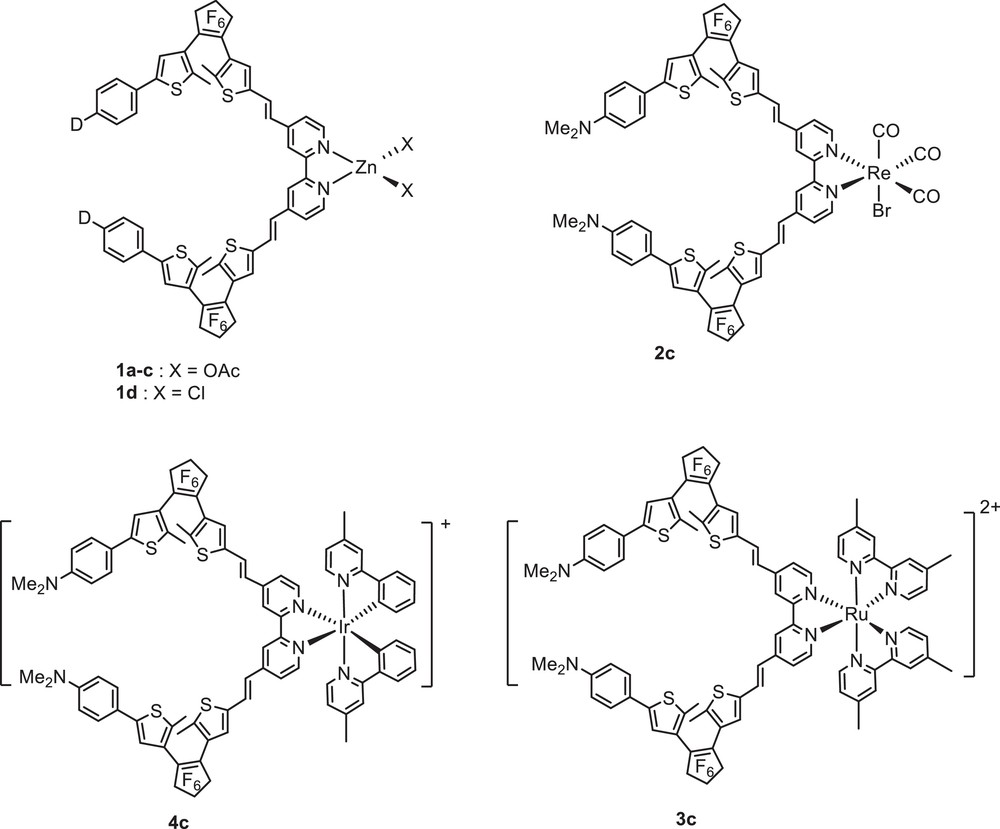
Chemical structures of photochromic bipyridine Zn(II), Re(I), Ru(II) and Ir(III) complexes.
The UV–visible spectra of the Zn complexes (in CH2Cl2) all show an intense band around 360 nm similar to that of the ligands, and slightly red-shifted by complexation (Table 2). The electronic absorption spectra of the Re, Ru and Ir complexes also show intense IL absorptions near 340 nm and 380–400 nm. In addition, the Ru complex displays another broad band in the visible at 490 nm corresponding to MLCT dπ(Ru) → π*(bipy) transitions, whereas the Re and Ir complexes show a moderately intense absorption shoulder at ca. 400–410 nm, which can be assigned as the dπ(M) → π*(bipy) transitions.
Main electronic absorption data for bipyridyl metal complexes in the open and closed (PSS) forms and % conversion at the PSS.
| Complex | λabs/nma open | λabs/nma closed (PSS) | % convb |
| 1a | 357 | 629 | 95% |
| 1b | 355 | 646 | 95% |
| 1c | 360 | 687 | 90% |
| 1d | 351 | 714 | 90% |
| 2c | 340, 380 (sh),410 (sh) | 705 | 65% |
| 3c | 338, 377 (sh), 491 | 716 | 65% |
| 4c | 341, 397 (sh) | 715 | 79% |
a At 298 K in CH2Cl2.
b Determined by 1H NMR spectroscopy.
3.2 Photochromic properties
For Zn complexes 1a–d, upon illumination at 365 nm, new absorption bands between 629 and 715 nm appear, originating from the formation of the corresponding closed isomers. As shown in Table 2, the absorption maxima of the closed-ring forms are dependent on the nature of the donor groups and metallic fragments: complexation of ligands La-c to Zn(OAc)2 induces a small bathochromic shift (Δλ = 6–18 nm) of the IL bands, whereas complexation to ZnCl2 (1d vs 1c) induces a much larger bathochromic shift (Δλ = 35 nm) than Zn(OAc)2, in agreement with the higher Lewis acidity of ZnCl2 vs Zn(OAc)2 [15,16,18]. According to 1H NMR analysis, integration of the methyl groups indicates a photocyclization yield of ca. 90%, showing that complexation to the Zn(II) ion does not perturb the photochromic properties of the bipyridyl ligands. Similarly to the free bipyridine ligands, photokinetics studies show that the photochromic ring-closure/opening reactions occur in a stepwise fashion with a decrease in the quantum yields [18]. Photocyclizations of the Re(I), Ru(II) and Ir(III) complexes 2–4c by irradiation with UV light at 350 nm, also give rise to lower-energy absorptions between 705 and 716 nm, corresponding to the ring-closed isomers (see Fig. 6) [16,19]. Thus, a substantial bathochromic shift of the IL band (Δλ = 36–47 nm) is also observed upon complexation of Lc to these organometallic fragments. According to 1H NMR, the ratio of the methyl signals between the closed and open-DTE units (ring closing) in the photostationary states (PSS) is lower (60–79%) than those found for the Zn(II) complexes. It is also interesting to note that the photochromic reaction can be triggered by irradiation of the Re, Ru and Ir complexes into the low-energy MLCT bands. This MLCT photosensitization suggests the intermediacy of the triplet state 3IL(DTE) in the photocyclization process. Finally, for all the complexes, excitation at 650 nm, in the absorption band of the closed forms leads to the quantitative regeneration of the open isomers, indicating the fully reversible closed-to-open photoisomerization.
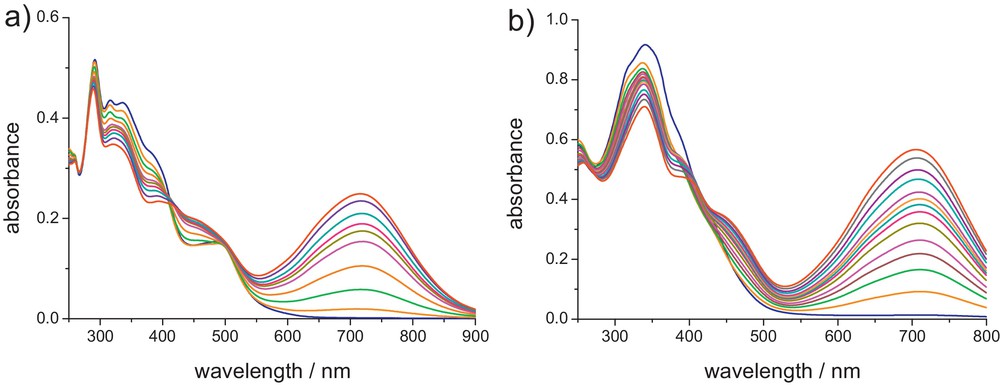
UV-vis absorption spectra changes of (a) 3c and (b) 2c in dichloromethane upon irradiation at 350 nm.
3.3 Photoregulation of the quadratic NLO properties
The second-order NLO responses of the dipolar Zn and Re complexes were determined in CH2Cl2 (concentration = 10−3 M) by the EFISH technique, working with an incident wavelength of 1.91 μm [15,16]. The values of EFISH μβ1.91, before and after UV irradiation, are reported in Table 3. The μβ1.91 values of 1c–d and 2c are quite weak, as expected by the absence of conjugation between the amino donor group and the bipyridyl metal acceptor moieties. A dramatic increase of the second-order NLO activity is observed after UV irradiation and subsequent accumulation of the ring-closed isomers: in the case of LdZnCl2 1d, the second-order NLO response is increased almost 50-fold after only 65% of ring closing [23]. Furthermore, it is also interesting to note that this latter EFISH μβ1.91 value is even higher than that found for LcZn(OAc)2 2c in the PSS (90% of ring closing) as expected by the higher Lewis acidity of ZnCl2 vs Zn(OAc)2. A similar, but somewhat less dramatic, behavior is observed for the Re(I) complex with an increase of μβ1.91 from 240 to 2920 × 1048 esu in the PSS (65% of ring closing). The lower efficiency of the Re(I) complex in comparison with that of the corresponding Zn(II) complexes has already been shown in dipolar donor-substituted styryl bipyridine metal chromophores [12], and can be explained by the presence of two vectorially opposed charge-transfer transitions (MLCT and ILCT) which contribute to a lowering of the total NLO response. The large enhancement of μβ1.91 values after ring-closure clearly reflects the delocalization of the π-electron system in the closed forms and, as expected, the largest NLO enhancement is observed with the complex featuring the harder ZnCl2 Lewis acid associated with the better NBu2 donor group, in agreement with the higher red-shift of the intra-ligand transition which dominates the second-order NLO response. This efficient ON/OFF switching markedly contrasts with the absence of photomodulation of the NLO response observed for the bis-cyclometallated phenylpyridine Ir(III) complex 3c featuring the same photochromic bipyridyl ligand Lc (Table 3) [19]. Whereas for the Zn(II) and Re(I) complexes the ILCT mainly contributes to the NLO response, for the Ir(III) complex the EFISH hyperpolarizability is dominated by L′LCT/MLCT processes, and thus, the nature of the π-conjugated substituents on the bipyridine ligand does not influence so much the μβ values.
EFISH μβ values and HLS β values determined at λinc = 1910 nm in CH2Cl2.
| Compound | EFISHa open μβ1.91a,b | EFISHa after UV irr. μβ1.91 a,c | HLS open <β1.91>d,e | HLS after UV irr. < β1.91>d,e | λem/nm (τ/μs)f Open | % Ring closing |
| 1c | 200 | 4220 | – | – | – | 90 |
| 1d | 113 | 5650 | – | – | – | 65 |
| 2c | 240 | 2920 | 229 | 965 | 652, 725(22) | 65 |
| 3c | – | – | 325 | 1113 | 680, 753(13) | 62 |
| 4c | –2190 | –2000 | – | – | 650, 717(23) | 79 |
a 1048 esu.
b Error ± 20%.
c Error ± 5%.
d 1030 esu.
e Error ± 15%.
f At 77 K in EPA (ether/isopentane/ethanol 2:2:1, v/v); λexc = 400 nm.
The Harmonic Light Scattering (HLS) technique was also used for the molecular first hyperpolarizability β measurements of the dicationic Ru complex 3c, and for comparison of the neutral Re complex 2c [16]. The HLS technique can be used also for ionic molecular species and for non-dipolar molecules, such as octupolar molecules. The measurements were performed in dichloromethane at a fundamental wavelength of 1.91 μm (Table 3). It has already been shown that the large β responses of bipyridine ruthenium chromophores, such as octupolar D3 tris-chelate ruthenium complexes are dominated by low-lying ILCT or MLCT excitations, depending on the electron-donating or -accepting nature of the substituents on the bipyridyl ligands [13,20]. It turned out that 3c in its open form also shows a fairly large <βHLS> value, larger than that of the Re complex, which can be reasonably attributed to the low-lying MLCT dπ(Ru) → π*(bipy) transition. Upon photocyclisation of the DTE, <βHLS> is found to increase by a factor of ca. 3.4, a result which is consistent with an NLO response mainly controlled by the red-shifted ILCT transition.
3.4 Photoregulation of the luminescence properties
In contrast to the free ligand Lc, which is non-emissive at room temperature in fluid solution and even in frozen glasses at 77 K, the corresponding Re(I), Ru(II) and Ir(III) complexes 2–4c in their open forms emit at 77 K (EPA), displaying structured luminescence spectra with vibronic progressions of 1400–1500 cm−1, typical of aromatic and/or CC bond vibrations [16,19]. The emission characteristics are summarized in Table 3 and the emission spectra of 2c and 3c and their PSS are shown in Fig. 7. The long lifetimes (13–23 μs), low-energy, and structured spectra are indicative of emission from a triplet IL state predominantly localized on the bpy–CC–Ar moiety, rather than from the MLCT state. Following conversion of solutions of 2–4c to the PSS at room temperature and re-freezing to 77 K, a substantial quenching of the 77 K luminescence is observed. The quenching can be attributed to intramolecular energy transfer from the triplet emissive state to the IL state of the closed-ring DTE part of the molecule: there is extensive overlap of the emission bands of the Re and Ru and Ir chromophores (λmaxem = 650–680 nm) with the low-energy absorption band of the acceptor photochromic unit in its closed form (λmaxabs = 705–716 nm). Thus, the combination of the photochromic DTE-based bipyridine ligand Lc with luminescent Re and Ru organometallic fragments allows the photoregulation of the emission and NLO properties of the resulting system. This study demonstrates for the first time that the photocontrol of two optical properties, linear and non-linear,could be achieved by using the same photochromic ligand.
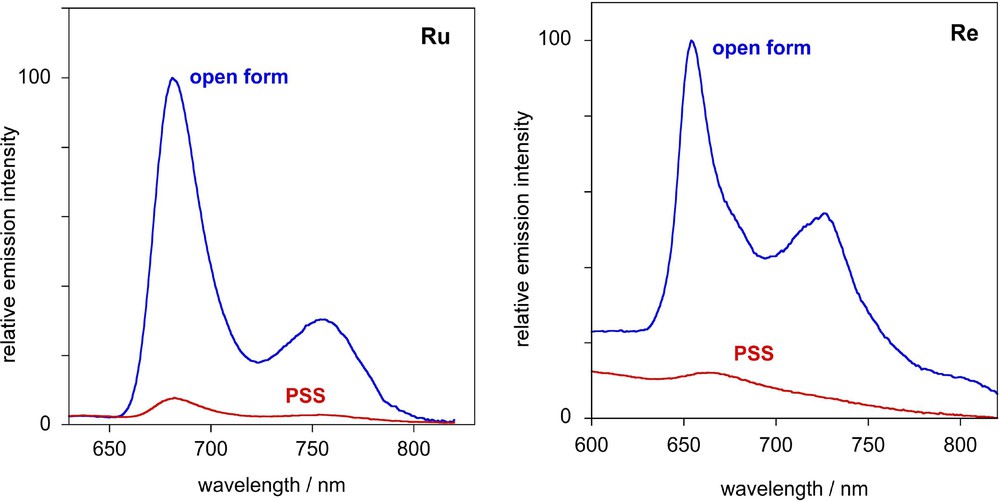
Left: Emission spectrum of 3c at 77 K in EPA (blue line) and the corresponding spectrum recorded for the photostationary state (red line). Right: Corresponding spectra of 2c and its PSS.
4 Photochromic and NLO properties of octupolar multi DTE-based metal complexes.
Having demonstrated the photoswitching of the quadratic NLO responses of dipolar metal complexes, we sought to study for the first time the photoregulation of the NLO properties of tetrahedral (Td, D2d) and octahedral (D3) octupolar chromophores. To this end, we have very recently designed new series of bis(bipyridyl) copper(I) complexes [17] and tris(bipyridyl) metal complexes featuring four and six DTE photochromic DTE units [21], respectively (Fig. 8). To the best of our knowledge, only a very few numbers of single molecules containing four DTE have been synthesised up to now [22], and only one example of star-shaped DTE-substituted molecule, in which six photochromic DTE units are connected around a central benzene core has been reported [23].
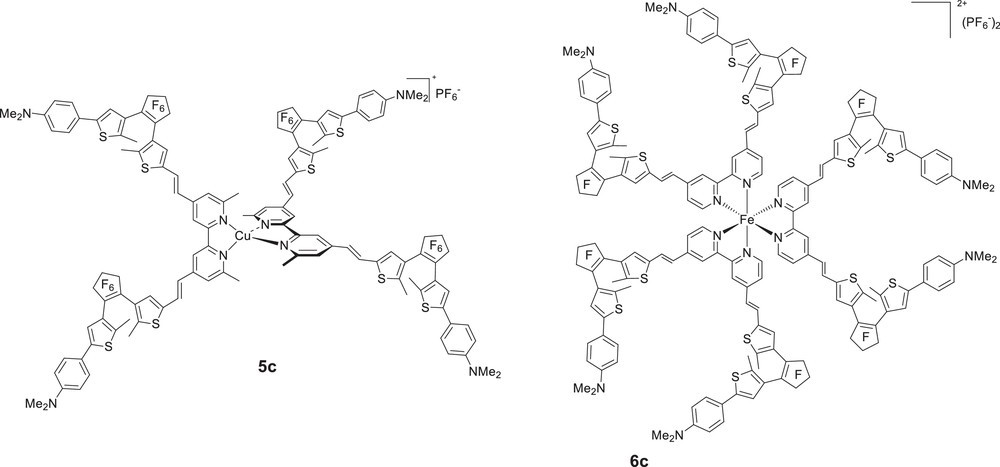
Chemical structure of the Cu(I) and Fe(II) complex salts (open forms).
4.1 Synthesis and photochromic properties
The Cu(I) complex 5c was formed almost quantitatively after mixing 2 equiv of L′c(o,o) with [Cu(MeCN)4]PF6 at room temperature in dichloromethane–acetonitrile solution. The tris(bipyridyl) iron(II) 6c was prepared in good yield by refluxing in ethanol the metallic precursors FeCl2·4H2O with 3 equiv of bipyridines Lc(o,o), followed by an anionic metathesis with sodium hexafluorophosphate. The 1H and 13C NMR spectra were in agreement with the proposed structures, and theoretical calculations confirmed both series of complexes belong to the D2d and D3 symmetry, respectively.
The UV–visible spectra of the fully open forms of 5c and 6c (in CH2Cl2) show like the free ligands an intense band around 340 nm assigned to (IL) π → π* transition of the bipyridyl moieties with some mixing of the DTE units, and another band in the visible at 511 nm and 583 nm, corresponding to the dπ(Cu) → π*(bipy) and dπ(Fe) → π*(bipy) MLCT transitions, respectively (Fig. 9, Table 4). The photocyclization process for both complexes was monitored by UV–vis (Fig. 9) and 1H NMR spectroscopy in dichloromethane. Upon irradiation at 350 nm, the red solution of 5c and the violet solution of 6c turn green, due to the ring-closure of the DTE units with a photostationary state (PSS) > 90% for the Cu(I) complex and > 60% for the Fe(II) complex. New broad bands emerge in the visible at 701 and 715 nm, respectively, which are attributed to intra-ligand (IL) transitions located on the closed-DTE centers. These results have been rationalized with the help of TD-DFT calculations, and the computed TD-DFT electronic spectra are in good agreement with experimental ones [17,21]. Upon irradiation at 650 nm, the quantitative regeneration of the fully open isomers is observed, indicating the fully reversible closed-to-open photoisomerization.
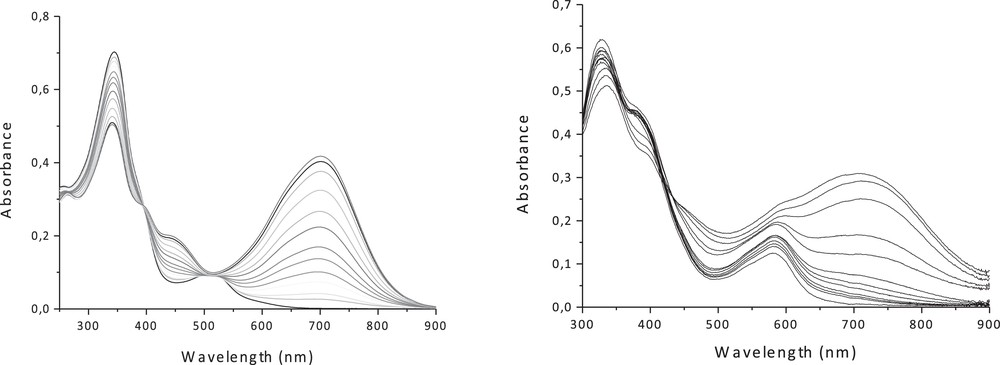
UV-vis absorption spectra changes of (left) 5c and (right) 6c in CH2Cl2 upon irradiation at 350 nm.
Experimental electronic absorption data for complexes in the open and closed (PSS) forms and measured quadratic hyperpolarizabilities (β) by HLS for the copper and iron derivatives.
| Compounda | λabs/nm (ɛ/M−1 cm−1)a Open | λabs/nm Closed (PSS) | HLS open <β1.91 > b | HLS after UV irr. < β1.91>b |
| 5c | 344(166000) 511(24000) | 343(119000) 399(sh) 450(47000) 701(95000) | 266 ± 52 | 1672 ± 160c |
| 6c | 331(206000) 387(sh) 583(42000) | 339(170000) 392(sh) 599(sh) 715(103000) | 250 ± 50 | 2000 ± 200d |
a Measured in CH2Cl2.
b 1030 esu.
c 65% Ring closing determined by 1H NMR.
d 60% Ring closing determined by 1H NMR.
4.2 Quadratic NLO studies
The Harmonic Light Scattering (HLS) technique was used for the molecular first hyperpolarizability β measurements of the Cu(I) and Fe(II) complexes. Measurements were carried out in dichloromethane (10-4 M) with an incident wavelength of 1.91 μm. Complexes 5c and 6c in their open forms shows fairly large <βHLS> values of 250–300 × 1030 esu (Table 4), which can be reasonably attributed to the low-energy MLCT dπ(M) → π*(bipy) transitions (M = Cu, Fe). A substantial enhancement of the second-order NLO activity is then observed after UV irradiation and subsequent accumulation of the ring-closed isomer, a result which is consistent with an NLO response mainly controlled by the red-shifted Intra-Ligand transition with a 6–8-fold enhancement of the dynamic first hyperpolarizability after only 60–65% of ring closing to reach extremely large βHRS values exceeding 1600 × 1030 esu. Theoretical calculations of the dynamic (1907 nm) and static first hyperpolarizabilities of the fully open and fully closed structures of 5c and 6c also predict a large enhancement of the non-linear response by a factor between 8 and 12, when switching all the four and six DTE units, respectively [17,21]. This agreement confirms that the increase of the NLO response is related to the additional IL transitions rather than in variations of the MLCT bands.
5 Conclusions
In this account, we summarize our successful strategies toward the synthesis of photochromic organometallic and coordination compounds based on metal-bipyridyl chemistry. We have shown that coordination chemistry is a versatile tool for the design of multi-photochromic molecules incorporating from two to six dithienylethene (DTE) units, and their access can be easily achieved by combining bipyridyl ligands with different metal ions. These metal systems, which can be either dipolar or octupolar, allowing the photomodulation of optical properties, such as quadratic non-linear optics and phosphorescence. This study also demonstrates for the first time that the photocontrol of two optical properties, linear and non-linear, could be achieved by using the same photochromic ligand. This coordination chemistry approach should open up new perspectives for the design of multi-photochromic metallo-switches with improved switching efficiency and NLO contrast upon varying the metal ion. Finally, the integration of such photochromic molecules into polymeric matrices in order to develop switchable NLO materials and devices should be another interesting issue. We are currently working in that direction.
Acknowledgements
We are grateful to the very talented graduate students and co-workers, Dr V. Aubert, Dr L. Ordronneau, Dr H. Nitadori and Dr J. Boixel for their outstanding contributions. We are greatly indebted to Prof I. Ledoux, Prof J. Zyss (LPQM, ENS Cachan), and Prof D. Roberto (University of Milano, Italy) for carrying out the NLO measurements. We thank Prof K. Nakatani, Dr R. Métivier (PPSM, ENS Cachan) and Prof E. Ishow (University of Nantes) for their contribution to the photokinetics studies, and Prof J. A. G. Williams (Durham University, UK) for performing the photophysical studies. We gratefully acknowledge Prof A. Boucekkine (University of Rennes) and Prof D. Jacquemin (University of Nantes) for the computational studies. This work was supported by the CNRS, the ANR BLAN-COMET and the Région Bretagne.