

1 Introduction
Nowadays, multi-component reactions (MCRs) have proven to be appreciated in organic, medicinal chemistry and other industries [1–4]. Such protocols can be used for drug design and diversity-oriented synthesis of heterocycles because of their simplicity, efficiency, and high selectivity [5]. This environmentally friendly process can avoid waste of time as well as energy consumption, and permits to reduce the number of steps. MCRs under solvent-free conditions have attracted much interest from chemists, particularly from the viewpoint of green chemistry. The possibility of performing MCR reactions under solvent-free conditions with a catalyst could enhance their efficiency from an economic as well as ecological point of view [6–10].
Nitrogen-containing heterocycles are widely found in nature and gain much importance due to their applications in biologically active pharmaceuticals, agrochemicals and medicinal chemistry [11]. Pyrimidinobenzimidazoles possess antimicrobial activity, molluscidal activities, antidiabetic, herbicidal activity, platelet anti-aggregant antiulcer activity, DNA-gyrase inhibitors and anti-inflammatory activities as well as anticancer efficacy; they can play the role of antagonists of the paralyzing action of antidiabetic activity and are important core motifs in a broad range of biologically active compounds that are frequently used in pharmaceutical and drug research, agricultural science, and dye industry [12–14]. N-Fused heterocycles of imidazole and pyrimidines frameworks, imidazopyrimidine derivatives are important biological molecules, and they exhibit more biological activity [15], such as anti-inflammatory, antiviral, antimicrobial, anticonvulsant, anti-asthma, anti-diabetes, anti-hepatitis B, anti-vascular hypertension, anti-arrhythmia, and anticancer activities [15d]. Only a few methods have been reported for the one-pot synthesis of imidazopyrimidine derivatives [16]. However, these procedures suffer from relatively long reaction times and low yields. Therefore, the development of an easy and efficient method for the preparation of 2-amino-4-substituted-1,4-dihydrobenzolo[4,5]imidazo[1,2-a]pyrimidine-3-carbonitriles is still a challenging task.
p-Toluenesulfonic acid (p-TSA) has received considerable attention as a catalyst for various organic transformations due to its non-toxicity, ease of handling, commercially availability, inexpensiveness, air and water compatibility, and strong organic acidic nature [17]. Solvent-free reaction-endorsed p-TSA reactions are well known as environmentally benign methods that also usually provide improved selectively, enhanced reaction rates, cleaner products, and manipulative simplicity [18].
In a continuation of our interest in developing more efficient and environmental benign methodologies [19], we described a method for the synthesis of 2-amino-4-substituted-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitriles by a one-pot, multi-compound condensation of 2-aminobenzimidazole, aldehydes, and malononitrile catalyzed by p-TSA under neat conditions at 80 °C (Scheme 1).
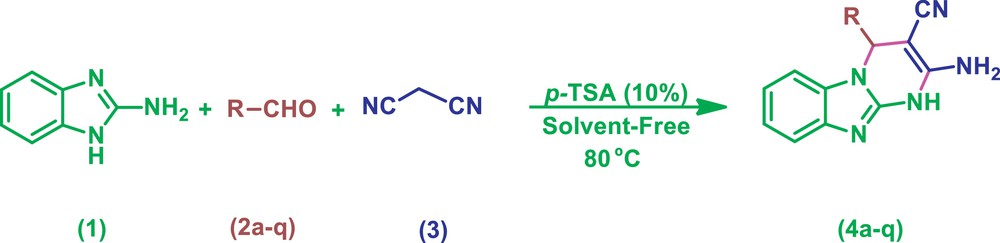
Synthesis of 2-amino-4-substituted-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitriles catalyzed by p-TSA under neat conditions.
2 Results and discussion
In our initial study, the reaction of 2-aminobenzimidazole 1, 4-bromobenzaldehyde 2a, and malononitrile 3 was chosen as a model reaction to optimize the reaction conditions at 80 °C. The results are summarized in Table 1. The reaction was first carried out in ethanol in the absence and presence of several acid catalysts. The reaction did not proceed even after prolonged reaction time and no desired product was formed in the absence of catalyst. But under vigorous reaction conditions, a small amount of product was observed (Table 1, entry 1). Therefore, our efforts focused on the search for a suitable catalyst. Some Lewis acid (Table 1, entries 2–7) can catalyze this reaction with moderate yields. With these merits, like reaction time, product yield, the p-TSA is the desired protocol for MCR reactions (Table 1, entry 8) and for the model reaction. In addition, methanol, water, DMF, and CH3CN (Table 1, entries 9–12) were also tested as the solvents. In these cases, product 4a was formed in slightly lower yields.
Optimization of catalysts and solvents in the synthesis of 4aa.
| Entry | Catalyst (%) | Solvent | Time | Yield (%)b |
| 1 | None | Ethanol (10 mL) | 6 h | 20 |
| 2 | InCl3 (10) | Ethanol (10 mL) | 3 h | 65 |
| 3 | InF3 (10) | Ethanol (10 mL) | 4 h | 56 |
| 4 | GaCl3 (10) | Ethanol (10 mL) | 3 h | 45 |
| 5 | FeCl3 (10) | Ethanol (10 mL) | 4 h | 65 |
| 6 | ZnCl2.H2O (10) | Ethanol (10 mL) | 5 h | 45 |
| 7 | Li(OTf) (10) | Ethanol (10 mL) | 2 h | 70 |
| 8 | p-TSA (10) | Ethanol (10 mL) | 1 h | 80 |
| 9 | p-TSA (10) | Methanol (10 mL) | 1.5 h | 70 |
| 10 | p-TSA (10) | Water (10 mL) | 4 h | 45 |
| 11 | p-TSA (10) | DMF (10 mL) | 2 h | 65 |
| 12 | p-TSA (10) | Acetonitrile (10 mL) | 3 h | 55 |
| 13 | InCl3 (10) | Neat | 2 h | 75 |
| 14 | InF3 (10) | Neat | 3 h | 65 |
| 15 | GaCl3 (10) | Neat | 2 h | 54 |
| 16 | FeCl3 (10) | Neat | 1 h | 70 |
| 17 | ZnCl2.H2O (10) | Neat | 3 h | 55 |
| 18 | Li(OTf) (10) | Neat | 2 h | 75 |
| 19 | p-TSA (10) | Neat | 30 min | 92 |
| 20 | p-TSA (2) | Neat | 60 min | 60 |
| 21 | p-TSA (5) | Neat | 45 min | 85 |
| 22 | p-TSA (20) | Neat | 30 min | 93 |
a Reaction of 2-aminobenzimidazole 1 (1 mmol), 4-bromobenzaldehyde 2a (1 mmol) and malononitrile 3 (1 mmol) at 80 °C.
b Isolated yield.
With respect to the solvent system, the best results were achieved using ethanol (Table 1, entry 8). In recent years, the synthesis of compounds under solvent-free conditions is an important task in heterocyclic synthesis. Therefore, we decided to test a solvent-free version with various catalysts (Table 1, entries 13–18). We found that the reaction was rapid and gave excellent yields of the products when using p-TSA (Table 1, entry 19). Most excitingly, when p-TSA was used, the reaction proceeded very smoothly and gave the product 4a in 92% yield (Table 1, entry 19). Moreover, we found that the yields were obviously affected by the amount of p-TSA loaded. When 2 mol%, 5 mol%, 10 mol%, and 20 mol% of p-TSA were used, the yields were 65, 85, 92, and 93%, respectively (Table 1, entries 19–22). Therefore, 10 mol% of p-TSA was sufficient and excessive amounts of catalyst did not increase the yields significantly.
To find the optimum reaction temperature, the reaction was studied at different temperatures for the preparation of 4a with 10 mol% p-TSA under neat conditions (Table 2). It was observed that the reaction did not proceed at room temperature. We were pleased to find that the reaction proceeded smoothly and that almost complete conversion of reactants was observed at 80 °C to afford the desired product (4a) in 92% yield within 30 min (Table 2 entry 4). Further increase of the temperature did not affect the product yield.
Effect of temperature for the preparation of 4aa.
| Entry | Temperature (°C) | Time (min) | Yield (%)b |
| 1 | Room temperature | 120 | 20 |
| 2 | 50 | 75 | 78 |
| 3 | 70 | 45 | 85 |
| 4 | 80 | 30 | 92 |
| 5 | 100 | 30 | 93 |
a Reaction of 2-aminobenzimidazole 1 (1 mmol), 4-bromobenzaldehyde 2a (1 mmol) and malononitrile 3 (1 mmol) using p-TSA (10%) under neat condition.
b Isolated yield.
Under these appropriate reaction conditions (neat, 80 °C, 10 mol% p-TSA), a series of 2-amino-4-substituted-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitriles (4) were synthesized. The results are summarized in Table 3.
p-TSA catalyzed multi-component synthesis of 4a–q.
| Entry | Product | Aldehyde R | Yielda (%) | Time (min) | Mp (°C) | ||
| Found | Reported | Found | Reported | ||||
| 1 | 4a | 4-Br–C6H4 | 92 | 30 | 317–319 | – | |
| 2 | 4b | 4-Cl–C6H4 | 93 | 70 [16c] | 30 | 234–236 | 238[16c] |
| 3 | 4c | 2-F–C6H4 | 90 | 35 | 224–226 | – | |
| 4 | 4d | 3-F–C6H4 | 90 | 32 | 234–236 | – | |
| 5 | 4e | 4-F–C6H4 | 91 | 85[16b] | 28 | 266–268 | 270[16b] |
| 6 | 4f | 3-Br–C6H4 | 89 | 83[16c] | 36 | 238–240 | 240[16c] |
| 7 | 4g | 4-isopropyl–C6H4 | 92 | 33 | 230–232 | – | |
| 8 | 4h | 4-OEt–C6H4 | 93 | 31 | 202–204 | – | |
| 9 | 4i | 3,4-5-OMe–C6H2 | 89 | 38 | 230–232 | – | |
| 10 | 4j | 2-Cl–C6H4 | 85 | 69[16c] | 40 | 236–238 | 237[16c] |
| 11 | 4k | 2-F-4-OMe–C6H3 | 88 | 32 | 204–206 | – | |
| 12 | 4l | 3-F-4-Me–C6H3 | 90 | 34 | 228–230 | – | |
| 13 | 4m | 2-Me–C6H4 | 88 | 72[16c] | 40 | 233–235 | 236[16c] |
| 14 | 4n | 90 | 31 | 203–205 | – | ||
| 15 | 4o | C2H5 | 88 | 40 | 198–200 | – | |
| 16 | 4p | CH(CH3)2 | 89 | 40 | 225–227 | – | |
| 17 | 4q | C3H7 | 85 | 45 | 238–240 | – |
a Isolated yield.
A variety of substrates were submitted to the optimum reaction conditions and the desired products were obtained in excellent yields (Table 3). Various functional groups were found to be compatible under our reaction conditions. It is noteworthy that there are no report of the synthesis of 2-amino-4-substituted-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitriles from aliphatic aldehydes [16]. In general, the reactions were clean and no side products were detected. In all cases, the reactions proceeded efficiently at 80 °C in solvent-free conditions and with 10 mol% of the catalyst. All the new compounds were characterized by 1H NMR, 13C NMR, and HRMS.
A schematic presentation of the catalytic activity of p-TSA in the synthesis of title compounds (4a–q) should be postulated, as shown in Scheme 2. The reaction is thought to proceed in a stepwise manner. Firstly, we assumed that the reaction occurs via a Knoevenagel condensation between an enolic form like malononitrile, 3, and aldehyde, 2, to form the intermediate 5 in the presence of acidic p-TSA, which suffers immediate Michael addition of 2-aminobenzimidazole (1) to be the conjugated CC bond of 5, forming intermediate 6. Finally, the title compounds (4a–q) were formed by intramolecular concerted cyclisation by tautomerization of Michael adduct 6.
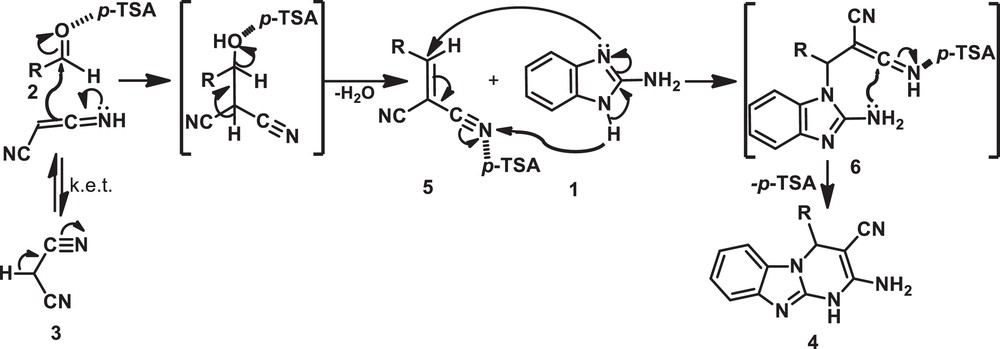
Schematic mechanism for the catalytic activity of p-TSA in the synthesis of title compounds (4a–q).
3 Conclusion
In conclusion, we have developed an efficient and environmentally benign methodology for the preparation of 2-amino-4-substituted-1,4-dihydrobenzolo[4,5]imidazo[1,2-a]pyrimidine-3-carbonitriles in the presence of an inexpensive p-TSA catalyst under neat conditions. The advantages of this method over other existing ones are reduced reaction times, high to excellent yields, simple experimental procedure, low cost, easy handling, and economic viability of the catalyst. The procedures provide access to compounds that are useful in heterocyclic synthesis.
4 Experimental
4.1 General
Chemicals were purchased from Aldrich and Alfaaesar Chemical Companies. NMR spectra were recorded in ppm in DMSO-d6 on a Jeol JNM ECP 400 NMR instrument using TMS as an internal standard. Mass spectra were recorded on a Jeol JMS-700 mass spectrometer. All melting points were determined using open capillaries on an Electrothermal-9100 (Japan) instrument. Infrared (IR) spectra were obtained using a Shimadzu IR Prestige-21 FT IR spectrophotometer.
4.2 Synthesis of 2-amino-4-(4-bromophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4a)
A mixture of 2-aminobenzimidazole (1, 1 mmol), 4-bromobenzaldehyde (2a, 1 mmol), malononitrile (3, 1 mmol) and p-TSA (10 mol%) was stirred at 80 °C under solvent-free conditions for 30 min (Table 3, entry 1). The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was washed with water and ethanol, and the residue recrystallized from ethanol to afford the pure product 4a (92%).
4.2.1 2-Amino-4-(4-bromophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4a)
Yield 92%; yellow powder; mp 317–319 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.62 (s, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 8.5 Hz, 2H), 7.24 (t, J = 8.5 Hz, 3H), 7.11 (t, J = 7.5 Hz, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.87 (s, 2H), 5.24 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 151.2, 149.2, 143.5, 142.2, 131.6, 129.2, 128.2, 123.4, 121.0, 119.9, 119.0, 116.1, 112.4, 61.4, 52.6. HRMS (ESI, m/z): calcd for C17H12BrN5 (M + H+) 366.025; found: 366.0038.
4.2.2 2-Amino-4-(4-chlorophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4b)
Yield 93%; yellow powder; mp 234–236 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.60 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.43–7.38 (m, 2H), 7.30 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 7.5 Hz, 1H), 7.14 (d, J = 8.2 Hz, 2H), 6.85 (s, 2H), 5.25 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 151.5, 149.3, 143.1, 134.3, 132.4, 130.8, 129.1, 128.5, 123.0, 121.8, 119.9, 119.0, 116.0, 112.8, 61.2, 55.5.
4.2.3 2-Amino-4-(2-fluorophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4c)
Yield 90%; yellow powder; mp 224–226 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.58 (s, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.37–7.10 (m, 5H), 7.01 (t, J = 7.9 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 6.86 (s, 2H), 5.48 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 160.7, 151.7, 149.4, 143.5, 130.0, 129.8, 129.3, 127.9, 124.7, 123.3, 119.9, 118.7, 116.5, 112.4, 60.7, 48.5. HRMS (ESI, m/z): calcd for C17H12FN5 (M + H+) 305.108; found: 305.103.
4.2.4 4.2.4. 2-Amino-4-(3-fluorophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4d)
Yield 90%; yellow powder; mp 234–236 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.65 (s, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.45–7.38 (m, 1H), 7.24 (d, J = 7.3 Hz, 1H), 7.14–7.08 (m, 4H), 7.02–6.98 (m, 1H), 6.89 (s, 2H), 5.28 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 161.0, 151.5, 149.3, 145.8, 143.8, 130.8, 129.2, 123.3, 121.9, 119.9, 119.0, 116.1, 112.9, 112.7, 112.4, 61.2, 52.5. HRMS (ESI, m/z): calcd for C17H12FN5 (M+H+) 305.108; found: 305.102.
4.2.5 42-Amino-4-(4-fluorophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4e)
Yield 91%; yellow powder; mp 266–268 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.23 (s, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.21–7.14 (m, 5H), 7.08 (t, J = 7.5 Hz, 1H), 6.95 (t, J = 7.5 Hz, 1H), 6.71 (s, 2H), 5.30 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 160.5, 151.9, 148.9, 143.8, 135.5, 130.8, 129.1, 123.3, 121.7, 119.8, 119.3, 116.7, 114.8, 112.9, 112.7, 60.2, 54.5.
4.2.6 2-Amino-4-(3-bromophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4f)
Yield 89%; yellow powder; mp 238–240 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.60 (s, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.5 Hz, 2H), 7.26(t, J = 8.5 Hz, 3H), 7.10 (t, J = 7.5 Hz, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.85 (s, 2H), 5.25 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 151.2, 149.1, 143.5, 141.2, 131.6, 130.5, 129.6, 128.5, 123.2, 121.5, 119.9, 119.0, 116.1, 112.2, 61.5, 52.8.
4.2.7 2-amino-4-(4-isopropylphenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4g)
Yield 92%; yellow powder; mp 230–232 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.22–7.16 (m, 5H), 7.10 (t, J = 7.3 Hz, 1H), 6.99 (t, J = 7.3 Hz, 1H), 6.80 (s, 2H), 5.15 (s, 1H), 2.86–2.80 (m, 1H), 1.65 (d, J = 6.6 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 151.7, 149.0, 148.0, 143.6, 129.2, 126.5, 125.8, 123.2, 119.8, 119.2, 116.0, 112.3, 62.0, 53.0, 33.0, 23.7. HRMS (ESI, m/z): calcd for C20H19N5 (M + H+) 329.164; found: 329.162.
4.2.8 2-amino-4-(4-ethoxyphenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4h)
Yield 93%; yellow powder; mp 202–204 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.49 (s, 1H), 7.96 (d, J = 8.8 Hz, 2H), 7.62 (t, J = 8.0 Hz, 1H), 7.20–7.15 (m, 1H), 7.10 (t, J = 7.3 Hz, 1H), 6.99 (t, J = 7.3 Hz, 1H), 6.88 (d, J = 8.8 Hz, 2H), 6.77 (s, 2H), 5.13 (s, 1H), 4.01–3.94 (m, 2H), 1.28 (t, J = 6.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 160.4, 151.2, 149.2, 143.5, 133.5, 133.4, 129.2. 127.2, 123.9, 123.2, 119.7, 115.5, 114.8, 113.9, 64.0, 62.9, 52.7, 14.3. HRMS (ESI, m/z): calcd for C19H17N5O (M+H+) 331.143; found: 331.142.
4.2.9 2-Amino-4-(3,4,5-trimethoxyphenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4i)
Yield 89%; yellow powder; mp 230–232 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.52 (s, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.11 (t, J = 7.3 Hz, 1H), 7.00 (t, J = 7.3 Hz, 1H), 6.83 (s, 2H), 6.62 (s, 2H), 5.18 (s, 1H), 3.69 (s, 6H), 3.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 152.8, 151.7, 149.2, 143.5, 138.1, 137.1, 129.2, 123.3, 119.9, 119.1, 116.0, 112.3, 103.6, 61.6, 59.9, 55.8, 53.3. HRMS (ESI, m/z): calcd for C20H19N5O3 (M+H+) 377.1490; found: 377.1485.
4.2.10 2-Amino-4-(2-chlorophenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4j)
Yield 85%; yellow powder; mp 236–238 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 1H), 7.65 (d, J = 8.2 Hz, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.35–7.20 (m, 5H), 7.08 (t, J = 7.5 Hz, 1H), 6.80 (s, 2H), 5.23 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 151.5, 149.5, 143.5, 138.3, 135.5, 132.5, 130.5, 129.1, 128.5, 127.5, 123.0, 121.8, 119.9, 116.0, 112.8, 59.5, 52.5.
4.2.11 2-Amino-4-(2-fluoro-4-methoxyphenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4k)
Yield 88%; yellow powder; mp 204–206 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.44 (s, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.23–7.09 (m, 4H), 7.00 (t, J = 6.9 Hz, 1H), 6.81 (s, 2H), 6.75 (dd, J = 2.5, 11.0 Hz, 1H), 5.38 (s, 1H), 3.73 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.6, 151.7, 149.2, 143.3, 129.3, 128.6, 123.3, 121.2, 119.8, 118.7, 116.0, 112.3, 110.4, 102.0, 61.1, 56.0, 55.6. HRMS (ESI, m/z): calcd for C18H14FN5O (M+H+) 335.1184; found: 335.1184.
4.2.12 4.2.12. 2-Amino-4-(3-fluoro-4-methylphenyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4l)
Yield 90%; yellow powder; mp 228–230 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.60 (s, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.27–7.21 (q, 2H), 7.11 (t, J = 7.5 Hz, 1H), 7.04–7.00 (m, 3H), 6.86 (s, 2H), 5.22 (s, 1H), 2.17 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 161.7, 151.6, 149.2, 143.5, 142.9, 131.9, 129.2, 123.8, 123.6, 121.6, 119.9, 116.1, 115.0, 112.6, 61.4, 52.4, 13.8. HRMS (ESI, m/z): calcd for C18H14FN5 (M+H+) 319.1234; found: 319.1236.
4.2.13 2-Amino-4-(o-tolyl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4m)
Yield 88%; yellow powder; mp 233–235 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.21 (d, J = 8.8 Hz, 1H), 7.14–7.15 (m, 3H), 7.05 (t, J = 7.5 Hz, 1H), 6.95 (t, J = 7.5 Hz, 1H), 6.68 (s, 2H), 5.30 (s, 1H), 2.25 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 151.3, 149.2, 143.5, 137.5, 135.5, 131.2, 129.5, 128.5, 125.6, 122.9, 119.6, 116.8, 112.1, 59.4, 55.2, 17.4.
4.2.14 2-Amino-4-(benzo[d][1,3]dioxol-5-yl)-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4n)
Yield 90%; yellow powder; mp 203–205 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.34 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.21–6.81 (m, 6H), 6.21 (s, 2H), 5.97 (s, 2H), 5.15 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 160.3, 152.9, 148.3, 129.9, 125.5, 123.3, 119.8, 116.0, 113.7, 112.3, 109.2, 106.4, 102.4, 62.0, 52.9. HRMS (ESI, m/z): calcd for C18H13N5O2 (M+H+) 331.1070; found: 331.1065.
4.2.15 2-Amino-4-ethyl-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4o)
Yield 88%; yellow powder; mp 198–200 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.24 (s, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.08 (t, J = 7.3 Hz, 1H), 6.95 (t, J = 7.3 Hz, 1H), 6.68 (s, 2H), 4.01 (t, J = 5.4 Hz, 1H), 1.60–154 (m, 2H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 152.7, 150.2, 144.3, 129.8, 123.5, 120.6, 119.5, 116.3, 112.2, 60.7, 51.2, 29.2, 7.6. HRMS (ESI, m/z): calcd for C13H13N5 (M + H+) 239.1175; found: 239.1170.
4.2.16 2-Amino-4-isopropyl-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4p)
Yield 89%; yellow powder; mp 225–227 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.23 (s, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.08 (t, J = 7.3 Hz, 1H), 6.95 (t, J = 7.3 Hz, 1H), 6.69 (s, 2H), 3.83 (d, J = 5.1 Hz, 1H), 1.76–172 (m, 1H), 0.83 (t, J = 5.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 152.3, 149.8, 143.7, 129.2, 123.1, 119.9, 119.5, 115.7, 112.2, 59.0, 55.8, 35.4, 17.4. HRMS (ESI, m/z): calcd for C14H15N5 (M+H+) 253.1335; found: 253.1330.
4.2.17 2-Amino-4-propyl-1,4-dihydrobenzo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitrile (4q)
Yield 85%; yellow powder; mp 238–240 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.24 (s, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.20 (d, J = 8.8 Hz, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.95 (t, J = 7.5 Hz, 1H), 6.69 (s, 2H), 3.83 (d, J = 5.1 Hz, 1H), 1.77–1.69 (m, 2H), 1.29–1.27 (m, 2H), 0.83 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 152.3, 149.8, 143.7, 129.2, 123.2, 119.9, 115.8, 112.2, 59.0, 55.8, 35.4, 20.8, 17.4. HRMS (ESI, m/z): calcd for C14H15N5 (M+H+) 253.1335; found: 253.1330.
Acknowledgements
This research work was supported by the second stage of BK21 Program.