

1 Introduction
La gomme de caroube, connue dès l’Égypte ancienne, est un composé dont l’utilisation industrielle remonte au début du 20e siècle. Dans les années 1940, lorsque les restrictions liées à la guerre rendaient difficile son approvisionnement, il fut nécessaire de lui trouver un substituant, ce qui a conduit à l’émergence de la gomme de guar [1]. La gomme de caroube a retrouvé par la suite ses lettres de noblesse et est, à l’heure actuelle et grâce à sa capacité d’auto-association, un additif alimentaire (E410) utilisé industriellement pour ses propriétés texturantes (épaississantes et viscosifiantes) et stabilisantes [2]. Elle entre notamment dans la composition des crèmes glacées, des soupes, des friandises, des aliments pour bébés, des produits laitiers, des boissons alcoolisées et non alcoolisées, des aliments pour bétail, etc. [3–12]. La gomme de caroube possède également la propriété de former des gels par association à d’autres polysaccharides (carraghénanes, xanthane, agarose, etc.), à des concentrations bien inférieures à une utilisation de ces polysaccharides seuls. Il est, dès lors, économiquement intéressant d’y avoir recours comme agent co-gélifiant pour des applications telles que les crèmes laitières, les crèmes fouettées, les crèmes glacées sans lait, le surimi, les boulettes de viande allégées, les analogues de mozzarella, etc. [13–16]. Dans les produits de boulangerie, l’adjonction de gomme de caroube prévient ou retarde la rétrogradation des amidons et des amidons modifiés et influe sur la viscosité de ces derniers [17]. La gomme de caroube est également utilisée en tant qu’additif pour des applications non alimentaires dans l’industrie papetière, cosmétique, textile et pharmaceutique [4,18–21], où ses propriétés d’agent de libération contrôlée de médicament sont prisées [22] ainsi que sa capacité de co-gélification, utilisée en encapsulation de principes actifs [23]. La gomme est fabriquée à partir de l’endosperme des graines du caroubier (Ceratonia sliliqua L.). Cet endosperme est composé de polysaccharides de réserve (hémicelluloses) appelés galactomannanes. Ils sont constitués d’une chaîne principale de mannose sur laquelle sont greffés des substituants latéraux : les résidus galactosyles (Fig. 1). Les galactomannanes, une fois extraits et purifiés, confèreront à une solution aqueuse de gomme de caroube les propriétés texturantes tant recherchées par l’industrie. Les propriétés physiques de la gomme en solution sont intimement liées à sa structure moléculaire fine, qui est elle-même dépendante du procédé de fabrication et de purification de la gomme de caroube.
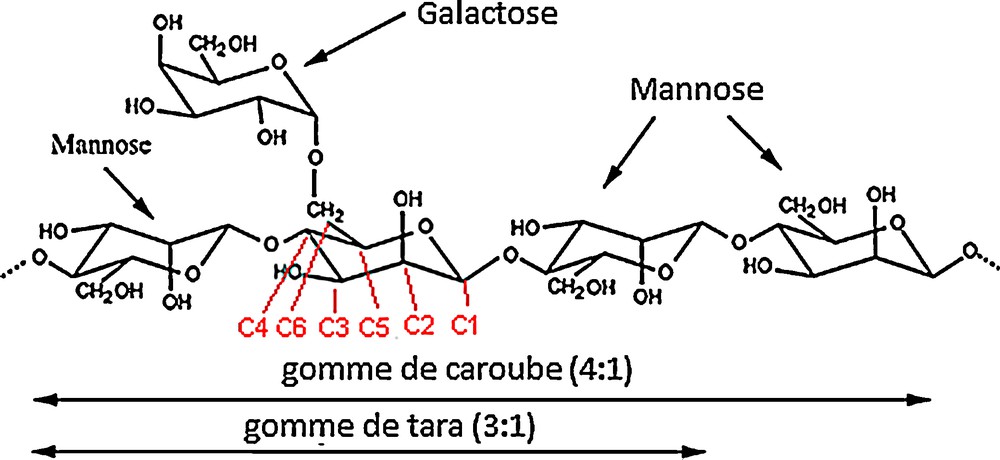
Structure des galactomannanes de la gomme de caroube et de la gomme tara. Adapté d’après [24].
Il existe donc, dans la littérature scientifique, plusieurs théories visant à mettre en relation la structure chimique et le comportement en solutions aqueuses, et vice versa [7–9,25–38]. Malheureusement, bien souvent, les seuls paramètres pris en compte dans ces modèles sont la masse moléculaire et le degré de substitution en galactose. Pourtant, le motif de distribution des résidus galactosyles latéraux semble impacter profondément les caractéristiques physiques d’une solution de galactomannanes.
Le présent article a donc pour objectif de réintégrer ce paramètre à la discussion, tout en dressant un état des lieux, le plus complet possible, des relations structures–fonctions des galactomannanes de caroube.
2 La caroube : origine, situation et description
Le caroubier, ou C. sliliqua L., de la famille des Caesalpiniaceae, est un arbuste dioïque à feuilles persistantes et de croissance lente [4,19]. C’est une essence thermophile cultivée en climat méditerranéen, mais originaire des pays arabes [39]. Elle est souvent utilisée pour lutter contre la déforestation et l’érosion des sols [40]. Le C. siliqua atteint une taille de 15 à 17 mètres et a une durée de vie de 200 à 500 ans. Ses fleurs, petites et rouges, donneront après fécondation naissance aux fruits appelés gousses. La gousse du caroubier, nommée caroube, est composée de pulpe et de graines. La composition de la pulpe de caroube dépend de la variété, du climat et des techniques de cultures. Toutefois, on peut avancer que la pulpe de caroube représente 90 % de la masse du fruit. Elle est riche en tanins et en sucres, dont le saccharose représente 65 à 75 % des sucres totaux [39]. Les graines constituent environ 10 % de la masse de la gousse. Elles sont composées de trois parties : la coque (téguments), l’endosperme et la radicelle.
L’endosperme se trouve sous les téguments. Il représente 40 à 50 % de la masse de la graine [41] et renferme des polysaccharides de réserve appelés « galactomannanes ». Au centre de l’endosperme se trouve la radicelle (ou embryon), représentant entre 15 et 30 % de la masse sèche de la graine [41]. Celle-ci possède une valeur énergétique élevée due à son taux important de protéines principalement solubles dans l’eau et de lipides majoritairement insaturés. Elle est employée pour la nutrition du bétail et comme aliment diététique [18]. La radicelle contient également un certain nombre d’enzymes : des endo-1,4-β-mannanases, des α-galactosidases et des β-mannosidases, susceptibles d’hydrolyser les galactomannanes lors, par exemple, d’un processus de germination [42].
3 Extraction et purification de la gomme de caroube
L’endosperme de la graine de caroube peut être extrait et broyé pour donner naissance à une poudre appelée « gomme de caroube ». Le procédé d’extraction utilisé doit veiller à ne pas dégrader les galactomannanes afin que leurs propriétés technofonctionnelles demeurent intactes, tout en garantissant une certaine pureté à la gomme obtenue. Les enzymes résiduelles doivent également être dénaturées afin de ne pas hydrolyser les galactomannanes lors de la mise en solution aqueuse de la gomme. La première étape du procédé général de fabrication de la gomme de caroube consiste à extraire mécaniquement les graines des cosses [43]. La seconde étape, nommée « décutilation », a pour but d’éliminer les téguments. Quatre procédés principaux existent : le traitement chimique à l’acide [4,5,43,44] ; le procédé thermique par rôtissage [5,44,45] ; le procédé mécanique utilisant des broyeurs à marteaux et des décortiqueuses [43] ; le traitement par trempage et congélation [46]. L’étape suivante consistera en l’élimination du germe. Celui-ci sera donc écrasé par broyage mécanique et enlevé principalement par tamisage. Il ne reste alors plus que les endospermes qui seront broyés en fines particules pour obtenir la gomme de caroube [47].
Des traitements de purification ont ensuite lieu dans le but d’éliminer les odeurs, les impuretés et les enzymes endogènes [9]. La clarification, le processus le plus répandu, consiste en une solubilisation de la gomme dans de l’eau [5], de la soude ou de l’acide acétique [1] suivie d’une étape de filtration [5] ou de centrifugation [1] pour éliminer les insolubles. Les galactomannanes seront ensuite précipités à l’aide de solvants tels que l’éthanol [48], l’isopropanol [49–51] ou le méthanol [52]. La précipitation des galactomannanes peut également être obtenue par la formation de complexes cuivriques [48,53] ou de baryum [54]. La précipitation dans du tampon borate est également citée dans la littérature scientifique [9,55]. L’état de purification a une grande influence sur la composition et les propriétés de la gomme de caroube. Lors de la clarification, la température de dissolution influence considérablement la composition de la gomme obtenue et les propriétés physicochimiques qui en découlent. Ainsi, une gomme de caroube purifiée à froid présente des propriétés significativement différentes de celle extraite à chaud [56]. La température n’est pas le seul facteur impactant sur les propriétés de la farine de caroube clarifiée. La proportion de solvant utilisé lors de la précipitation joue également un rôle important.
4 Les galactomanannes de caroube, leurs structures fines et leurs conformations en solutions aqueuses
Les galactomannanes sont constitués d’une chaîne principale de résidus d-mannopyranoses liés en β-(1→4) sur laquelle se greffent des résidus d-galactopyranoses uniques grâce à une liaison de type α-(1→6) (Fig. 1) [34,57]. Cette structure est basée principalement sur les résultats d’analyses de méthylation et des expériences d’hydrolyse partielle. Ainsi, la méthylation et l’hydrolyse subséquente génèrent des solutions mixtes de 2,3,4,6-tetra-O-methyl-d-galactose, de 2,3,6-tri-O-methyl-d-mannose et de 2,3-di-O-methyl-d-mannose, tandis que des hydrolyses partielles produisent des disaccharides de 6-O-α-d-galactopyranosyl-d-mannose et de 4-O-β-d-mannopyranosyl-d-mannose [1]. Cette structure de base est communément admise pour tous les galactomanannes. Trois éléments vont cependant permettre d’établir leurs structures fines et ainsi de distinguer les différents types de galactomannanes : le rapport mannose/galactose, la distribution des résidus galactose le long de la chaîne de mannose et la longueur des chaînes.
4.1 La composition en galactosyls latéraux
Le rapport mannose/galactose (M/G) est généralement déterminé par la méthode de Blakeney [58]. Il peut, en fait, varier de 10 à 1 en fonction de l’espèce végétale [59]. Les galactomannanes constituant une gomme de caroube brute possèdent, en moyenne, un M/G de 4. Ce rapport est toutefois à nuancer car il peut varier selon les conditions de culture (conditions climatiques, composition du sol, variété et âge de l’arbre, etc.) et le procédé de fabrication de la gomme [19,60]. La gomme de guar et la gomme tara – deux autres gommes commercialement importantes – possèdent un rapport moyen de 2 et 3 respectivement [61]. On utilise également dans la littérature le degré de substitution en galactose (DSGal) à la place du rapport M/G. Il correspond à la masse de galactose divisée par la masse de mannose [DSGal = G/M = 1/(rapport M/G)].
4.2 La structure fine de la gomme
L’intérêt commercial porté aux galactomannanes a débouché sur l’étude de la distribution des résidus d-galactopyranoses sur la chaîne principale de résidus d-mannopyranoses. Malgré l’utilisation relativement ancienne de ces gommes, l’élucidation de la structure fine des galactomannanes n’a débuté que bien plus tard (années 1960–1970), avec l’apparition et la généralisation de certaines techniques de mesure. Les premières études réalisées ont mis en œuvre des α-d-galactosidases [62,63] et des β-d-mannanases [62]. Ces enzymes testées sur des galactomannanes, de même rapport M/G mais d’origines botaniques différentes, ont généré des cinétiques d’hydrolyse et des produits de dégradation forts différents. Il apparaît ainsi que l’arrangement des substituants galactosyl sur la chaîne de mannose est très différent d’un galactomannane à l’autre et est vraisemblablement lié à l’espèce végétale d’origine [34,56]. La distribution des groupes d-galactosyl le long de la chaîne principale de mannose serait donc la conséquence de mécanismes de synthèse in vivo, qui impliquent probablement des transférases d-mannosyl et d-galactosyl fonctionnant de concert et variant significativement d’une espèce à l’autre selon les différences structurales autour des sites actifs des enzymes [64]. Sur base de ces observations, différents types de répartition ont été proposés : la disposition régulière, la disposition aléatoire et la disposition en blocs uniformes (Fig. 2). Des intermédiaires entre ces trois structures « types » peuvent également exister dans la nature [1].
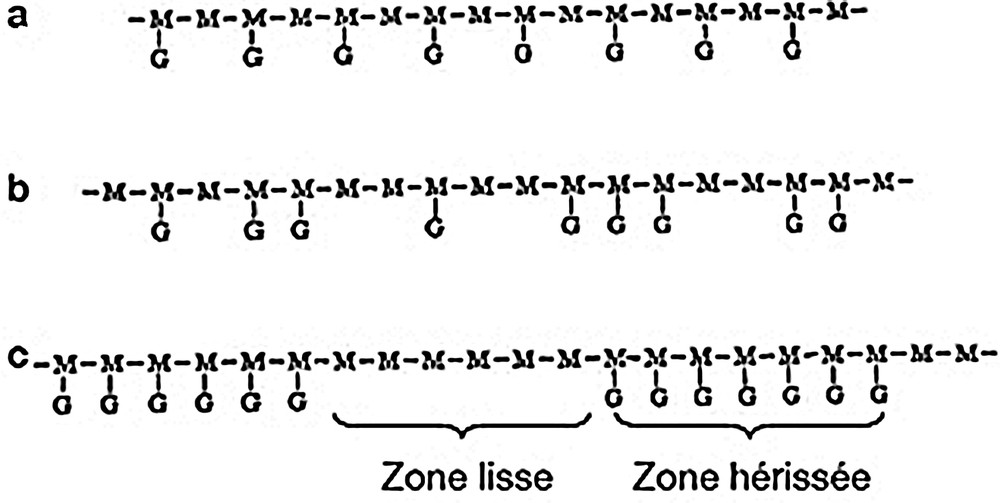
Distribution des résidus galactoses sur la chaîne de mannoses : (a) répartition régulière, (b) répartition aléatoire, (c) répartition en blocs uniformes. Adapté d’après [1].
L’élucidation spécifique de la structure de la gomme de caroube a été proposée pour la première fois en 1975 en utilisant une technique de dégradation alcaline (Fig. 3) [65]. Cette étude a pu démontrer que la gomme de caroube ne contient pas de chaînes latérales disposées de manière régulière. Ses résidus latéraux sont situés sur des blocs de résidus mannose, chaque bloc substitué étant composé de 25 résidus mannose. En considérant que la masse moléculaire de la gomme de caroube est d’environ 210 000 Daltons et que les blocs substitués sont séparés par des séquences d’unités de mannose non substituées de longueurs égales, on peut estimer le nombre d’unités mannose de chaque bloc non substitué à 85. La même méthode appliquée à la gomme de guar démontre, par contre, une structure primaire régulière [65]. La structure proposée pour la gomme de caroube a ensuite varié et été affinée avec l’évolution des techniques d’analyse. Plusieurs études complémentaires ont ainsi apporté certaines précisions quant à la structure chimique des galactomannanes issus de la gomme de caroube. La méthode d’oxydation au périodate a d’abord mis en évidence que la gomme de caroube était constituée de longs blocs de résidus mannose non substitués, de longs blocs dans lesquels chaque second résidu mannose est substitué par un galactose et de blocs plus courts dans lesquels il y a une haute densité en substituants galactose [66]. Par la suite, le recours à la résonance magnétique nucléaire du 13C [67–69] a pu informer sur la proportion entre les disaccharides de mannose di- mono- et non substitués en galactose et sur la proportion de triplets de mannoses tri- di- et monosubstitués en galactose. Il est ainsi possible d’estimer la répartition des galactoses sur la chaîne de mannose. Ces études accréditent la thèse d’une répartition des galactoses sous forme de blocs, du moins pour une certaine fraction de la gomme de caroube. Cette théorie semble confirmée par des études de résonance magnétique nucléaire en phase solide sur un gel de gomme de caroube. Le mannose y apparaît sous deux populations dynamiques différentes, traduisant des différences de mobilité au sein de la structure. La population la moins mobile pourrait correspondre aux blocs non substitués du galactomannane impliqués et bloqués dans des interactions intra- et intermoléculaires [70]. Plus récemment, des travaux de résonance magnétique nucléaire à deux dimensions prétendent que les substituants galactosyls sont eux-mêmes substitués par – au minimum – deux galactopyranoses liés en α (1-4) [71]. Ce modèle, en rupture avec la structure de base communément admise, n’est toutefois pas confirmé par les études de méthylation et d’hydrolyses partielles. Le dernier type d’études réalisées met en œuvre une hydrolyse enzymatique suivie d’une simulation par ordinateur [59,72,73]. Cette approche implique la caractérisation et la quantification des oligosaccharides produits par hydrolyse de galactomannanes par des β-mannanases hautement purifiées ainsi que la connaissance complète de la cinétique d’action de celles-ci. À partir de là, un modèle décrivant les besoins en sites de liaison des enzymes étudiées a été établi. En confrontant ce modèle aux oligosaccharides obtenus lors de l’hydrolyse, il est possible de déduire la structure originelle du polysaccharide étudié. Les résultats apportent des précisions supplémentaires à ceux obtenus jusque-là. La distribution des résidus galactose y est irrégulière, avec une haute proportion de doublets substitués, une proportion plus faible de triplets substitués et l’absence de blocs de substitution. La probabilité de séquences dans lesquelles des résidus de mannoses sont substitués alternativement est faible. La distribution probabiliste des tailles de blocs des résidus mannoses non substitués indique qu’il y a une plus grande proportion de blocs de taille intermédiaire que celle que l’on retrouverait dans des galactomanannes avec une distribution aléatoire des substituants. Il ne convient dès lors plus de parler de blocs entièrement substitués. La structure fine des galactomannes de caroube est donc constituée de zones « lisses », faiblement substituées et de zones « hérissées » beaucoup plus denses en galactoses latéraux, sans pour autant qu’ils soient systématiquement adjacents. Ces dernières années, aucune nouvelle étude sur la détermination structurale de la gomme de caroube n’a été publiée. La structure en zones « lisses » et zones « hérissées » est celle qui est donc communément admise (bien que difficile à démontrer avec certitude) et qui sert de base de discussion à la plupart des auteurs lors d’études de caractérisation des propriétés fonctionnelles de la gomme de caroube.

Dégradation alcaline par : (A) β-élimination et méthylation, suivie par (B) hydrolyse acide, réduction et acétylation [65]. Avec l’autorisation d’Elsevier.
4.3 La masse moléculaire des galactomannanes
La masse moléculaire est également un élément important à prendre en compte dans la caractérisation de la structure moléculaire d’un polysaccharide. Il est en effet susceptible de modifier les propriétés physiques du polymère en solution. Les galactomannanes, comme tous les polysaccharides naturels et polymères de synthèse, ont tendance à être polydispersés et, par conséquent, sont décrits par une masse moléculaire moyenne [31]. La valeur de la masse moléculaire moyenne est fortement dépendante des conditions de préparation des solutions de galactomannanes ainsi que de la technique de mesure utilisée. La température de solubilisation est le facteur clé qui va le plus influencer les valeurs des masses moléculaires. Plus elle est importante et plus les valeurs de masses moléculaires mesurées et les rendements de solubilisation seront également importants (Fig. 7). La durée de dissolution d’un échantillon est aussi un élément important. Au-delà de 2 heures de temps, elle n’aura toutefois pas d’influence sur la masse moléculaire mesurée [33]. La taille de la mouture de la poudre de caroube ne semble pas influencer la masse moléculaire des galactomannanes [33]. Deux types de techniques de mesures de masses moléculaires sont principalement utilisés.

Influence de la température sur la solubilité, après 2 heures de mise en solution, pour de la gomme brute et des particules de tailles différentes [33]. Avec l’autorisation d’Elsevier.
Les méthodes chimiques permettent d’estimer la masse moléculaire moyenne en nombre . Cette grandeur ne dépend que du nombre de molécules de polymère, indépendamment de leur forme et leur masse ; elle est définie par :
Des résultats différents sont observés lors du recours aux méthodes basées sur la transmission d’une onde lumineuse. Ces dernières déterminent la masse moléculaire moyenne en masse Mw. Cette grandeur ne dépend pas que du nombre de molécules, mais aussi de la masse des molécules de polymère :
Les phénomènes d’agrégation ont été mis en évidence, même à des très faibles concentrations (C = 0,1 %), dans certains solvants d’analyse, tels que l’eau. Une même gomme de caroube mesurée, par chromatographie d’exclusion stérique, dans de l’eau et du tampon phosphate donnera ainsi des valeurs de Mw forts différentes, respectivement de 820 000 Daltons et 370 000 Daltons [36]. La valeur obtenue de la masse moléculaire est donc fortement affectée par la conformation de la molécule en solution aqueuse et la concentration, responsables des propriétés d’auto-interaction. Ce phénomène est particulièrement marqué pour les polymères possédant de nombreuses séquences non substituées [76].
4.4 La conformation des galactomannanes en milieu aqueux
La conformation la plus probable des galactomannanes en solution aqueuse semble être en ruban (ribbon-like) [57]. Dans cette structure en trois dimensions, le C6 du résidu mannosyl est localisé à l’extérieur de la chaîne principale (Fig. 4 et 5). La liberté de rotation au niveau des liens glycosidiques est très réduite et, par conséquent, les chaînes latérales de d-galactosyl de mannoses adjacents ou séparés par un nombre pair d’unités, vont se situer sur des côtés opposés de la chaîne principale. Ceux qui sont séparés par un nombre impair d’unités mannose resteraient du même côté de la chaîne principale [57]. Dans d’autres travaux, la conformation des galactomannanes en solution aqueuse est moins ordonnée et de type pelote (random coil) (Fig. 5) [24,26]. Enfin, d’après une autre étude, les galactomannanes peuvent tantôt adopter une conformation plus ordonnée en ruban, tantôt adopter une conformation en pelote aléatoire (Fig. 5) [77]. La conformation plus ordonnée serait privilégiée lors de la formation de gels, tandis que la pelote est favorisée en solutions aqueuses. La structure de la pelote est, en outre, susceptible de varier selon le mode de distribution des d-galactosyl latéraux. Ainsi, une étude de prédiction informatique informe que plus le nombre de zones lisses est important, plus la pelote sera étendue en solution aqueuse [78]. Ces résultats sont assez contradictoires avec d’autres modèles existants, où l’existence de zones lisses est présentée comme favorisant les interactions intra-chaînes qui réduisent le volume hydrodynamique (Fig. 6) [49,79].

Conformation possible des galactomannanes en solution aqueuse et arrangement des chaînes latérales de galactosyles [57]. Avec l’autorisation d’Elsevier.
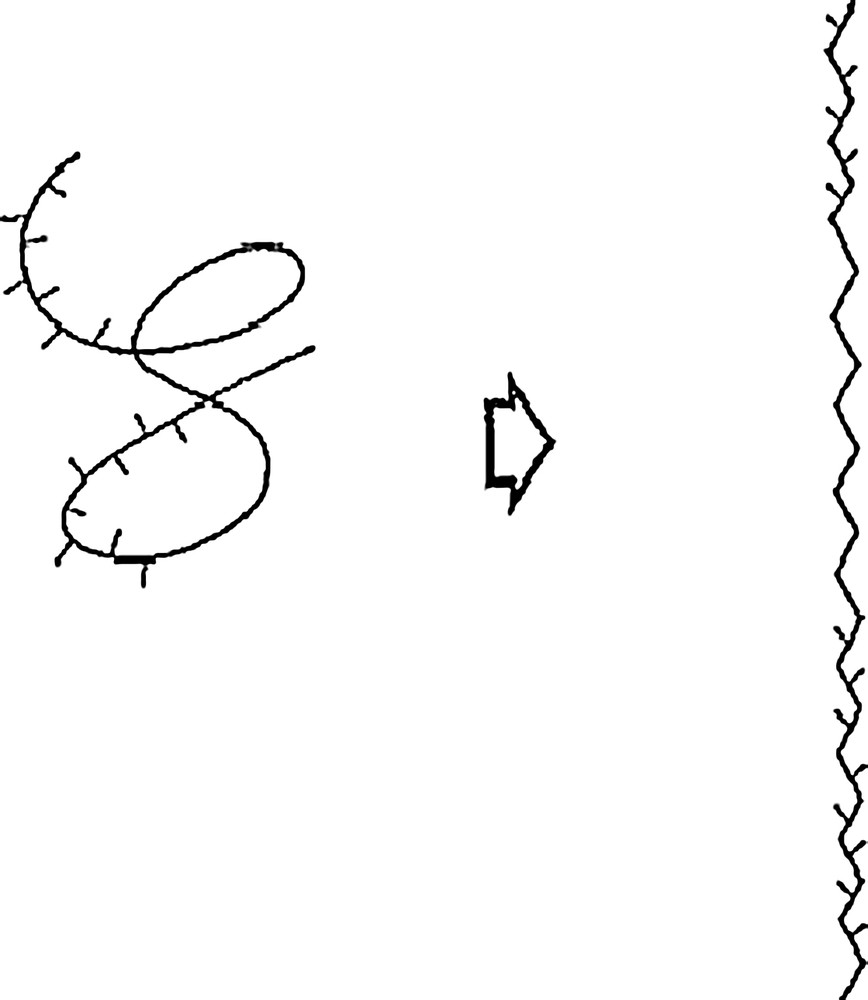
Les deux conformations des galactomannanes – en hérissé, les zones substituées – à gauche, la pelote aléatoire ; à droite, la forme en ruban [77]. Avec l’autorisation d’Elsevier.
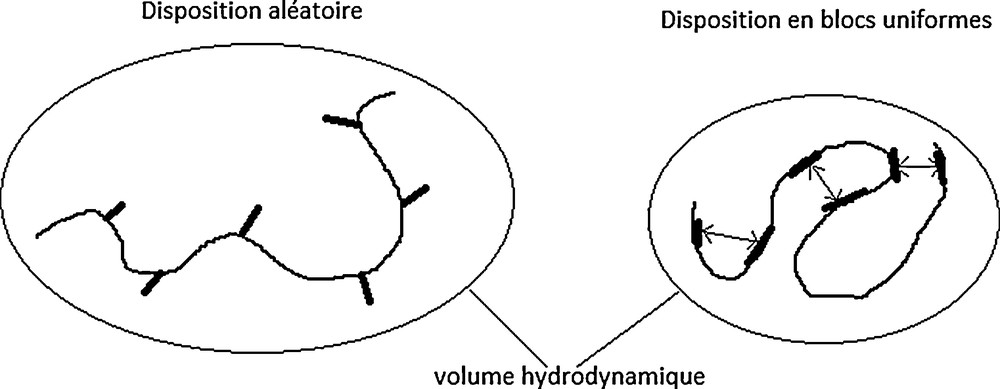
Intervention des zones « lisses » dans les interactions intramoléculaires. Adapté d’après [49].
5 Les propriétés des galactomannanes en solutions aqueuses
Les propriétés physiques des galactomannanes de caroube en solutions aqueuses qui sont développées dans ce chapitre sont celles qu’il est possible de mettre en relation avec la structure. Il s’agit de la solubilité, de la viscosité et viscoélasticité, de la formation d’hydrogels et de gels associés à d’autres polyholosides. L’impact de la gomme de caroube dans des formulations plus complexes (de type alimentaire) n’apporte pas d’éléments nouveaux de réflexion à la question de la relation structure–fonction. Il ne sera donc pas abordé dans ce travail.
5.1 Solubilité et influence de la température sur les caractéristiques de la gomme de caroube
5.1.1 La solubilisation des galactomannanes
L’étude de la solubilité des polysaccharides en milieu aqueux est très importante car elle est intimement liée aux autres propriétés que ces polymères développent en solution. D’un point de vue pratique, la solubilité en solution aqueuse des galactomannanes de caroube s’exprime par le biais d’un pourcentage mesurant la proportion du produit restant en solution suite à une centrifugation. Cette solubilité dépend des dissolutions préalables, de l’équilibre entre les interactions molécules–molécules et molécules–solvant, de la nature physicochimique des molécules ainsi que de l’état physique de la matière. La solubilité dans l’eau de la gomme de caroube est relativement faible. Plusieurs techniques ont donc été avancées pour augmenter cette solubilité. La carboxylation, la formation d’éther, l’hydroxyalkylation, l’estérification et la formation de dérivés phosphates en font partie [38]. Certains auteurs se sont intéressés à l’étude de la solubilité de la gomme de caroube.
Des analyses de diffraction aux rayons X aux grands angles ont ainsi montré que la poudre de caroube brute ou clarifiée avait, classiquement, un profile amorphe [68]. Par opposition, d’autres galactomannanes commerciaux, tels que le fenugrec, présentent un profil cristallin. En réalité, plus le DSGal augmente, plus la tendance à la cristallinité augmente. Dans le cas de la gomme de caroube, la solubilité n’est donc pas liée à une quelconque fusion de zones cristallines. La dissolution correspond principalement à une extraction polymère–solvant [32]. Il s’agit d’un partitionnement thermodynamique des chaînes de polymères entre la phase solide amorphe du polymère, gonflée par le solvant, et la phase de solvant pure environnante. Ce partitionnement dépend de la concentration et de la température de la solution mais également des caractéristiques structurales des polymères. Le degré de substitution en galactose (DSGal) et la masse moléculaire (Mw) sont les seules caractéristiques structurelles dont l’influence sur le comportement de solubilité des galactomannanes a été régulièrement étudiée. Contrairement à la distribution des substituants galactosyls, ces deux paramètres présentent l’avantage de pouvoir être déterminés assez facilement.
Sans une proportion de groupements latéraux (DSGal = 0), un polysaccharide isolé est instable en solution et va cristalliser ou précipiter [33]. Les galactomannanes avec un DSGal généralement au-dessus de 0,4 tels que la gomme de guar (0,5) ou de fenugrec (0,9) sont considérés comme solubles dans des solutions aqueuses diluées ou semi-diluées et thermodynamiquement stables. Ils présentent une structure aléatoirement enroulée dans des solutions diluées. Comme les groupes ionisables sont absents, la solubilité dans l’eau et la qualité du solvant semblent être directement liées à la densité de liaisons hydrogène (eau-galactomannanes) fournie par les groupements de galactoses latéraux [31]. Les résidus galactopyranoses représentent les parties hydrophiles du galactomannane. Plus le degré de substitution en galactose est élevé, plus la solubilité dans l’eau des galactomannanes est importante [8,34]. Les galactomannanes de DSGal faibles à intermédiaires (< 0,4) sont constitués de chaînes qui affichent une solubilité partielle qui dépend de la valeur exacte de DSGal [31]. Les galactomannanes de caroube font partie de cette catégorie et sont généralement considérés comme partiellement solubles [10], voire faiblement à basse température [27]. Les zones non substituées en galactose sont le siège d’interactions intra- et intermoléculaires générées par la présence de ponts hydrogènes entre ces zones conduisant, dans les cas extrêmes, à l’agrégation et à la précipitation [27,80]. La présence de groupements galactosyls ne jouerait donc pas uniquement le rôle de « partie hydrophile », elle pourrait simplement favoriser la solubilisation en empêchant la formation de liaisons coopératives entre les segments de mannose. Une certaine proportion de la gomme de caroube va donc se dissoudre directement dans de l’eau froide, mais une dissolution totale accompagnée d’une viscosité importante nécessite un long trempage à haute température [31] ou une forte agitation à haute température [8].
5.1.2 L’influence de la température sur la solubilisation
Approximativement la moitié d’une gomme de caroube brute est soluble après deux heures d’agitation à 5 °C [33]. Le rendement d’extraction augmentera ensuite de façon linéaire avec la température pour atteindre 85–90 % à 85 °C (Fig. 7). La gomme de caroube purifiée présente une solubilité supérieure dans les mêmes conditions [9]. En effet, la purification permet l’élimination des composés insolubles et la gomme purifiée devient totalement soluble avec la température (98 % contre 89 % pour la gomme non clarifiée).
Plusieurs études montrent que le DSGal est inversement lié aux températures de solubilisation (Fig. 8) : les échantillons solubilisés à des températures de plus en plus hautes contiennent de moins en moins de galactose [28,29,35–37,72]. À l’inverse, ceux dissous à froid contiennent le plus de galactose [33]. Un échantillon soluble à haute température possède également un nombre plus important de régions non substituées que la gomme brute dont il est issu [81].

Degré de substitution en galactose de fractions de galactomannanes obtenues sur une large source de gomme de caroube, parfois par fractionnement [31]. Avec l’autorisation d’Elsevier.
Il existe également une tendance à obtenir de plus hautes masses moléculaires (Mw) par chromatographie d’exclusion stérique couplée à une triple détection, lorsque la température de dissolution augmente (Fig. 9) [33]. Les composants polysaccharidiques qui se dissolvent avec une augmentation progressive de la température possèdent des chaînes de plus en plus longues, de l’ordre de 1 000 000 Daltons, qui contiennent également moins de galactose [32]. Ils fournissent donc une contribution positive à la masse moléculaire globale. Par conséquent, une gamme assez large de valeurs peut être observée, dans le même matériel, selon la température de dissolution [33]. De même, par méthode chimique, certains auteurs ont estimé, dans les mêmes conditions, la masse moléculaire d’une gomme de caroube solubilisée à froid à 150 000 Daltons et celle solubilisée à chaud à 650 000 Daltons [82].
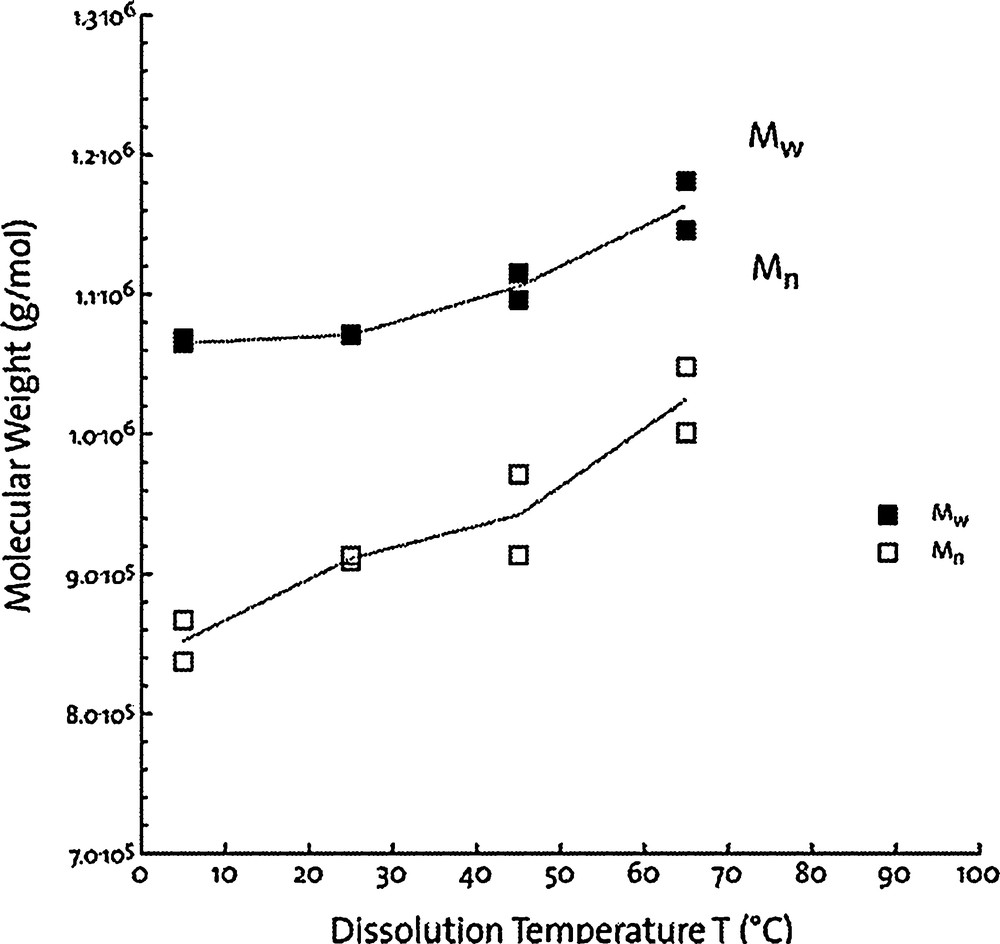
Poids moléculaire (déterminé par SEC) de fractions de gomme de caroube commerciale, solubilisée entre 5 °C et 65 °C [32]. Avec l’autorisation d’Elsevier.
5.1.3 Le fractionnement de la gomme de caroube
Les propriétés de solubilité de la gomme de caroube peuvent donc être utilisées pour la séparer en fractions de structure fine différentes. Plusieurs auteurs proposent de fractionner la gomme de caroube en jouant sur la température de solubilisation en milieu aqueux, sans longue préparation des échantillons [9,35,38,44]. Le pouvoir de solvatation de l’eau augmente avec la température, fournissant un moyen de fractionner le polysaccharide original. Ainsi, comme dans les solutions de polymères synthétiques, l’extraction isotherme génère une fraction polysaccharidique avec des distributions étroites de masses moléculaires et de composition chimique [32]. Les fractions de gomme de caroube générées présenteront, selon la température de travail, une gamme de rapports M/G assez large (2,5 à 5) autour du rapport moyen de la gomme de caroube brute (4) [35]. Il en va vraisemblablement de même pour la masse moléculaire et la distribution des substituants. En réalité, la tendance sera la même que celle observée lors de la variation de la température de dissolution, mais avec un caractère plus marqué. Les portions issues d’un fractionnement possèdent une distribution en galactomannanes plus étroite que celle de la gomme brute. Deux techniques de fractionnement sont principalement recensées dans la littérature. La première (intégrale) se réalise de la même manière que l’étape de clarification d’un procédé industriel de purification de la gomme de caroube. La farine brute d’endosperme de caroube est dissoute à des températures données et rigoureusement contrôlées puis les fractions solubles et insolubles à ces températures sont séparées par centrifugation. Enfin, le surnageant soluble sera ensuite précipité par un volume d’éthanol afin de récupérer la fraction spécifique [9,35,38]. Une étude met toutefois en évidence le fait que le volume d’éthanol utilisé influencera la composition de la fraction récoltée [44]. En effet, pour une température de solubilisation donnée, un surnageant d’après centrifugation donnera une gomme contenant une proportion croissante de galactose au fur et à mesure que la quantité d’éthanol utilisée lors de la précipitation augmente. L’adjonction de grands volumes d’éthanol permet donc de précipiter une fraction plus soluble de la gomme (car plus riche en galactoses latéraux). La seconde, une technique par épuisement (soustractive), consiste à ré-extraire successivement le même échantillon dans des volumes d’eau de températures croissantes, pour générer les fractions suivantes, jusqu’à ce que plus aucune matière supplémentaire ne puisse être extraite [9,31,37,83]. Elle donne également des fractions de caractéristiques plus marquées que dans le premier cas de fractionnement, car la matière récupérée correspond à celle possédant les caractéristiques structurales la rendant soluble dans une gamme étroite de température. En utilisant la première technique de fractionnement, de grandes quantités de galactomannanes solubles à de plus basses températures viennent enrichir la fraction concernée. Une autre technique de fractionnement consiste en une solubilisation de la gomme de caroube à une température de 95 °C suivie d’une précipitation par ajout d’un gradient de sulfate d’ammonium de 20, 30, 45, 80 et 100 % de saturation [69]. Les polymères contenant de hautes masses moléculaires, un faible degré de substitution et une répartition de type blocs précipitent à de faibles degrés de saturation. Cette fraction générera les solutions les plus visqueuses. La structure fine et la viscosité semblent donc étroitement liées.
5.2 Les propriétés visqueuses, viscoélastiques et l’auto-association des galactomannanes de caroube
5.2.1 La viscosité intrinsèque
En solution diluée, la viscosité spécifique ηsp d’une solution de polymère est une grandeur sans dimension définie de la manière suivante : .
ηs est la viscosité du solvant en l’absence de polymère dissous et η0 la viscosité de la solution contenant une concentration C de polymère dissous. De la viscosité spécifique, il est possible de déterminer la viscosité intrinsèque [η] : , qui possède la dimension de l’inverse d’une concentration. Expérimentalement, la viscosité intrinsèque [η] peut être estimée en réalisant des mesures de viscosité à basses concentrations en polymères. Ensuite, une extrapolation est réalisée, en faisant tendre C vers 0, à l’aide des équations de Huggins : et Kraemer : , où ηr est la viscosité relative (ηr = η0/ηs) et k ' et k ' ' des constantes [26].
Dans des conditions de régime « dilué » (Fig. 10a), la viscosité intrinsèque [η] va être influencée par la masse moléculaire, le DSGal et le motif de distribution des galactoses. La relation de Mark–Houwink exprime ainsi une relation de proportionnalité entre viscosité intrinsèque [η] et masse molaire moyenne viscosimétrique : . K (cm3·mol1/2/g3/2) est une mesure de la rigidité d’une chaîne de polymère [36], et v, l’exposant de Mark–Houvink, est égal à 0,8 dans le cas de la gomme de caroube [84]. Un accroissement de masse moléculaire générera donc une viscosité intrinsèque plus importante. Ce modèle un peu simpliste a été revu par certains auteurs pour tenir compte du DSGal. Plus ce dernier sera important et plus la viscosité intrinsèque [η] sera faible [1,7,28,29]. Il a également été adapté pour intégrer le volume hydrodynamique occupé par unité de masse. Dans ce modèle, la viscosité intrinsèque [η] est proportionnelle au cube du rayon de giration [26], lui-même lié à la distribution des galactoses latéraux.
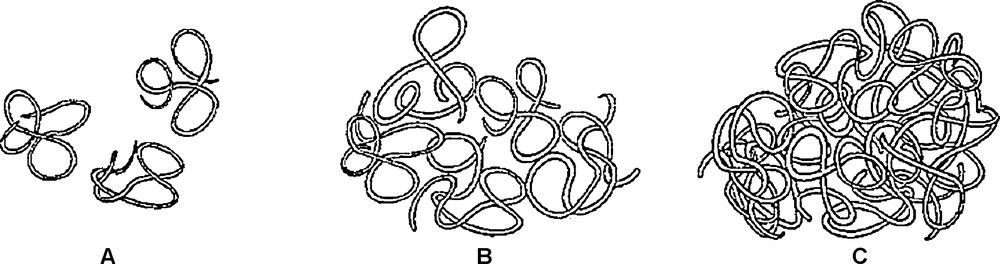
Illustration de différents régimes de concentration : A, solution diluée avec c < c* (molécules individuelles) ; B, molécules montrant un début de recouvrement c ≈ c* ; C, molécules enchevêtrées c > c* [84]. Avec l’autorisation d’Elsevier.
Lorsque la concentration du milieu augmente, la concentration critique C* (occupation totale du volume par les macromolécules, Fig. 10b) est franchie et le régime est dit : « semi-dilué » (Fig. 10c). Dans une telle situation, un nouveau phénomène susceptible d’influencer la viscosité intrinsèque [η] apparaît : l’interpénétration des chaînes macromoléculaires. Il conduit à la création de recouvrements « physiques » plus ou moins denses [79]. Le développement des enchevêtrements ne peut toutefois se faire que si la concentration est suffisante pour obliger les pelotes individuelles à s’interpénétrer [85,86]. Dans de telles conditions, certains auteurs indiquent que la viscosité intrinsèque initiale théorique d’une solution de polymère ne dépend que de la concentration et du cube de la masse moléculaire [87]. Pourtant, les galactomannanes de caroube présentent une viscosité intrinsèque supérieure à celles des prédictions théoriques, des autres polymères et même des autres polysaccharides de conformation pelote aléatoire. Ce phénomène s’explique par la présence d’interactions plus spécifiques entre les macromolécules, en plus des recouvrements physiques non spécifiques. Ces interactions sont appelées hyper-enchevêtrements [24]. Ces phénomènes contribuent fortement au développement de la viscosité des galactomannanes et expliquent, par rapport à d’autres polymères, la plus forte dépendance de la viscosité à la concentration.
Les hyper-enchevêtrements observés résultent de phénomènes d’agrégation intermoléculaire entre les chaînes de galactomannanes. En effet, une baisse réversible de la viscosité intrinsèque de la gomme de caroube est observée lorsque le caractère alcalin du milieu augmente. Les hydroxyles des polysaccharides neutres sont ionisés à un pH élevé. Cela génère l’apparition de répulsions électrostatiques entre les constituants des chaînes [88]. L’agrégation moléculaire est également influencée par les caractéristiques structurales de la gomme [51]. Une étude a ainsi montré qu’en augmentant le rapport M/G, par sélection de variétés de caroubiers différents, la viscosité intrinsèque augmentait également progressivement [25]. Des fractions pauvres en galactose généreront donc une viscosité intrinsèque élevée. Cette constatation a également pu être établie sur des galactomannanes d’origines botaniques différentes. En réalité, cette analyse est un peu incomplète car il est fortement probable que la quantité de régions « lisses » ait plus d’importance dans la viscosité en solutions semi-diluées que le simple rapport M/G [1]. D’autres auteurs ont ainsi déterminé en régime semi-dilué la viscosité spécifique d’échantillons de caroube fractionnées à haute et basse températures [35]. La fraction extraite à chaud présente une viscosité spécifique plus importante que celle extraite à froid car elle possède une quantité de zones « lisses » plus importantes. D’autres études mettent également en évidence un accroissement de la viscosité intrinsèque de solutions de gomme de caroube lorsque le contenu en zones lisses augmente [69,80]. Le phénomène d’agrégation moléculaire est ainsi d’autant plus marqué que le polysaccharide possède des séquences non substituées (lisses) capables d’intervenir dans des associations intermoléculaires (Fig. 11) [81]. Ces zones, faiblement substituées en galactose, deviennent évidement de plus en plus abondantes au fur et à mesure que le rapport M/G augmente [36,89]. Les phénomènes d’agrégation et l’hyper-enchevêtrement qui en résulte seraient, par conséquent, dus à des interactions chaîne–chaîne subvenant entre les segments de mannanes non substitués en galactose [26]. Les zones non substituées des chaînes de galactomannanes seraient donc à la base des interactions intramoléculaires (la molécule se replie alors sur elle-même) et des associations intermoléculaires (la molécule s’associe à une autre molécule). Le phénomène qui contrôle l’agrégation de ces polymères neutres hydrosolubles trouverait son origine dans l’établissement de ponts hydrogène coopératifs entre les zones des chaînes dépourvues de résidus galactose [27].
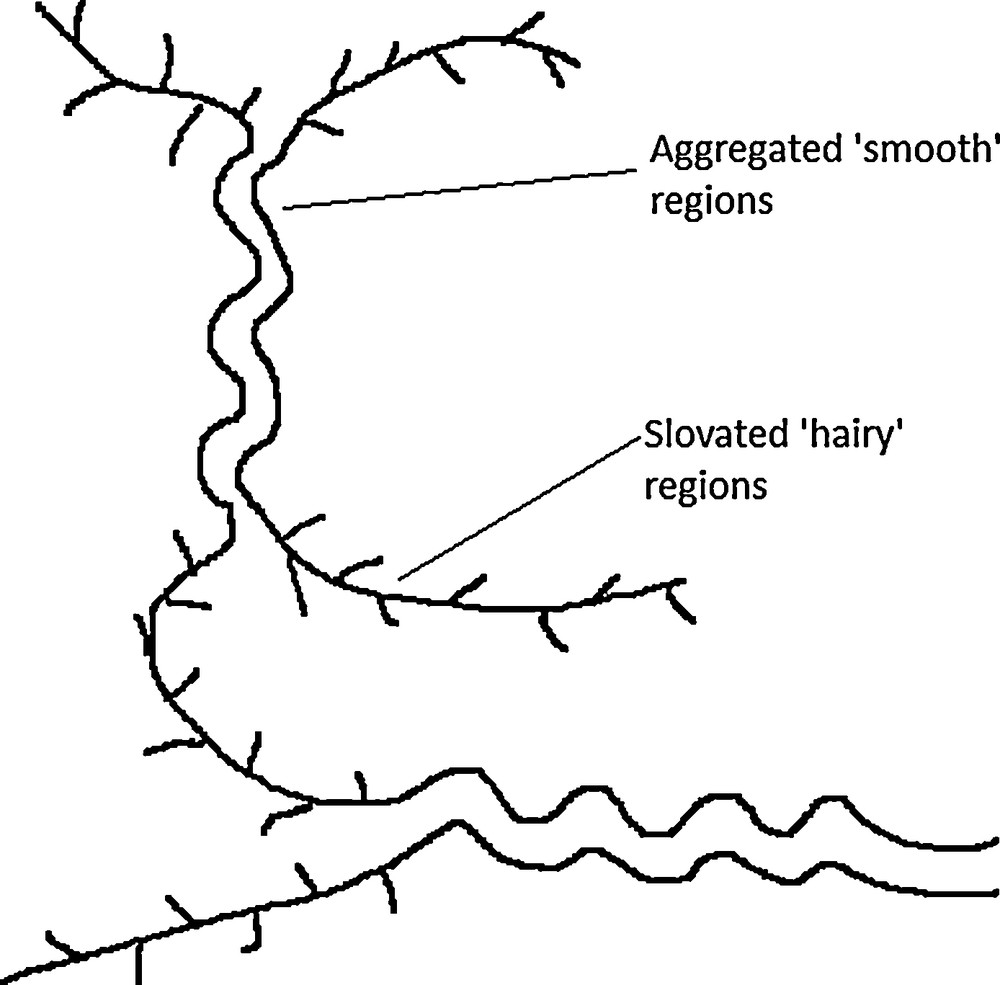
Structure proposée pour les gels de galactomannanes. Les régions non substituées de la chaîne de mannane sont associées, comme dans un état solide, pour former des jonctions interchaînes, tandis que les régions substituées sont fortement hydratées, comme en solution, et empêchent la précipitation complète. Adapté d’après [88].
5.2.2 La viscosité dynamique
Pour caractériser le comportement visqueux de polysaccharides, la viscosité dynamique η (Pa·s) des solutions semis-diluées est également étudiée. Ces mesures se réalisent au rhéomètre. Les galactomannanes en solution présentent un comportement de liquides rhéofluidifiants, c’est-à-dire que la viscosité apparente (Pa·s) diminue lorsque la vitesse de cisaillement (s−1) augmente. En effet, sous la contrainte de cisaillement imposée, les macromolécules de galactomannanes se déchevêtrent plus ou moins rapidement suivant la densité des enchevêtrements et la force des interactions pour s’orienter dans la direction du flux. Cette réorganisation des macromolécules se traduit par une baisse de la viscosité [24,80]. L’effet rhéofluidifiant est réversible [60]. Certains auteurs n’observent toutefois pas de diminution franche de la viscosité apparente dans la région de faibles vitesses de cisaillement. Ce pallier est suivi d’une chute rapide de viscosité apparente dans les régions de plus hautes vitesses de cisaillement (Fig. 12A). Ils en concluent donc que les galactomannanes de caroube possèdent un comportement typiquement pseudoplastique [38,76]. Plusieurs paramètres influencent la viscosité dynamique η de solutions semi-diluées. L’augmentation de la concentration en galactomannanes de caroube d’une solution augmentera la viscosité initiale du liquide. Elle accentuera également le caractère rhéofluidifiant de la solution [24]. Outre l’effet de la concentration, la viscosité apparente d’une solution semi-diluée reste liée à la structure moléculaire, qui influence les possibilités d’interactions [26,27,86]. La présence de zones lisses influence donc le comportement visqueux. Plus celles-ci sont présentes, plus la viscosité apparente initiale de la solution sera importante et plus le caractère rhéofluidifiant sera marqué [69]. Cela démontre une fois de plus le rôle crucial joué par les zones peu substituées dans les interactions moléculaires.

A. Courbe d’écoulement de concentrations croissantes en galactomannanes. B. Spectre mécanique de solutions de galactomannanes de caroube, pris à 25 °C [76]. Avec l’autorisation d’Elsevier.
Les solutions visqueuses de galactomannanes ne sont affectées que légèrement par le pH, les sels et les variations thermiques [9,59]. La viscosité apparente d’une solution reste donc relativement constante sur une gamme de pH de 1 à 10,5 [1]. Néanmoins, un maximum de viscosité est observé à pH 5 [25]. Toutefois, une dégradation des polymères peut apparaître dans des conditions hautement acides ou alcalines, surtout si elles sont chauffées [1]. La viscosité apparente évolue tout de même sur une large gamme de température. Elle est gouvernée, lors d’une montée progressive de la température, par trois facteurs :
- • une diminution réversible de la viscosité apparente de la gomme de caroube avec l’augmentation de la température, due à une augmentation des mouvements macromoléculaires (20–40 °C) ;
- • une augmentation de la viscosité apparente due à une augmentation de la solubilisation (40–70 °C) ;
- • une perte de viscosité apparente due à la diminution de masse moléculaire, résultant d’une dégradation thermique (70–120 °C).
Lors du refroidissement, la gomme de caroube retrouve une viscosité apparente plus élevée que sa viscosité initiale [8]. La température de préparation de la solution revêt donc une importance capitale lors des mesures de viscosité dynamique. Certains auteurs montrent ainsi que la viscosité apparente d’une même gomme peut passer, dans certains cas, du simple au double, selon qu’elle est solubilisée à 25 ou 80 °C [90]. Ces données sont évidement à interpréter sous l’angle des propriétés de solubilité. Une augmentation de la température permet de solubiliser davantage de polymères riches en « zones lisses », qui peuvent s’agréger et s’enchevêtrer, formant ainsi un réseau plus dense et donc plus visqueux.
5.2.3 Les propriétés viscoélastiques
Les solutions marcomoléculaires semi-diluées peuvent également être analysées au rhéomètre sur base de leurs propriétés viscoélastiques. Ces dernières sont plus ou moins marquées du fait des interactions se produisant entre chaînes. C’est une des voies principales utilisées pour mettre en évidence les enchevêtrements. Le spectre mécanique d’une solution de gomme de caroube est présenté sur la Fig. 12B. On peut y observer que la gomme de caroube en solution semi-diluée est un fluide typiquement viscoélastique. Le module visqueux G” (Pa) est plus important que le module élastique G’ (Pa) à de basses fréquences d’oscillations, tandis que l’inverse est observé à de plus hautes fréquences. Le comportement viscoélastique d’une solution est également influencé par la structure des polymères. À concentration égale, un déplacement du point d’intersection vers les plus basses fréquences est observé lorsque la masse moléculaire moyenne augmente. Cela signifie que les chaînes plus longues ont tendance à générer plus vite un comportement élastique, car les enchevêtrements se dissocient moins facilement [76]. D’autre part, une étude réalisée sur des gommes de caroubes non purifiées indique que le module élastique semble être d’autant plus important que le rapport M/G de la gomme de caroube l’est également [29]. La tendance est la même pour une gomme fractionnée à différentes températures : des températures élevées de solubilisation généreront des rapports M/G plus importants et un module élastique G’ accru [28]. Par ailleurs, une étude montre qu’une fraction de gomme de caroube contenant peu de galactose et beaucoup de « zones lisses » génère un spectre mécanique au caractère élastique bien plus marqué, même à de basses fréquences [69]. Ce comportement serait également dû aux interactions intermoléculaires entre les régions non substituées de la chaîne de mannane qui génèrent la formation d’un réseau plus dense. La concentration influence toutefois fortement les propriétés viscoélastiques d’une solution de galactomannanes. Il est important d’en tenir compte lors de la comparaison de résultats. Ainsi, l’augmentation de la concentration en gomme de la solution va générer la transition d’un comportement plus visqueux vers un comportement plus élastique [24,85] et donc, un déplacement du point d’intersection vers des fréquences plus basses. Cette observation est logique car l’augmentation de la concentration favorise l’apparition d’un réseau interpénétré et enchevêtré, difficile à dissocier, et donc au comportement élastique.
5.2.4 La formation d’hydrogels
L’auto-association des galactomannanes peut également être étudiée par gel–dégel. En effet, une solution visqueuse de gomme de caroube peut former un hydrogel contracté lorsqu’elle est soumise à plusieurs cycles de congélation-décongélation [70,89,91]. L’analyse calorifique différentielle (DSC) et l’analyse thermomécanique (TMA) indiquent que les hydrogels de caroube sont thermiquement irréversibles et aucune transition gel–sol n’est observée [92]. Il est supposé que la formation des hydrogels résulte d’une agrégation des chaînes, entre les régions de mannoses linéaires non substitués. Il semble, en effet, y avoir une bonne corrélation entre la longueur moyenne des zones non substituées (estimées par simulation informatique) et le pourcentage de précipitation sous forme d’hydrogel, obtenu par un traitement gel–dégel [64,81]. Ces résultats sont toutefois à prendre avec prudence car la détermination du précipité repose sur une centrifugation à température ambiante. Elle n’est donc pas le seul fait de l’auto-association mais aussi celui des propriétés de solubilité. Ces résultats sont cependant corroborés par une étude de RMN en phase solide qui démontre, dans de tels gels, la plus grande mobilité des segments composés de galactose et de mannose par rapport à ceux contenant du mannose seul [70]. Ces derniers seraient donc impliqués dans les jonctions.
Ainsi, les résultats caractérisant le comportement de la gomme de caroube en solutions diluées et semi-diluées, indiquent clairement que la taille des molécules et le degré de substitution ne sont pas les seuls paramètres influençant la viscosité, la distribution des résidus d-galactosyl sur la chaîne principale y joue également un rôle prépondérant.
5.3 Formation de gels par association à d’autres polyholosides
L’association de galactomannanes avec d’autres molécules est utilisée depuis longtemps en industrie. Majoritairement, il s’agit d’autres polysaccharides tels que l’amidon [2,17], l’agar, les carraghénanes ou le xanthane [1]. Les protéines sont toutefois occasionnellement utilisées [93]. L’intérêt de cette association est lié au fait que la gomme de caroube est moins coûteuse que les autres saccharides avec lesquels elle interagit à des degrés divers avec celle-ci. Par cette technique, il est possible de former des gels de force spécifique pouvant également avoir des propriétés originales pour certaines applications. L’étude de la formation de ces gels apporte de précieuses informations sur la relation structure moléculaire–fonctionnalité des galactomannanes et consiste par conséquent en un moyen de détermination de la structure « fine » de la gomme de caroube. En réalité, ici aussi, la concentration et la répartition statistique des groupements galactose latéraux vont contrôler le comportement de solubilité et les propriétés d’interactions avec les autres saccharides [32].
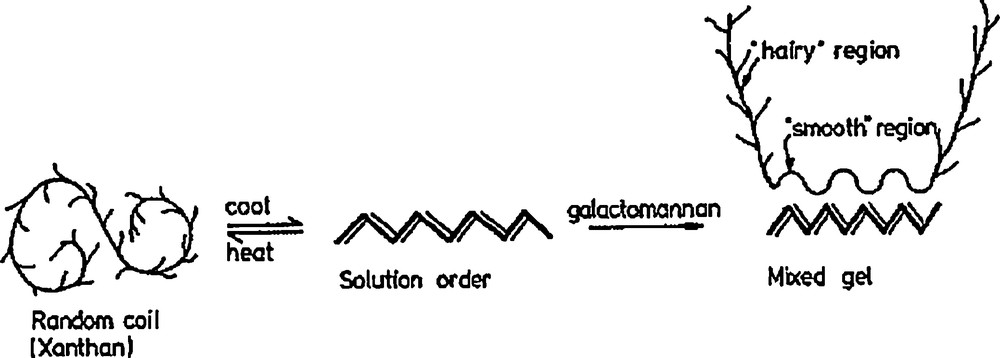
Le modèle le plus étudié d’interaction entre galactomannanes et un autre polysaccharide est le gel qu’il forme avec le xanthane. Le comportement rhéologique d’une solution de xanthane a été attribué à la structure moléculaire ordonnée adoptée sous la majorité des conditions environnementales. À haute température et faible force ionique, les molécules de xanthane sont vraisemblablement présentes dans une conformation désordonnée, mais en refroidissant, une transition désordre-ordre prend place et une conformation hélicoïdale rigide est adoptée (Fig. 13). Dans de l’eau distillée, cette température de transition conformationnelle (Tm ; °C) a été déterminée à 50 °C, mais peut varier avec la force ionique du milieu [37]. Le xanthane seul est incapable de former un gel. Par contre, les xanthanes et les galactomannanes de caroube sont capables d’interagir en solution pour donner une augmentation synergétique significative de la viscosité ou de la force d’un gel [89]. Dans une telle structure, la microscopie de polarisation semble indiquer que les molécules de xanthanes ne sont concentrées dans des zones spécifiques, mais uniformément distribuées dans le milieu [94]. L’accroissement du degré d’interaction avec le xanthane évolue linéairement et de façon inversement proportionnelle au contenu en d-galactose [28,95], des galactomannanes de caroube fortement substitués se révélant même incapables d’interagir avec le xanthane [69]. De même, une gomme de guar de rapport M/G 2:1 ne formera jamais de gel avec du xanthane, quelle que soit sa concentration [96]. Cela semble indiquer que les sites d’interaction entre xanthane et galactomannanes sont les régions les moins substituées. Partant de ce constat, de nombreux modèles ont, dès lors, été proposés pour tenter d’expliquer les mécanismes d’interaction.
Origine moléculaire des interactions du xanthane avec les galactomannanes. Les zones de jonctions pourraient être formées par des associations coopératives entre des régions de la chaîne de mannane et la structure native du xanthane [84]. Avec l’autorisation d’Elsevier.
Un modèle de base propose une association entre la chaîne principale de l’hélice de xanthane et les régions non substituées du galactomannane (Fig. 13 et 14) [89,95]. Ce modèle découle de l’observation expérimentale d’une diminution de l’ampleur de l’interaction avec une augmentation de la substitution en galactose et par l’apparente capacité des galactomanannes à stabiliser les hélices de xanthane. Ce modèle a été adapté pour tenir compte des interactions constatées entre xanthane et certains galactomannanes hautement substitués en galactose [30,97]. Il a donc été suggéré que les régions où plusieurs substituants sont situés du même côté de la chaîne de mannose sont également capables d’intervenir dans les interactions avec les xanthanes [64]. Les interactions ne nécessitent donc plus uniquement de longues sections de résidus mannoses non substitués continus. Les « zones de jonction » peuvent également se situer sur des sections plus courtes où tous les résidus galactosyles sont situés du même côté de la chaîne principale (Fig. 14). L’existence de « zones de jonction » a pu être établie dans des gels refroidis après chauffage, lors d’études de diffraction aux rayons X [98]. Les jonctions ne peuvent toutefois s’établir que si le xanthane est dans une conformation désordonnée (non hélicoïdale). Cependant, lors de l’étude de mélanges entre du xanthane et des galactomanannes de haut rapports M/G (5), une association des polysaccharides à des températures sous Tm a été observée, c’est-à-dire lorsque le xanthane est en conformation ordonnée [99]. Les auteurs suggèrent que les interactions ont lieu entre la chaîne de mannane de la gomme de caroube et certaines sections désordonnées du xanthane hélicoïdal ordonné. Une autre étude basée sur l’utilisation de galactomannanes hautement substitués en galactose a montré qu’ils pouvaient également interagir avec du xanthane à des températures de gélification pourtant inférieures à Tm [30].
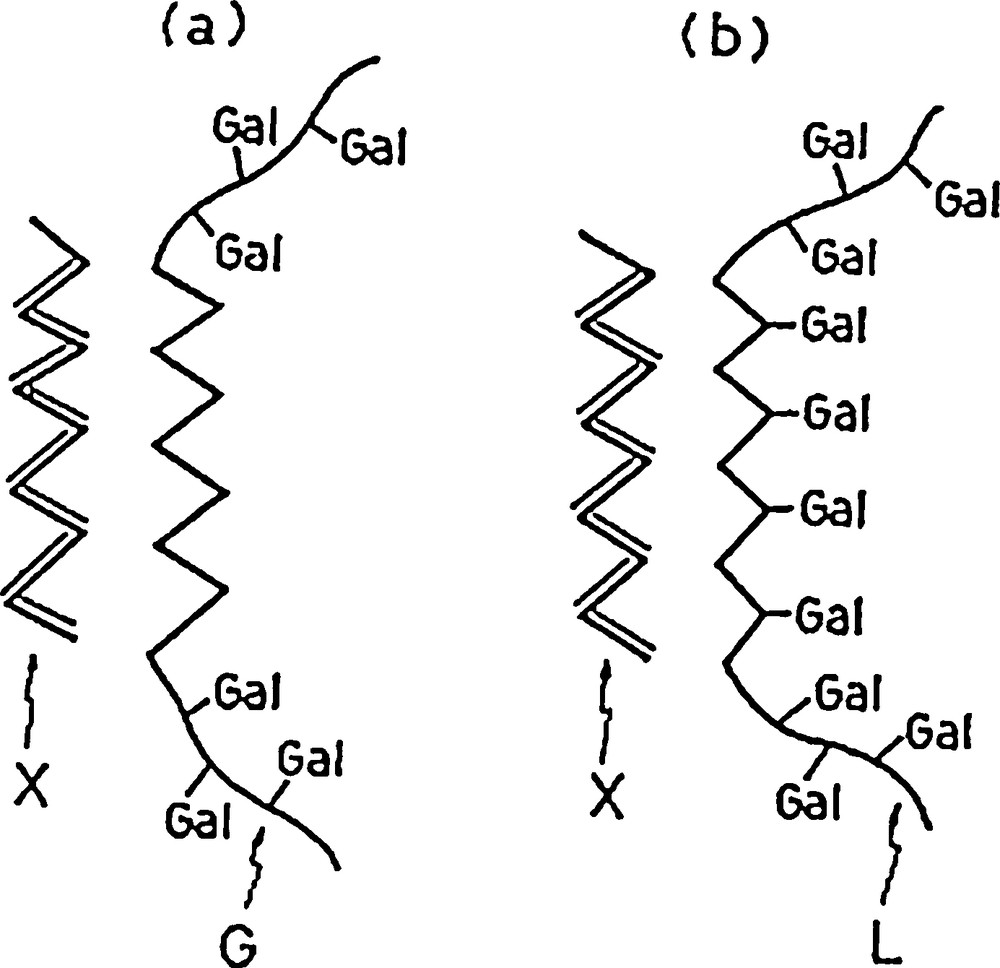
Modèle proposé pour l’association de xanthane et A) de gomme de caroube ; B) de galactomannane de Leucaena leucocephala [96]. Avec l’autorisation d’Elsevier.
En réalité, les interactions entre xanthanes et galactomananes pourraient être dues à deux mécanismes distincts. Le premier, prenant part à température ambiante, donne des gels faibles et cassants, et est relativement indépendant du contenu en galactose du galactomannane. Le second requiert des températures élevées pour former des gels forts et hautement dépendants de la composition en galactose du galactomannane [37]. Dans le cas du premier mécanisme, le mélange à froid des solutions de polysaccharides ne libérerait pas suffisamment de sites de liaisons sur les chaînes de xanthane, de sorte que tous les galactomannanes – faiblement ou fortement substitués – possèdent suffisamment de régions « lisses » pour occuper tous les sites de liaisons du xanthane. Selon d’autres auteurs, les deux mécanismes sont capables de coexister, du moins pour des galactomannanes de rapport M/G = 3 et 3,5, tout en étant influencés par la concentration saline du milieu [100]. La formation de gels à plus haute température que Tm est donc directement reliée au rapport M/G et à la distribution des galactoses sur les chaînes de mannose. Des travaux plus récents démontrent ainsi qu’il existe une corrélation entre le module visqueux G’ (déterminé au rhéomètre) d’un gel mixte gomme de caroube/xanthane et le nombre de résidus mannose successivement non substitués [101]. La même étude indique également une augmentation du module visqueux G’ avec l’accroissement de la masse moléculaire. Le comportement d’association au xanthane est donc fortement lié à la structure fine du galactomannane. La présence de zones « lisses » apparaît toutefois comme le critère principal qui favorise l’apparition de gels « forts » entre xanthane et galactomannanes, bien que les propriétés rhéologiques du gel soient également influencées par la composition du xanthane en acétates et en pyruvates [102].
Les galactomannanes sont également susceptibles de former des gels avec les carraghénanes, d’autant plus que le rapport M/G est important. Les études menées sur le sujet s’accordent également sur le fait que les zones à faible contenu en galactose ou « lisses » sont impliquées dans la jonction aux κ-carraghénanes. Les galactomannanes qui en sont pourvus sont donc plus fortement connectés aux κ-carraghénanes et forment des gels plus forts [1,29,83,103–106]. Les mêmes constatations découlent aussi de l’étude des gels galactomannanes/agarose [1]. Enfin, il est à signaler que le degré d’interaction aux protéines est également inversement proportionnel au DSGal [102].
6 Conclusions
Le procédé d’extraction et de purification utilisé lors de la fabrication de la gomme de caroube (ou son fractionnement ultérieur) va conditionner les caractéristiques de celle-ci et, par conséquent, ses potentialités d’applications. Lors d’un tel procédé, la température de solubilisation des polysaccharides est le paramètre critique à contrôler (Tableau 1).
Impact de la température de purification/fractionnement sur la relation structure–fonction des galactomannanes de caroube (méthode soustractive)a.
| Type de gomme | Auteurs | ||
| Brute | Basse température (≈25 °C) | Haute température (≈80 °C) | |
| Structure | |||
| Rapport M/G = 4,17 | Rapport M/G = 2,8 | Rapport M/G ≈ 4–5 | [9,35,37] |
| Mn (méthode chimique) = 300 000 Da | Mn (méthode chimique) = 150 000 Da | Mn (méthode chimique) = 650 000 Da | [1] |
| Nombre de résidus Mannoses successivement non substitués = 6,4 | / | Nombre de résidus Mannoses successivement non substitués = 9,4 | [81] |
| Propriétés | |||
| Viscosité intrinsèque [η] de solutions diluées = 15,3 | Viscosité intrinsèque [η] de solution dilués = 12,4 | Viscosité intrinsèque [η] de solutions diluées = 16,0 | [84] |
| / | ηsp d’une solution concentrée de galactomannane ≈ 8 | ηsp d’une solution concentrée de galactomannane ≈ 31 | [55] |
| Module élastique (G’) d’un gel mixte : galactomannane–xanthane = 505 Pa et galactomannane–carraghénanes = 716 Pa | Module élastique (G’) d’un gel mixte : galactomannane–xanthane = 199 Pa et galactomannane–carraghénanes = 455 Pa | Module élastique (G’) d’un gel mixte : galactomannane–xanthane = 710 Pa et galactomannane–carraghénanes = 827 Pa | [28,29,84] |
| Précipitation (%) après un cycle gel–dégel = 37 % | / | Précipitation (%) après un cycle gel–dégel = 88 % | [81] |
a Des résultats similaires sont obtenus par la méthode intégrale, mais sont moins contrastés.
Une température relativement basse sélectionnera des galactomannanes de faibles masses moléculaires et de contenu en galactose important. Ces fibres ont un caractère hydrophile plus marqué. A contrario, une température de travail plus importante permettra de solubiliser des galactomannanes plus longs et très pauvres en galactose, qui apporteront leurs contributions aux caractéristiques globales de la gomme obtenue. Celle-ci possédera une masse moléculaire moyenne plus importante, un degré de substitution en galactose plus faible, ainsi qu’un motif de distribution du galactose présentant plus de zones lisses. Ces deux types de gomme présenteront des propriétés physiques différentes en solutions aqueuses.
Les gommes clarifiées ou fractionnées « à chaud » nécessitent une montée en température pour atteindre la solubilisation. Elles présenteront une conformation en solutions aqueuses de type pelote, plutôt étendue, induite par la présence de nombreuses zones lisses. Leurs volumes hydrodynamiques sont donc plus importants, ce qui génère une viscosité intrinsèque plus importante. Leurs structures favorisent également, en régime semi-dilué, les phénomènes d’agrégations moléculaires et les hyper-enchevêtrements qui en résultent. Après le chauffage nécessaire à la mise en solution, la viscosité initiale de telles solutions sera donc plus importante et les caractères rhéofluidifiants et élastiques plus marqués que ceux de solutions de gomme clarifiées ou fractionnées « à froid ». La présence de zones lisses accroît les possibilités de formation de gels par association à d’autres saccharides, à travers de nombreuses zones de jonction. Après une phase de montée en température, les gels obtenus seront relativement forts et solides.
Les gommes obtenues « à froid » présentent l’avantage d’être solubles en solution aqueuse, même à de faibles températures. Bien que leurs caractéristiques structurales leur confèrent une conformation plus enroulée et a fortiori une viscosité intrinsèque plus faible, elles génèrent toutefois des solutions directement visqueuses, sans étapes de chauffage préalables. Leurs structures plus faiblement pourvues en zones lisses limitent les possibilités d’agrégation. Les solutions préparées avec de telles gommes présentent donc, même après chauffage, des viscosités plus faibles, un caractère rhéofluidifiants moins marqué et un comportement plus visqueux qu’élastique. Leurs structures globalement plus « hérissées » limitent, mais sans les exclure, les possibilités de formation de gels mixtes.
Dans le cas des galactomannanes de caroube, structure chimique et propriétés physiques sont donc étroitement liées. Si le degré de substitution en galactose et la masse moléculaire ont un impact indéniable sur les propriétés de la gomme, le motif de distribution des substituants galactosyles semble cependant être le paramètre qui conditionnera le plus le comportement du polymère en solution aqueuse. L’impact des zones non substituées sur la solubilisation n’a pas été abondamment étudié. Il est toutefois admis que la présence de galactose limite l’agrégation inter- et intramoléculaire ainsi que la précipitation. Les fractions riches en zones lisses seront donc mises en solution avec plus de difficulté et nécessiteront probablement une montée en température pour atteindre une solubilisation complète. La mise en solution est une étape primordiale au développement des propriétés physiques. Une fois cet état atteint, les galactomannes de structure en blocs présenteront cependant des capacités d’agrégations moléculaires beaucoup plus importantes, mettant en exergue la responsabilité des zones lisses dans les interactions chaînes/chaînes. Ils développeront donc des viscosités (et viscoélasticités) nettement plus marquées. Le motif de distribution des résidus galactosyls conditionne également l’aptitude à la co-géléfication. Les zones lisses semblent ainsi clairement impliquées dans les zones de jonctions avec les autres polysaccharides. La connaissance précise de la structure fine permettra donc de prédire les propriétés de la gomme (ou d’une de ses fractions) et de lui cibler des applications spécifiques. Il faudrait donc, à l’avenir, privilégier préférentiellement l’étude de ce paramètre, lors des études sur le comportement des galactomannanes. Le degré de substitution en galactose, si souvent cité, n’apparaît pas être une caractéristique suffisante. C’est une donnée utile, mais incomplète.