

1 Introduction
Electrochemical detection of analytes is a very elegant method in analytical chemistry [1]. The interest in developing electrochemical-sensing devices for use in environmental monitoring, clinical assays, and etc. Electrochemical sensors satisfy many of the requirements for such tasks, particularly owing to their inherent specificity, rapid response, sensitivity, and simplicity of preparation for the determination of organic molecules, including drugs and related molecules in pharmaceutical dosage forms and biological fluids [2,3].
Dopamine (DA) is an important neurotransmitter molecule of catecholamines and its deficiency leads to brain disorders, such as Parkinson's disease and schizophrenia [4–6]. Thus, detecting and determining the concentrations of dopamine and their metabolites in the presence of interfering species is an important goal in electrochemical analysis. Much attention has been paid to the design and development of novel materials coated on electrode surfaces with improved molecular recognition capabilities [7,8]. Different types of electrode materials are tailored to identify and measure the neurotransmitter molecules [9–13]. Measurements at bare carbon electrodes are complicated due to the coexistence of high concentrations of ascorbic acid (AA) and other related species, which are oxidized in the same potential region [11]. Uric acid (UA) also attenuates the detection of neuromolecules, such as dopamine (DA).
Several types of electrode modifications are reported in the literature to detect dopamine by electrochemical methods, such as surface coating [9], carbon paste electrodes [13], self-assembled monolayers [14]. The electroanalytical techniques have been excellently applied to the determination of various electroactive species [15–20]. However, each type of modification has its own advantages and limitations. Carbon electrodes, especially paste electrodes, are widely used in electrochemical investigations [21–25].
Electrochemical sensors based on carbon nanotubes (CNTs) represent a new and interesting alternative for quantification of different analytes. Carbon nanotubes can be used to promote electron transfer reactions when used as electrode material in electrochemical devices [26], electrocatalysis and electroanalysis processes due to their significant mechanical strength, high electrical conductivity, high surface area, good chemical stability, as well as relative chemical inertness in most electrolyte solutions and a wide operation potential window [27]. The electronics properties of these nanomaterials have been exploited as means of promoting the electron transfer reaction for a wide range of molecules and biological species, including carbohydrates [28], hydrogen peroxide [29], glucose [30], norepinephrine [31], aminophenol [32], morin [33], cytochrome c [34], promethazine [35], thiols [36], methyldopa [37], epinephrine [1], and nicotinamide adenine dinucleotide [38].
Surfactants are a kind of amphiphilic molecule with a polar head on one side and a long hydrophobic tail on the other one. The applications of surfactants in electrochemistry and electroanalytical chemistry have been widely reported [39]. Many of the studies of modified electrodes were undertaken simply because electrochemists were curious about how new species attached to electrode surfaces behave compared to these species in solution [40]. Some less soluble surfactants were employed in the immobilization of macromolecules or other functional materials. Wu et al. [41] developed a stable carbon nanotube (CNT)-modified electrode based on the immobilization of CNT in the film of insoluble dihexadecyl phosphate (DHP) on a glassy carbon electrode. This electrode exhibited an electrocatalytic activity towards biomolecules and has been used as a sensor for the determination of these species [42,43]. We described initially the preparation and suitability of a SDSMCNTPE as a new electrocatalyst in the electrocatalysis and determination of dopamine, ascorbic acid, and uric acid in an aqueous buffer solution.
In this study, we fabricated carbon nanotube paste electrodes modified by SDS surfactant. DA, AA, and UA are important neurotransmitters in the mammalian central nervous system. They were selected as analytical molecules for electrochemical detection. These SDSMCNTPEs allow better sensitivity and selectivity for the determination of DA in the presence of AA and UA and proved better than unmodified electrodes. Thus, the fabrication of the SDSMCNTPE has been considered in detail. This SDSMCNTPE was used for the determination of DA, AA, UA and mechanisms also studied (see Scheme 1).

Oxidation reactions of dopamine, ascorbic acid and uric acid.
2 Experimental
2.1 Reagents
SDS, DA, AA, UA, and silicone oil were obtained from Sigma–Aldrich, Malaysia. SDS was prepared as a 25 × 10−3 M stock solution by dissolution in double-distilled water. DA, AA, UA, and the other chemicals were of analytical grade and used without further purification. DA was prepared as a 25 × 10−3 M stock solution by dissolution in a 0.1 M perchloric acid solution, AA was prepared as a 25 × 10−3 M stock solution by dissolution in double-distilled water and a 25 × 10−3 M stock solution of UA was obtained by dissolving it in a 0.1 M sodium hydroxide solution. In all the measurements, the supporting electrolyte used was 0.2 M PBS (pH 6.5). Spectroscopically pure multiwall carbon nanotubes (diameter 50–100 nm, 5–10 μm length) were obtained from Nanostructured and Amorphous Material Inc., Texas, USA.
2.2 Apparatus
Electrochemical measurements were carried out with a model-201 electrochemical analyzer (EA-201 Chemilink system) in a conventional three-electrode system. The working electrode was a SDSMCNTPE and SDSMCPE, having a cavity 3 mm in diameter. The counter electrode was a bright platinum wire, with a saturated calomel electrode (SCE) as the reference electrode completing the circuit.
2.3 Preparation of the electrochemical sensor electrodes
The CNTPE was prepared by mixing in a mortar multiwall carbon nanotubes and silicone oil in a ratio 60.0% w/w carbon nanotubes and 40.0% w/w silicone oil [44]. The classical carbon (graphite) paste electrode (CPE) was prepared by grinding 70% of graphite powder (particle size 50 mm) and 30% of silicone oil to produce a homogeneous carbon paste electrode. The paste was then packed into the cavity of a homemade electrode and smoothed out on a weighing paper. SDS modified graphite paste electrode (SDSMCPE) and SDSMCNTPE were prepared respectively by immobilizing 10 μL of SDS on the surface of the electrodes for 5 min. This method is described elsewhere [22,25,45–49].
2.4 Electrochemical measurements
Electrochemical determination of DA, AA, and AA were carried out in a voltammetric cell with supporting solution. A standard stock solution was added to the cell according to the requirement. Cyclic voltammograms and differential pulse voltammograms from –200 mV to +600 mV were recorded in 0.2 M PBS (pH 6.5) at a 100 mV/s scan rate for the determination of DA, AA, and UA; all electrochemical measurements were carried out at room temperature.
3 Results and discussion
3.1 Morphological characterization of CNT, BCNTPE and SDSMCNTPE
The surface morphology of the samples obtained using CNT, BCNTPE and SDSMCNTPE was examined using FESEM. Fig. 1a shows the morphological characteristics of CNT, CNTPE, before and after modification has been shown in Fig. 1b and c, which clearly indicates the deposition of SDS on the surface of the BCNTPE. The surface of the BCNTPE appears to be rough, whereas in the case of the SDSMCNTPE, the deposition of SDS with some agglomeration is observed with FESEM. It is well known that surfactants not only endow the electrode/solution interface with different electrical properties, but also adsorb at the electrode surface or aggregate into supermolecular structures to change the electrochemical process.

FESEM images of (a) the CNT (b) the BCNTPE (c) the SDSMCNTPE.
Typical EDX spectra obtained from BCNTPE and SDSMCNTPE are also presented in the figures. In Fig. 2a, the BCNTPE shows only the presence of C, O, and Si elements, but the modified SDSMCNTPE shows the presence of the C, O, Na, Si, and S elements in the spectrum (Fig. 2b shows that the SDSMCNTPE is successfully modified by SDS).
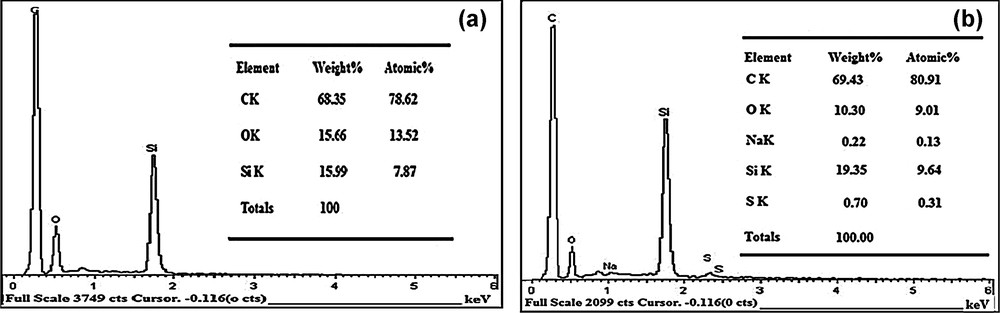
EDX spectrum of (a) the BCNTPE (b) the SDSMCNTPE.
3.2 Optimization of the electrochemical behavior of SDSMCNTPE
The variations of peak currents and of peak-to-peak separations for different amounts of SDS taken in the SDSMCNTPE were investigated using the well-known DA system in PBS at pH 6.5. It was found that the peak currents increased as the SDS content in the SDSMCNTPE increased. With more than 10 μL of SDS, the voltammograms recorded were drawn out, increasing the capacitance background current with a larger peak-to-peak separation in the case of the DA system (data will be given if asked). This caused a decreased reversibility as well as a positive shift in the oxidation potential of the analyte as compared to the SDSMCNTPE (10 μL SDS). The amount of the modifier, SDS, was optimized as 10 μL in the SDSMCNTPE to get the best voltammograms.
3.3 Stability and reproducibility of SDSMCNTPE
The main advantage of using the modified electrode is that the electrode surface can be renewed after every use by extrusion of approximately 0.5 mm of carbon nanotube paste from the cavity of the Teflon rod and by replacing it with a new paste. Indeed, five successive renewals of a SDSMCNTPE resulted in an RSD of 4.93%. The stability of the modified film was evaluated by examining the cyclic voltammetric peak currents of SDS after continuously scanning for 47 cycles in PBS (pH 6.5) (data not shown). The percentage degradation of SDSMCNTPE was calculated by the following equation:
3.4 The nature of cyclic voltammograms
The effects of increasing the amount of the modifier, SDS, in the SDSMCNTPE on the electrochemical behavior of DA, AA and UA were also investigated in order to optimize the conditions. Here, the focus was to get a maximum peak separation between DA, AA, and UA. The voltammograms for AA, UA and DA were recorded separately. The conditions were optimized so as to achieve better sensitivity and the best separation between the oxidation peaks of AA, UA, and DA. The optimum response in this case also was at 10 μL of the modifier, SDS, in the matrix of SDSMCNTPE. Voltammograms of the blank were recorded in PBS (pH = 6.5) as supporting electrolyte with a scan rate of 100 mV/s at BCNTPE and SDSMCNTPE. After optimizing the conditions, five replicates were performed under the same conditions to calculate RSD for anodic peak currents. Voltammograms of DA, AA and UA in the same buffer solution were recorded separately.
3.5 Electrochemical behavior of DA at SDSMCPE and SDSMCNTPE
The electrochemical behaviors of 0.2 mM DA at SDSMCPE and SDSMCNTPE were studied in 0.2 M PBS as the supporting electrolyte at pH 6.5, and the resulted cyclic voltammograms are displayed in Fig. 3. DA showed a quasi-reversible behavior with well-defined redox peaks at both electrodes. The anodic peak potential (Epa) and cathodic peak potential (Epc) of DA at the SDSMCPE were observed at 221 mV and 151 mV (solid line), while the same peaks were observed at the SDSMCNTPE at 230 mV and 150 mV (dashed line). Since the anodic and cathodic peak potentials and peak potential difference (ΔEp) – 70 mV and 80 mV, respectively – remain almost the same, redox kinetics on both electrode surfaces are similar. Peak currents (ipc and ipa) of DA at SDSMCNTPE increased by about 2.1 times compared to those at the SDSMCPE. These results confirmed that the SDSMCNTPE improved the sensitivity by enhancing peak currents. Possibly the large pore volume of carbon nanotubes provides a large specific area, leading to an enhancement in the peak current.
Cyclic voltammograms of 0.2 mM DA in pH 6.5. PBS at the SDSMCPE (solid line), at the SDSMCNTPE (dashed line).
3.6 Electrochemistry of DA at the SDSMCNTPE
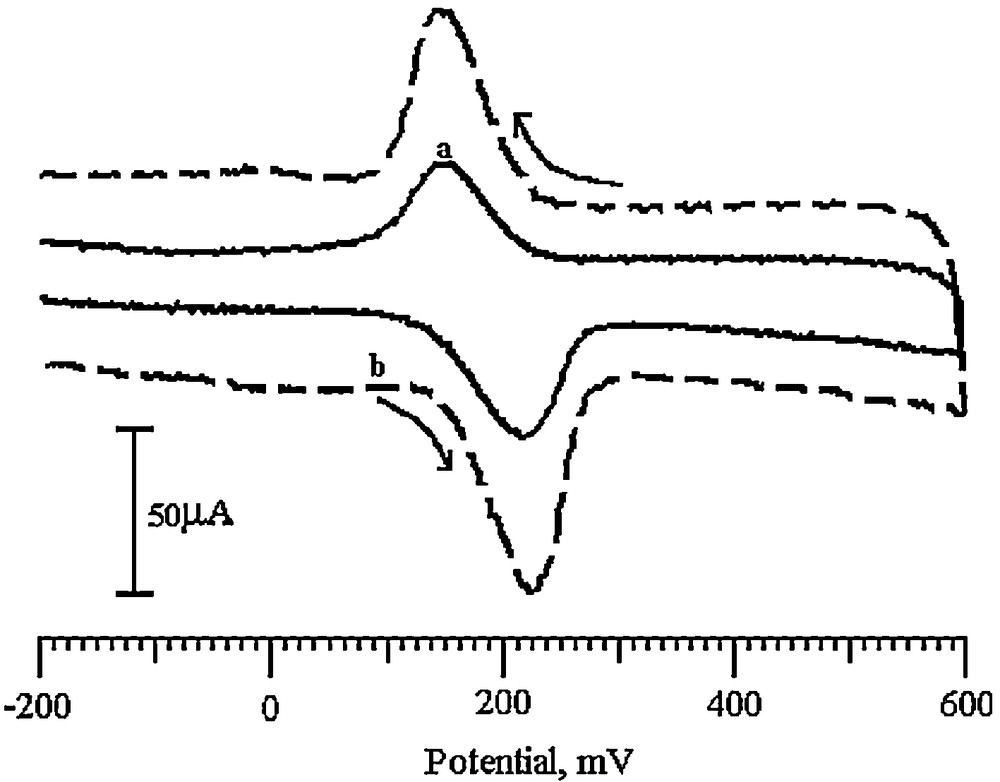
The electrochemical behavior of dopamine-o-quinone that was released from dopamine was studied in the presence of SDSMCNTPE. Fig. 4 shows the cyclic voltammograms of the sensor in the presence and absence of 0.2 mM DA in 0.2 M PBS (pH 6.5) at a scan rate of 100 mV/s. In the absence of DA, the SDSMCNTPE gave no peak response and only a background current was observed (solid line). When the DA was added to the buffer solution, a relatively larger anodic current at potentials of 230 mV was observed. In the present electrochemical approach, the electrode response was proportional to the oxidation of the electroactive species produced. The reaction mechanism has been shown in Scheme 1.
A typical cyclic voltammogram of SDSMCNTPE with 0.2 mM DA in pH 6.5. PBS (curve b) without 0.2 mM DA at pH 6.5. PBS blank (curve a).
3.7 Interaction of the SDSMCNTPE surface with potassium ferrocyanide
K4[Fe(CN)6] was used as the electrochemical redox probe to investigate interfacial properties of SDSMCNTPE. Usually modification of electrodes with charged species has remarkable effects on the electrochemical behavior of redox probe reactions. These effects depend on the charge of both electrode surface and redox probe. A more reversible behavior is observed for the charged probe redox reactions at the modified electrodes with opposite charge, and a less reversible behavior for the charged probe redox reactions at the modified electrodes with similar charge [50,51]. Fig. 5 shows the electrochemical response of a 1 mM potassium ferrocyanide solution at the BCNTPE (solid line) and at the SDSMCNTPE (dashed line) in 1 M KCl. The anodic and cathodic peaks of ferrocyanide shifted towards more positive and more negative values, respectively giving a less reversible behavior at the SDSMCNTPE compared to BCNTPE. It is apparent that the electron transfer of a negatively charged probe is partially blocked at a modified electrode. This may be due to the repulsion interaction between negatively charged SDSMCNTPE and the negatively charged ferrocyanide redox probe. The current response of potassium ferrocyanide at SDSMCNTPE has been noticeably improved, indicating that the surface coverage property of the SDSMCNTPE has changed significantly, resulting in a favorable and stable electrochemical behavior.
Cyclic voltammograms of a 1 mM potassium ferrocyanide/1 M KCl solution at the BCNTPE (solid line) and at the SDSMCNTPE (dashed line).
3.8 Electrochemical response of DA on the SDSMCNTPE
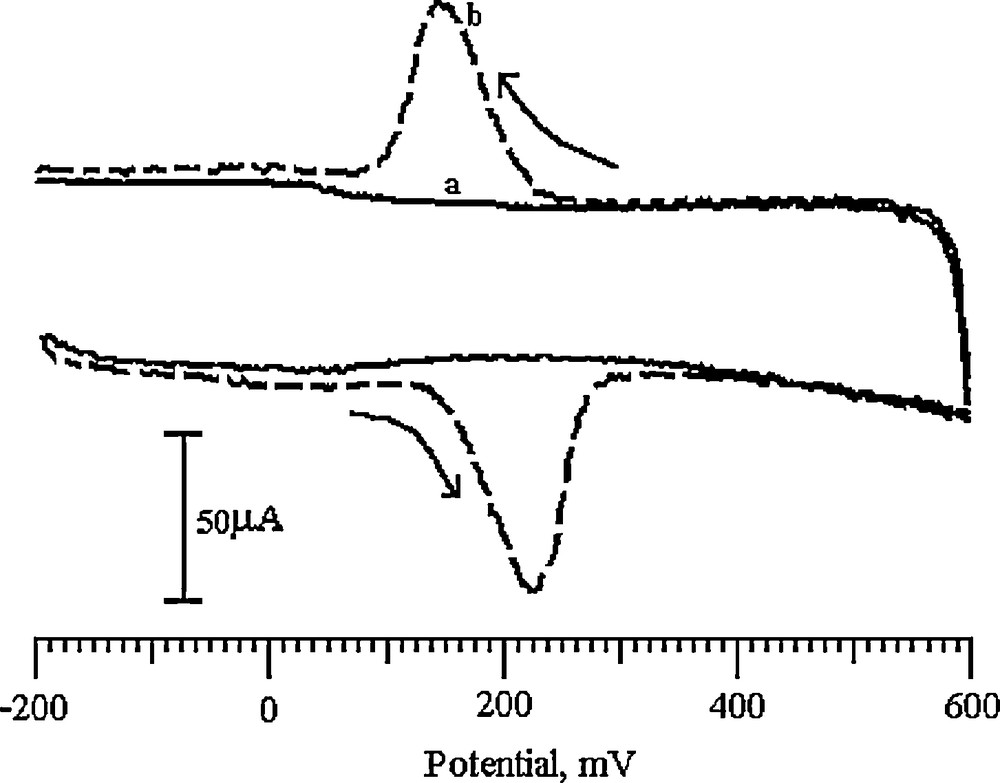
DA being an easily oxidizable catecholamine, its voltammogram was recorded in the potential range from −200 to 600 mV with a 0.2 M PBS solution as the supporting electrolyte (pH 6.5) at a 100 mV/s scan rate. Fig. 6 shows a pair of redox peaks for 0.2 mM DA on the BCNTPE (solid line), with an anodic peak potential (Epa) at 186 mV and a cathodic peak potential (Epc) at 138 mV (vs. SCE) in 0.2 M PBS (pH 6.5) as the supporting electrolyte. The peak-to-peak separation (ΔEp) was found to be 48 mV. However, for the SDSMCNTPE, a pair of redox peaks is obtained, with a strong increase in both anodic and cathodic peak currents (dashed line) with a small shift in redox peak potentials. The Epa was located at 230 mV and the corresponding Epc was located at 150 mV (vs SCE). The peak-to-peak separation was calculated as 80 mV, which is characteristic of a quasi-reversible electrode process.
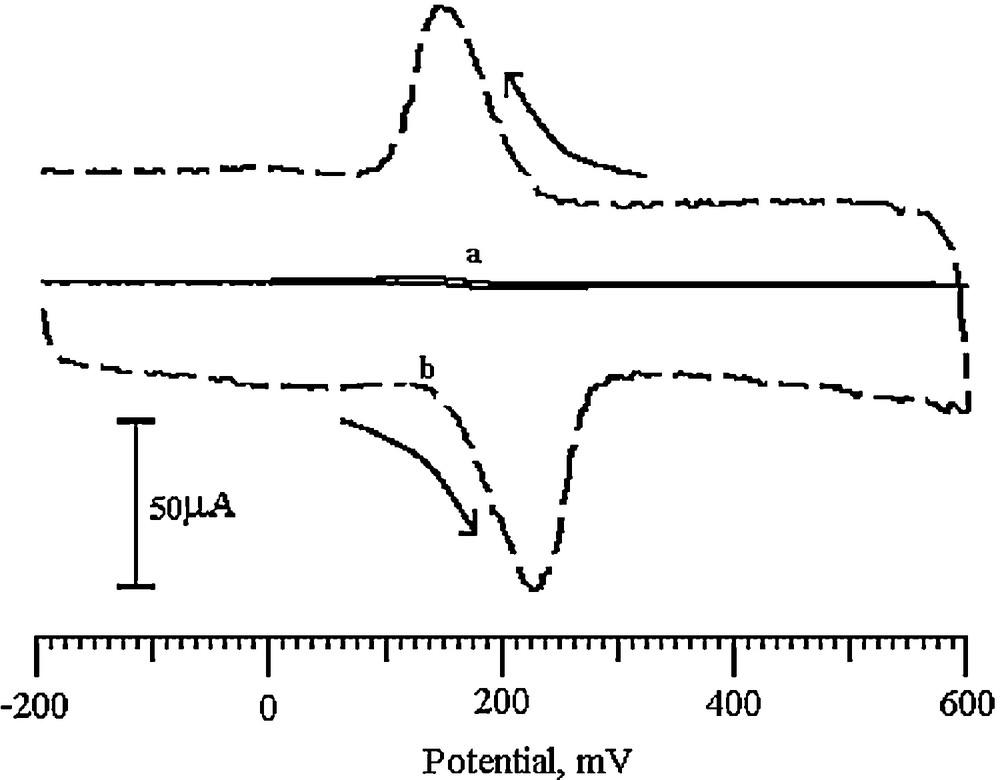
Cyclic voltammograms of 0.2 mM DA in pH 6.5 PBS at (a) the BCNTPE (solid line), (b) the SDSMCNTPE (dashed line).
3.9 Effect of scan rate of DA
Fig. 7a shows cyclic voltammograms of DA, recorded at different scan rates. Our results indicated that there is a linear relationship between the peak currents and the scan rate (v) in the range from 100 to 400 mV/s. Also, there is a linear relationship between the peak current (ip) and the square root of the scan rate (v1/2) for scan rate from 100 to 400 mV/s for DA. The SDSMCNTPE exhibited correlation coefficients r2 = 0.99316 and 0.9991, as shown in Fig. 7b and c, and the linear regression equations is given by [52]:
| ipa (μA) = –19.98214 − 0.9632 v (r2 = 0.99316) |
| ipc (μA) = 7.05 + 0.96786 v (r2 = 0.99416) |
| ipa (μA) = 180.61 − 911.44899 v1/2 (r2 = 0.99819) |
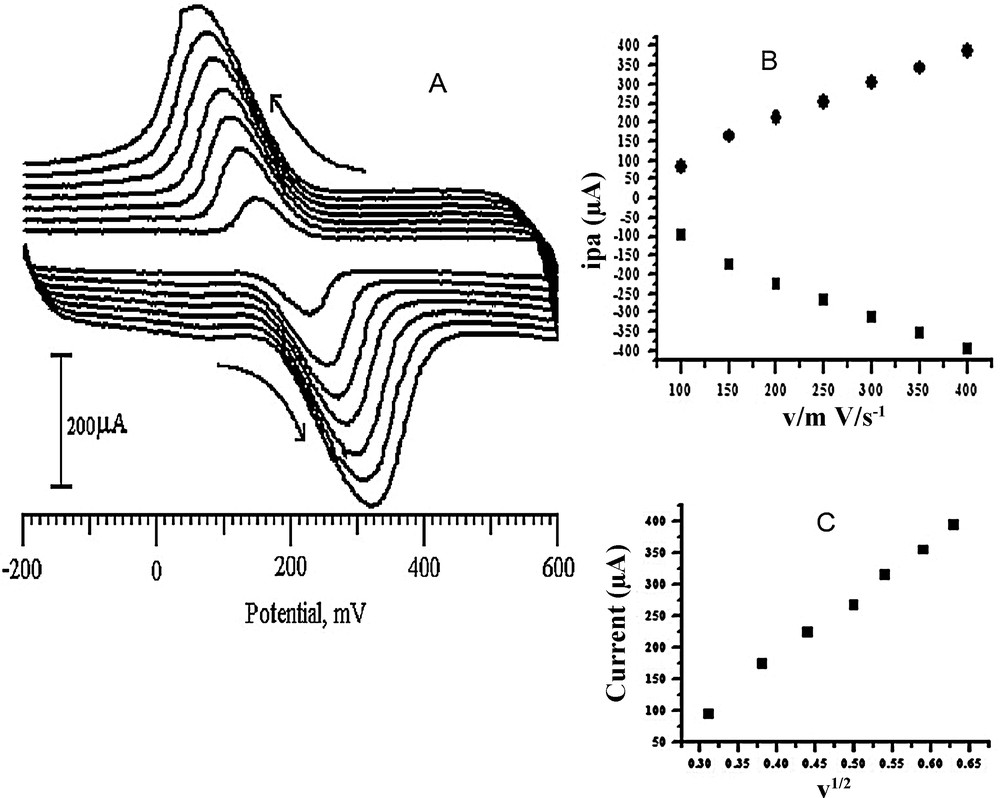
A. Cyclic voltammograms of 0.2 mM DA at the SDSMCNTPE in pH 6.5 PBS at various scan rates, 100, 150, 200, 250, 300, 350 and 400 mV/s. B. Plot of the anodic and cathodic peak currents of DA as a function of the scan rate. C. Plot of the anodic peak current of DA as a function of the square root of the scan rate.
The surface area available for electron transfer to the species in the solution can be estimated using the Randles–Sevcik equation [53]. This indicates that in addition to the existence of a diffusion-controlled mechanism, surface controlled reaction mechanisms are prevailing for DA electrochemical reactions at slow scan rates. This equation relates the peak current for an electron transfer-controlled process with the square root of the scan rate:
| ip = 2.69 × 105 n3/2A D1/2C v1/2 |
3.10 Relationship between pH values, peak potentials and peak current
The effect of pH on the electrochemical response of the SDSMCNTPE towards the 0.2 mM DA over the pH range 5.5 to 8.0 in a 0.2 M PBS solution is illustrated in Fig. 8a. The resulting profile showed the maximum sensitivity of the SDSMCNTPE at pH 6.5 (Fig. 8b). Thus, a solution of pH 6.5 was taken for the determination of DA. The SDSMCNTPE showed an optimum pH range of 6.0–7.0 [54]. The potential diagram was constructed by plotting the anodic peak potential Epa vs solution pH (Fig. 8c). The pH dependence of the oxidation peak potential of DA is Epa = 0.65182 − 0.05964 pH (r2 = 0.96444). The graph has a slope of 59.64 mV/pH, which obeys the Nernst equation for equal numbers of electrons and protons in the transfer reaction [55].
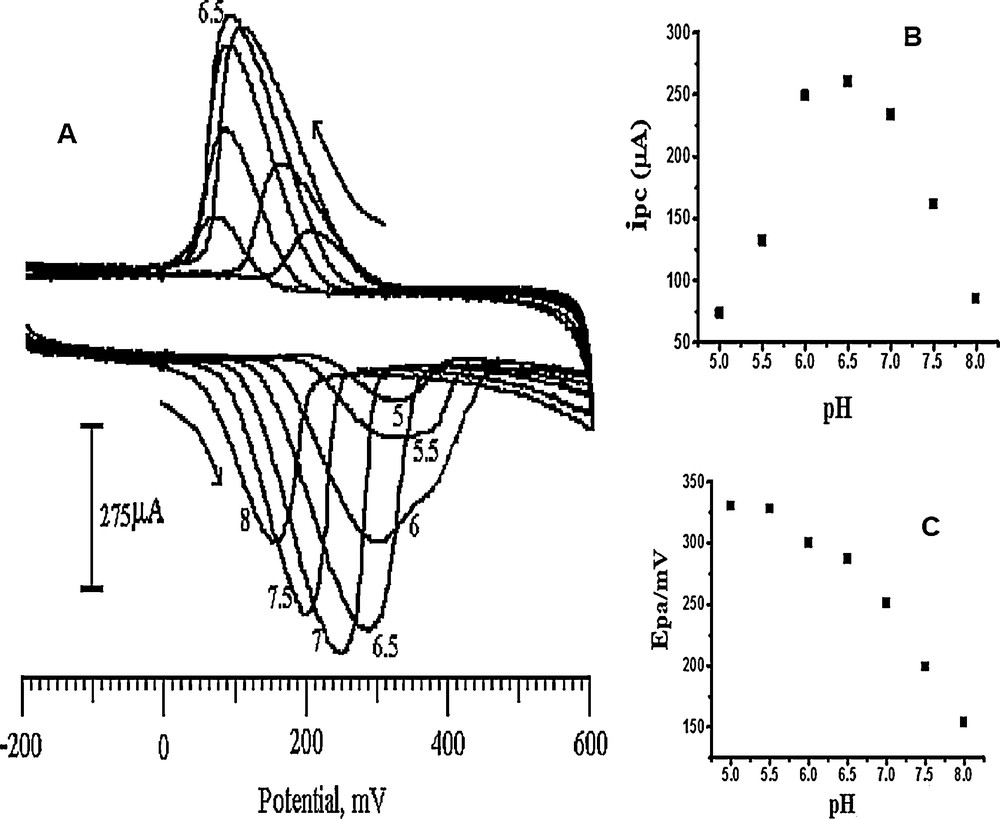
A. Cyclic voltammograms obtained at the SDSMCNTPE in 0.2 M PBS in pH values, (a) 5 (b) 5.5 (c) 6 (d) 6.5 (e) 7 (f) 7.5 (g) 8 containing 0.2 mM DA. B. Plot of the cathodic peak current vs pH (5.0–8.0) of 0.2 mM DA at the SDSMCNTPE. C. Plot of Epa vs pH for DA.
3.11 Calibration plot and limit of detection for DA
Cyclic voltammetry (CV) is a sensitive electrochemical technique that can be applied to analytic measurements; this method was used to determine the linear range and the detection limit of DA. The CV of DA at SDSMCNTPE in the concentration range from 1 × 10−6 to 2.8 × 10−5 M in PBS (pH 6.5) was recorded. The variation of the catalytic peak current versus the DA concentration represents two calibration curves corresponding to two concentration ranges. The first calibration equation is ipa (A) = 2.46 × 10−5 + 2.32364C [55] with a regression coefficient r2 = 0.99016 for the concentration range from 1 × 10−6 to 1 × 10−5M. The second calibration equation is ipa (A) = 3.54317 × 10−5 + 1.35135C with a regression coefficient r2 = 0.99482 for the concentration range from 1 × 10−5 to 2.8 × 10−5 M (Fig. 9). The detection limit (according to DL = 3 sb/m, where sb is the standard division of the blank response and m is the slope of the calibration plot) was obtained as 3.3 × 10−7 M using the calibration equation of the first linear segment. Table 1 shows the linear range and detection limit for DA at SDSMCNTPE in comparison with some sensors of other research groups. As it can be seen, the linear range for DA at SDSMCNTPE is better than all those of other sensors, and the detection limit is better than in other works [56–61], but the sensor proposed in the present work is cheaper and easier to use than other ones.

Calibration plot for the determination of DA at the SDSMCNTPE in pH 6.5 PBS (scan rate: 100 mV/s).
The comparison of SDSMCNTPE with some modified electrodes for the determination of dopamine.
| Electrode | Modifier | Analyte | Detection limit (M) | Linear range (M) | Refs |
| Carbon paste electrode | Pyrog allol red | DA | 7.8 × 10−7 | 1.0 × 10−6to 7 × 10−4 | [53] |
| Gold electrode | 2-Amino-5-mercapto-(1,3,4) triazole | DA | 8 × 10−7 | 2.5 × 10−6 to 5 × 10−4 | [54] |
| Glassy carbon electrode | Indole-3-carboxaldehyde | DA | 17 × 10−7 | 10 × 10−6 to 1 × 10−4 | [55] |
| Carbon paste electrode | Cobalt salophen | DA | 7 × 10−7 | 1 × 10−6 to 1 × 10−4 | [56] |
| Composite electrode | Phosphorylated zirconia-silica | DA | 1.7 × 10−6 | 6 × 10−6 to 1 × 10−4 | [57] |
| Glassy carbon electrode | Poly (caffeic acid) | DA | 4 × 10−7 | 1 × 10−6 to 4 × 10−5 | [58] |
| Multiwall nanotube paste | SDS | DA | 3.3 × 10−7 | 1 × 10−6 to 2.8 × 10−5 | This working electrode |
3.12 Oxidation of UA
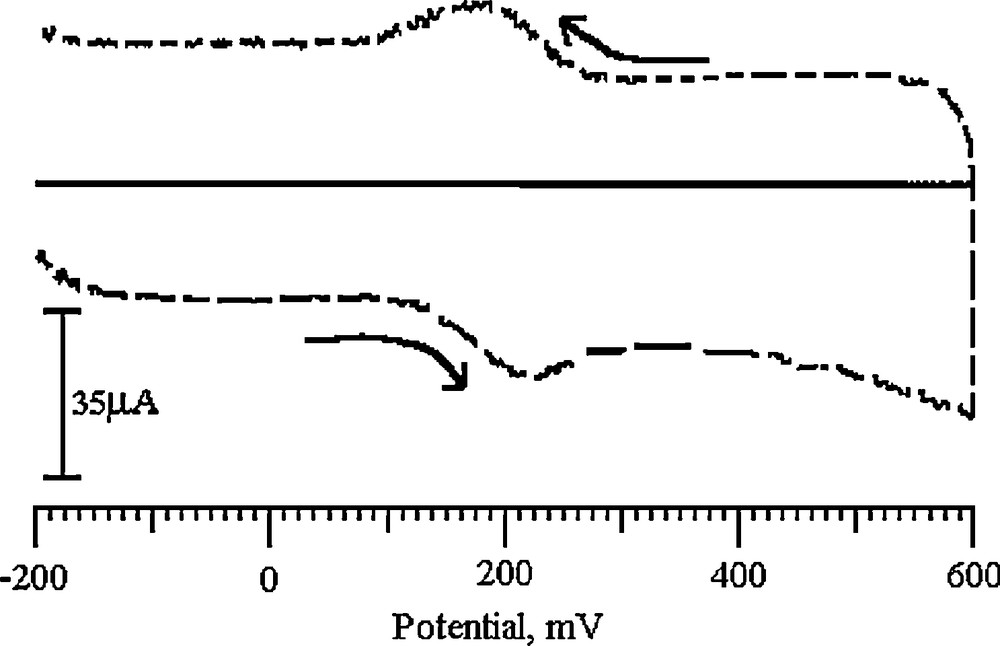
Cyclic voltammograms of UA at the BCNTPE and the SDSMCNTPE are shown in Fig. 10a, which shows that the current response of UA at the BCNTPE is weak, Epa = 321 mV, ipa = –7.6 μA and the current response of UA at the SDSMCNTPE is much better, Epa = 324 mV, Epa = –67.2 μA. The oxidation peak current of UA at the modified electrode is more than 10 times the current response at the BCNTPE, which indicates that SDSMCNTPE can significantly catalyze the UA oxidation process and that the electron transfer rate of UA in SDSMCNTPE is much faster. This may be attributed to the special nano-mesh structure of the SDSMCNTPE, which has a large specific surface area and a large number of defects. These defects result from the redox preparation process and serve as highly active reaction sites, making the UA activity at the SDSMCNTPE significantly improved and the response signal greatly increased. From the CVs of UA at the modified electrode, we can see that there is no reduction peak of UA: only its oxidation peak is observed, which evidences that the reaction is irreversible. Fig. 10b gives the cyclic voltammetry curves of UA at different scan rates, which shows that the oxidation peak potential is positively shifted when the scan rate is increased, and that the oxidation peak current is proportional to the square root of scan rates when the latter are between 100 and 500 mV/s. The linear equation is ipa (μA) = 34.5933 + 0.400047 v (r2 = 0.9979; Fig. 10c). This shows that the electrode reaction is controlled by the diffusion process, which is the typical characteristic of irreversible reactions. Kumar et al. reported the same electrode process of UA at a polymerized luminol film modified electrode [62]. A possible oxidation mechanism of UA at the electrode is proposed in Scheme 1. The main product of the electrochemical oxidation is 4,5-dihydroxyluric acid, which is unstable and mostly decomposes into allantoin. This explains why the cathodic peak is not obvious in the cyclic voltammogram [63].
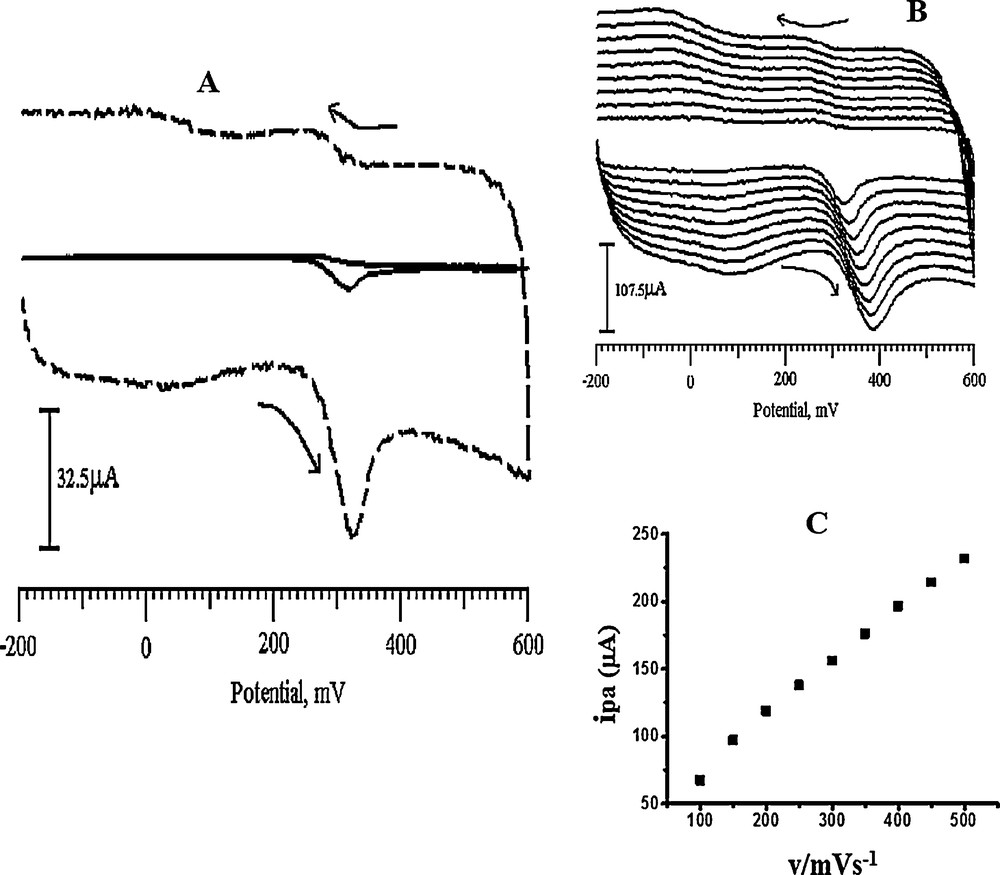
A. Cyclic voltammograms of 0.5 mM UA in pH 6.5 PBS at the BCNTPE (solid line) and at the SDSMCNTPE (dashed line). B. Cyclic voltammograms of 0.5 mM UA at the SDSMCNTPE in pH 6.5 PBS at various scan rates. From 100, 150, 200, 250, 300, 350, 400, 450, and 500 mV/s. C. Plot of the anodic peak current of UA as a function of the scan rate.
3.13 Electrochemical oxidation of AA at the SDSMCNTPE
Fig. 11a shows the cyclic voltammograms of 1 mM AA on the BCNTPE and the SDSMCNTPE with a 0.2 M PBS (pH 6.5) as the supporting electrolyte at a scan rate of 100 mV/s in the range from −200 to 600 mV. The Epa of the AA on the BCNTPE was found to be 113 mV, with a high voltammetric peak. However, on SDSMCNTPE, it was at 50 mV, with high enhancement. The shifting of Epa toward positive potentials on the SDSMCNTPE as compared to the BCNTPE suggests that AA has a high affinity for the modified electrodes. The cyclic voltammetric investigations at various potential sweep rates for AA were performed on the surface of the SDSMCNTPE (Fig. 11b). In these studies, a linear relationship with a correlation coefficient of ipa (μA) = 19.1133 + 0.3824 v, r2 = 0.99962, is observed between the anodic peak current and the potential sweep rate, which reveals that the oxidation of AA is a diffusion-controlled process (seen in Fig. 11c).
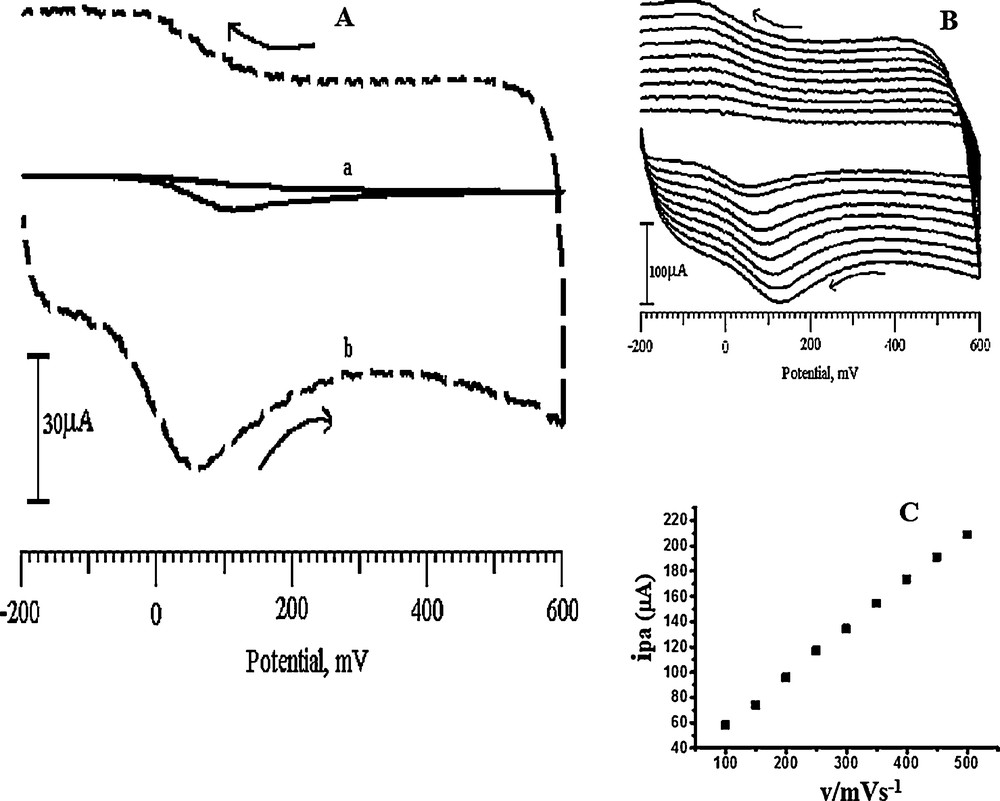
A. Cyclic voltammograms obtained for the oxidation of 1 mM AA at SDSMCNTPE (dashed line) and BCNTPE (solid line). B. Cyclic voltammograms for the oxidation of 1 mM AA at different scan rates (100, 150, 200, 250, 300, 350, 400, 450 and 500 mV/s) in 0.2 M PBS (pH 6.5). C. Plot of the anodic peak current of AA as a function of the scan rate.
3.14 Voltammetric resolution of DA, UA and AA using SDSMCNTPE
The objective of our work was the simultaneous determination of DA, UA, and AA. Fig. 12 shows the cyclic voltammograms of DA, UA and AA co-existing in PBS of pH 6.5 at the BCNTPE and the SDSMCNTPE. Curve (a) shows the cyclic voltammogram of a solution containing 2.5 mM AA, 1 mM UA, and 0.2 mM DA at the BCNTPE. It exhibits one broad peak for all analytes and indicates that the BCNTPE fails to separate the voltammetric signals of DA, UA, and AA. This is due to electrode fouling and the fact that their oxidation potentials are indeed quite close. The oxidized DA product, dopamine-o-quinone, can also be catalytically reduced to DA by AA, and UA becoming again available for oxidation [64,65]. Curve (b) shows the cyclic voltammogram of the oxidation of 2.5 mM AA, 1 mM UA and 0.2 mM DA mixture at the SDSMCNTPE. The SDSMCNTPE exhibits three well-defined oxidation peaks at 51, 278 and 372 mV, corresponding to the oxidation of AA, DA and UA, respectively. The difference between the oxidation peak potential of AA–DA, DA–UA and UA–AA was about 227, 94, 321 mV, showing more than 3.5 and 4.5 fold enhancement of the peak current for AA, DA and UA, respectively, compared to the BCNTPE. Therefore, the SDSMCNTPE can be effectively employed to separate DA in the presence of AA and UA.
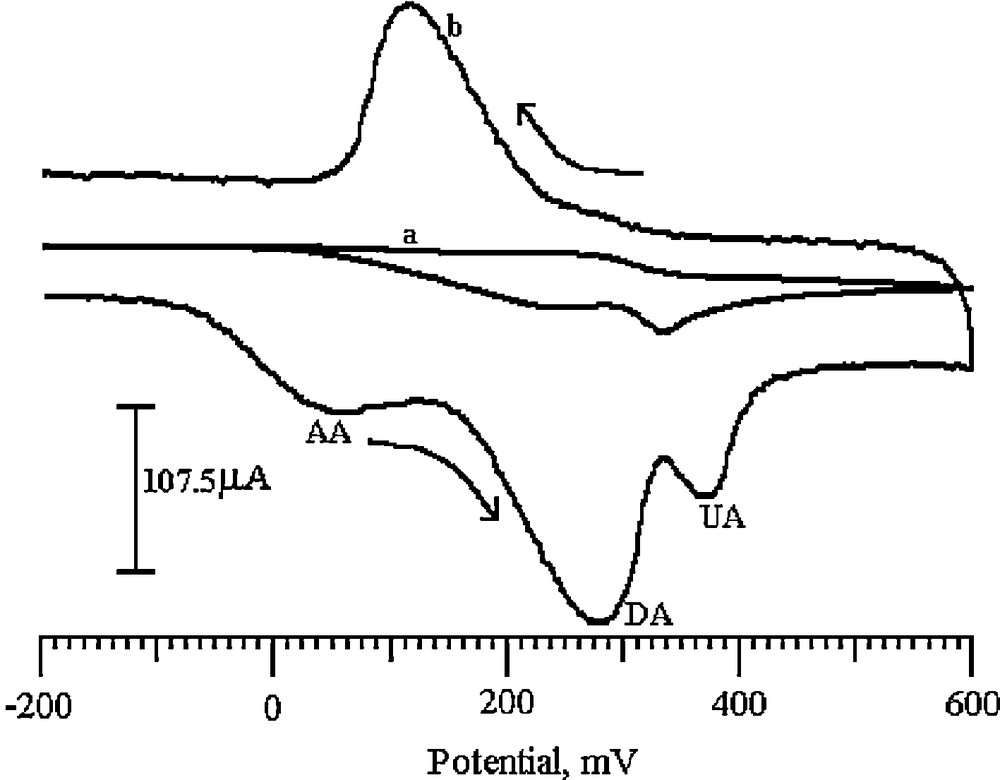
Cyclic voltammograms obtained at the BCNTPE (curve a) and the SDSMCNTPE (curve b) of a solution containing 0.2 mM DA, 2.5 mM AA and 1 mM UA in 0.2 M PBS (pH 6.5).
The simultaneous determination of DA, AA and UA in the mixture was carried out at the SDSMCNTPE when the concentration of one species changed, whereas the others were kept constant. From Fig. 13a, it can be seen that the peak current of DA was proportional to its concentration, which was increased from 0.2 to 0.6 mM when keeping the concentration of UA at 1.4 mM and that of AA at 3 mM. There was no change in the peak current and the peak potential occurred for AA and UA. Similarly, in Fig. 13b, it explains the concentration effect of UA from 1.2 to 2 mM when keeping the concentration of AA 2.5 mM and DA 0.2 mM. No change in the peak current and the peak potential occurred for AA and DA respectively. The corresponding UA linear regression equation can be expressed as, ipa (μA) = 57.16 + 107.4142 v, r2 = 0.99453 (Fig. 13c). These results show that the DA, AA and UA were present independently in their mixtures of samples.
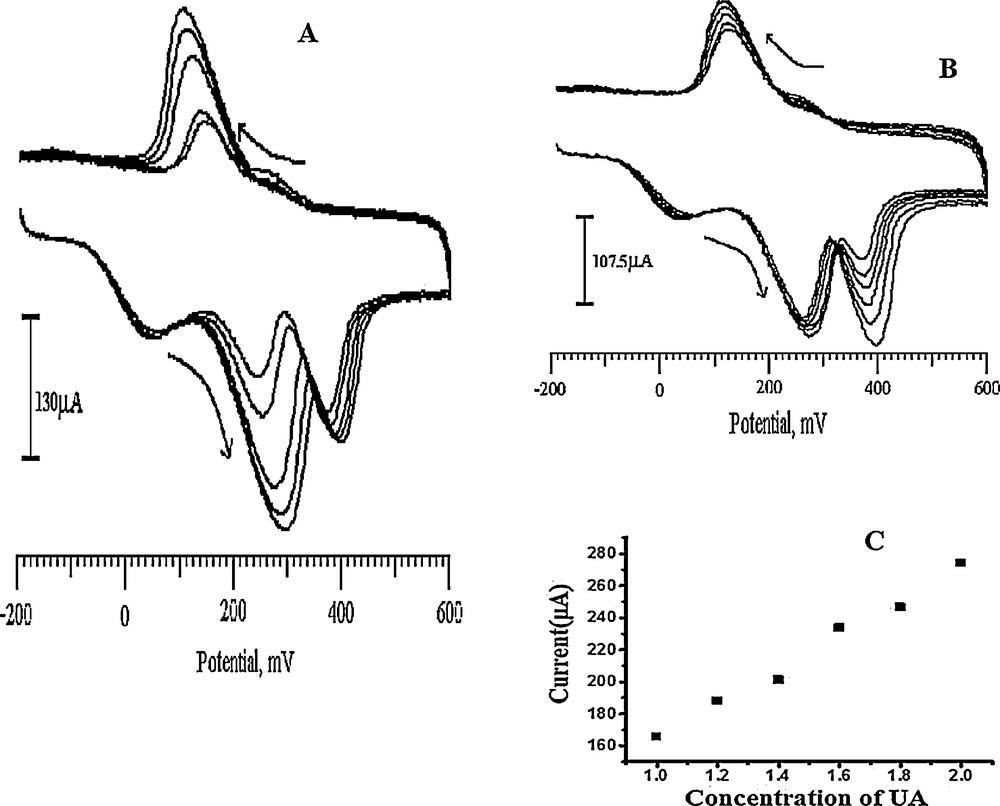
A. Cyclic voltammograms for DA at SDSMCNTPE in the presence of 3 mM AA and 1.4 mM UA in 0.2 M PBS (pH 6.5) for different concentrations of DA (pH 6.5, 0.2 to 0.6 mM DA). B. Cyclic voltammograms recordings for UA at the SDSMCNTPE in the presence of 2.5 mM AA and 0.2 mM DA in 0.2 M PBS of pH 6.5 for different concentrations of UA (1.2 to 2 mM UA) at pH 6.5. C. Plot of the redox peak current of DA versus concentration in pH 6.5 PBS (scan rate: 100 mV/s).
3.15 Interference studies by DPV
AA and UA were present along with DA in mammalian brain and the concentrations of these were much higher than that of DA. Since, the oxidation potential of both AA and UA was nearly the same as that of DA result in overlapped voltammetric responses at bare electrodes. However, our SDSMCNTPE faced this challenge and achieved the separation. The resulting voltammogram at the SDSMCNTPE had three well-defined peaks for DA, AA, and UA at different potentials. The oxidation peak potentials of DA, AA and UA were at 193, 11 and 304 mV, respectively (Fig. 14).
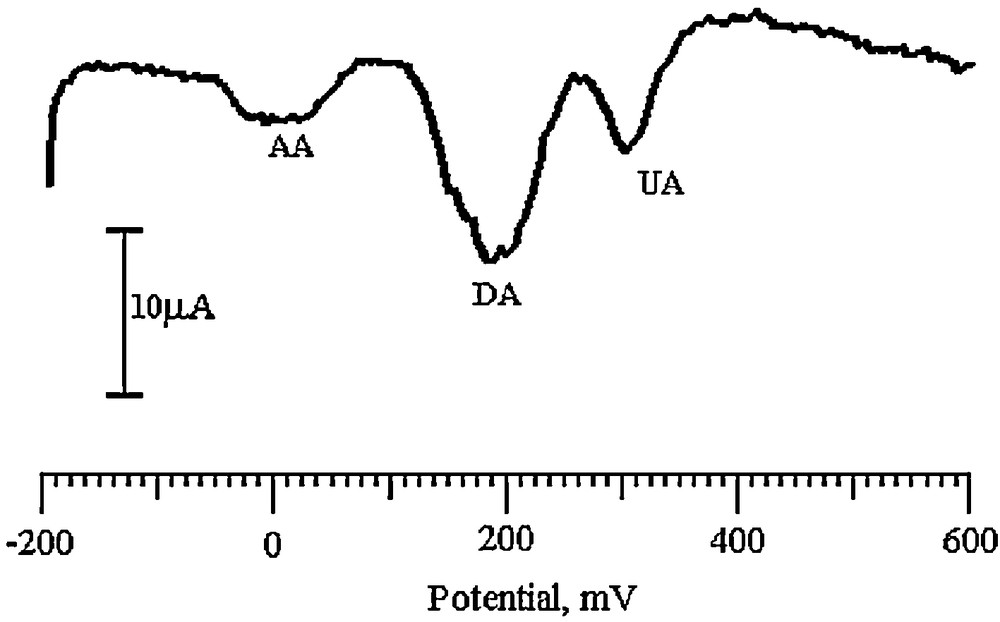
DPVs of a solution containing 0.2 mM DA, 2.5 mM AA and 1 mM UA in 0.2 M PBS (pH 6.5) at the SDSMCNTPE.
3.16 Sample analysis
The injections of DA were analyzed by the standard addition method. The results are shown in Table 2. The injection sample was used after suitable dilution, it contains 40.0 mg/mL DA. The recovery and RSD were acceptable (n = 5), showing that the proposed methods could be efficiently used for the determination of DA in DA injections with recovery in the range 98.8–103.8%, showing that the proposed method could be effectively used for the determination of DA in commercial samples.
Determination of DA in injections (n = 5).
| Sample | Added (mg/mL) | Found (mg/mL) | RSD (%) | Recovery (%) |
| 1 | 10 | 10.31 | 0.93 | 103.1 |
| 2 | 10 | 10.38 | 0.88 | 103.8 |
| 3 | 10 | 9.88 | 2.94 | 98.8 |
4 Conclusions
The SDSMCNTPE offers some important advantages, such as easiness of preparation, excellent reproducibility, low detection limit and wide linear dynamic range. Very small amounts of the modifier, SDS, were sufficient to get a good resolution of the voltammetric peaks for DA in a mixture containing DA, AA, and UA. The SDSMCNTPE exhibited excellent selectivity and sensitivity towards DA in the presence of an excess of AA and UA at pH 6.5. The SDSMCNTPE has been successfully applied to the determination of DA in real sample analysis. The low cost and simplicity of preparation makes this sensor a suitable candidate for commercial applications.
Acknowledgements
We acknowledge grants from the Universiti Kebangsaan Malaysia (UKM-GUP-216-2011, UKM-DLP-2012-022, UKM-DLP-2012-023), and the support of CRIM (Centre for Research and Innovation Management). The authors also thank Mr. Saini Sain for help with laboratory work.