



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Electrochemistry has a long history of surface modification ability, either by coating conductive surfaces with thin of thick films, or by covalent grafting of molecular moieties or supramolecular assemblies onto electrode materials. The concept of electrodeposition is known for more than a century when metal layers were deposited onto an electrode surface by electrolysis [1]. It was then extended to the deposition of other electroactive coatings on conductive surfaces and the field of electrodeposition expanded considerably in the 80’s [2], being still growing nowadays [3]. It is mainly based on electrochemically induced redox reactions involving electroactive precursors, which are intended to form solid deposits onto the electrode surface by electroplating (for metals [4, 5, 6], alloys [6, 7] or semiconductors [8]), electropolymerization (for, e.g., conducting polymers [9]) or other electrodeposition methods (for oxides [10, 11] or composite materials [3, 12]). More diverse and/or sophisticated materials can be also generated by electrodeposition (e.g., metal–organic frameworks [13], nanostructured catalysts [14], nanocomposites [15] or other functional (nano)materials [16, 17]). The resulting coatings are most often electroactive and/or conductive. In parallel, both oxidative and reductive electrografting methods have been developed to attach organic layers to solid conducting substrates [18] and such electrochemically induced surface modification can be followed further by post-functionalization processes to design advanced electrochemical interfaces with more complex molecular structures useful for targeted applications [19, 20]. Again, most of them were electroactive and/or conductive.
On the other hand, notwithstanding their insulating properties and non-electroactive nature, several inorganic materials have been used in connection to electrochemistry, such as clays [21, 22, 23, 24, 25, 26], zeolites [27, 28, 29, 30, 31, 32, 33], zeolitic imidazolate frameworks [34, 35, 36] or large band-gap metal oxides (most often silica gels or silica-based organic–inorganic hybrids [37, 38, 39, 40], sol–gel materials [40, 41, 42, 43, 44, 45, 46, 47], mesoporous silica [31, 47, 48, 49, 50, 51], or other nanostructured/nano-sized metal oxides [52, 53, 54]). They are attractive for the additional properties they can bring to an electrochemical interface (e.g., preconcentration and/or permselective features, host for biomolecules and/or catalysts). They can also be chemically modified in order to become electroactive (e.g., by impregnation/adsorption/ion exchange of redox-active species, or via the formation of hybrid materials made of organic moieties covalently attached to the inorganic structure). At the beginning, the intersection between electrochemistry and such materials was focusing mainly onto two parallel and complementary approaches [55]:
- the use of electrochemical characterization methods to get information on the materials properties (i.e., guest transport [56, 57, 58, 59, 60, 61, 62, 63], size/charge selectivity [63, 64, 65, 66, 67], charge transfer mechanisms in the insulating porous media [68, 69, 70, 71, 72], recognition and/or accumulation properties [73, 74, 75, 76, 77, 78, 79, 80, 81]);
- the exploitation of the remarkable properties of these materials used as electrode modifiers for selected applications, mainly in the field of electrochemical sensors and biosensors (see selected review papers [24, 25, 28, 30, 31, 37, 40, 48, 49, 50, 51, 52, 53, 54, 82, 83, 84, 85, 86]), but also in electrocatalysis [87, 88, 89, 90], electrochemical energy storage and conversion [34, 90, 91, 92], corrosion protection [93], electrochromism [94], and more recently molecular electronics [95, 96].
Because they are insulating materials, their use in connection to electrochemistry requires a close contact to a conductive electrode surface. This can be basically achieved by dispersing particles in a conductive composite matrix (e.g., carbon paste [97, 98, 99, 100, 101, 102, 103], carbon ink [103, 104, 105, 106]) or via the deposition of the materials as thin/thick films onto a solid electrode support (see examples of electrodes covered with clay platelets films [107, 108, 109, 110, 111], zeolite particulate or continuous films [31, 64, 112, 113, 114, 115, 116, 117, 118], silica-based or other sol–gel derived films [46, 47, 119, 120, 121], composite deposits containing mesoporous silica particles [31, 122, 123, 124, 125] or continuous mesoporous silica films [31, 126, 127, 128]). For film preparation, the approaches frequently used are solvent casting, spin-coating, dip-coating, layer-by-layer assembly, or via the Langmuir–Blodgett methodology.
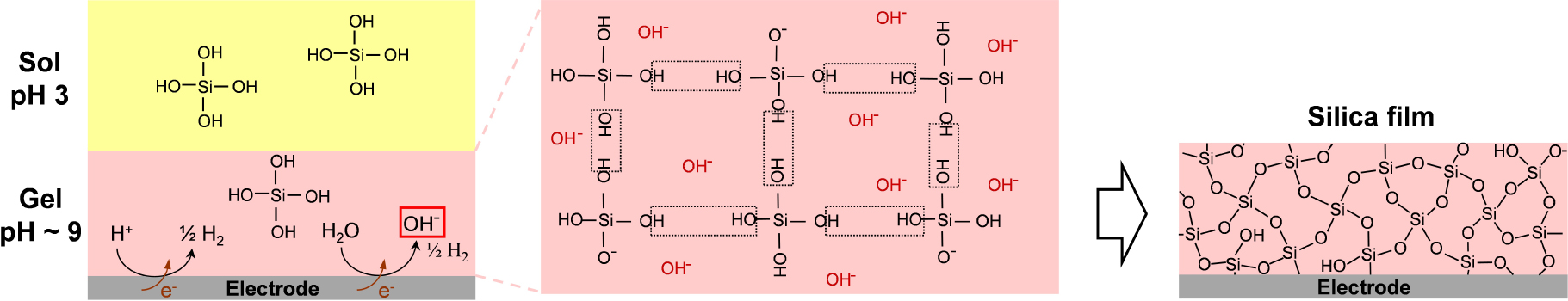
Schematic illustration of the electro-assisted deposition of a silica film onto an electrode surface from a hydrolysed sol at pH 3 (yellow part) by applying a cathodic potential likely to increase pH at the electrode/solution interface (pink part) to induce gelation of the sol and formation of the silica network by condensation of Si(OH)4 precursors catalysed by the electrogenerated OH− species.
In recent decades, significant efforts have been made to prepare thin films of non-conductive and non-electroactive materials by indirect electrochemically induced deposition. This method is based on the electrochemical generation of species that are likely to react with precursors in solution in the vicinity of the electrode, leading to the deposition or growth of solid phases onto the electrode surface mainly driven by local pH changes [51, 86, 89, 121, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137]. This is conceptually distinct from electrophoretic deposition, i.e., a technique involving the application of an electric field to a solution containing suspended matter, leading to the deposition of charged particles onto a conductive substrate [138], which was mainly applied to the formation of metal oxide and ceramic coatings [139, 140, 141, 142] or the deposition of other advanced (bio)materials [143, 144, 145]. In the present case, a reagent is generated via an electron transfer reaction at the electrode surface and so-generated species are involved in subsequent reactions inducing the transformation of solution-phase precursors (via precipitation or polycondensation) into a desired material coating the electrode support. Examples are available for metal oxides [129, 130], polymers [131, 132], sol–gel derived materials [133, 134], layered double hydroxides [89, 135], ordered mesoporous thin films [51, 86, 128, 136, 137]. They can be formed according to different driving forces. Precipitation of metal oxides and hydroxides or layered double hydroxides can be performed by electrogenerated OH− ions [89, 129, 130, 135], pH tuning in the electrode/solution interfacial region (as a result of water electrolysis [146]) to induce changes in solubility of polymeric materials in suspension due to variation in their hydrophilic/hydrophobic ratio [131, 132], or the electrogeneration of a catalyst that is likely to induce the growth of sol–gel films by accelerating the polycondensation reactions [133, 134]. Mesostructures are obtained if operating in the presence of surfactant templates [51, 86, 128, 136, 137]. In all cases, the electrogeneration of the catalyst leads to the indirect formation of a thin film, i.e., without direct electron transfer with the precursors, and such synthesis concept can be considered as counterintuitive as it enables the preparation of non-conductive and non-electroactive materials by an electrochemical way. This account intends to summarize the major advances primarily made by our group in this field, focusing mainly on electrochemically induced sol–gel bioencapsulation and electrochemically assisted self-assembly of oriented and functionalized mesoporous silica films, also considering recent applications developed by other groups.
2. The concept of electrochemically assisted deposition of sol–gel films by local pH tuning
The concept of sol–gel electrodeposition was born in 1999 when attempting to coat metal surfaces with hydrophobic organosilane for corrosion protection [147], and then extended to the formation of non-functionalized silica layers [133, 148] or organically modified silicate films [112, 149] on electrodes. As shown in Figure 1, it is based on the use of a silica sol containing tetraalkoxysilane precursors that are hydrolysed in moderate acidic medium (pH 3, Equation (1)) and an electrode biased at a suitable cathodic potential likely to generate OH− species by water reduction (imposing pH 9–10 at the electrode/solution interface) acting as a catalyst for polycondensation of the hydrolysed precursors (Equation (2)), leading to concomitant silica film growth onto the electrode surface.
| (1) |
| (2) |
The presence of nitrate ions in the electrosynthesis medium can contribute to promote further the cathodic alkalization at the origin of electro-assisted deposition of sol–gel films [150]. Although mainly applied to the generation of silica-based thin films, the method can also be implemented to obtain other metal oxide layers (zirconia [151], titania [152]) or for the co-electrodeposition of binary silica-metal oxide coatings [153, 154, 155].
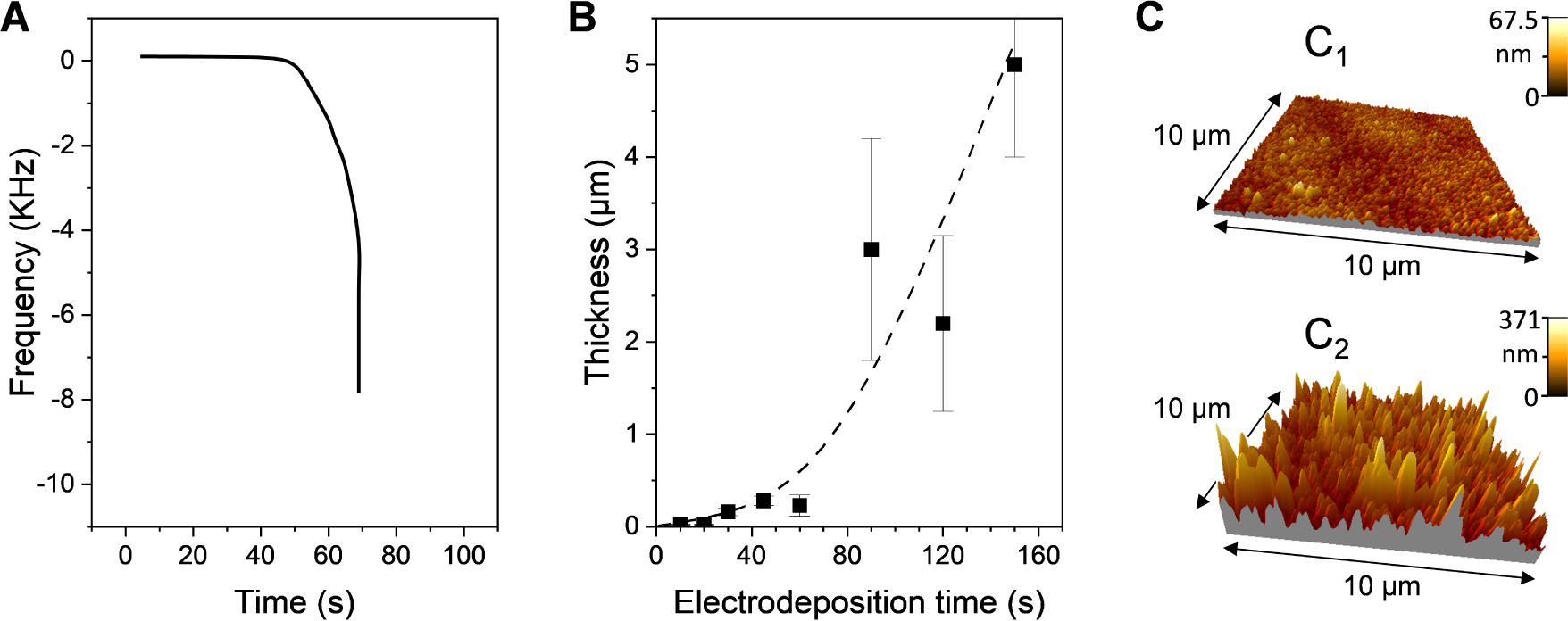
Electro-assisted deposition of amino-functionalized silica films onto a gold electrode at an applied potential of −1.2 V from a sol solution containing tetraethoxysilane (TEOS) and aminopropyltriethoxysilane (APTES) in 90:10 molar ratio. (A) Frequency–time plot obtained by quartz crystal microbalance. (B) Variation of the film thickness as a function of electrodeposition time. (C) Atomic force microscopy imaging of two films obtained respectively after 10 and 60 s electrodeposition. Reprinted and adapted from Sibottier et al. [149], Copyright (2006) American Chemical Society.
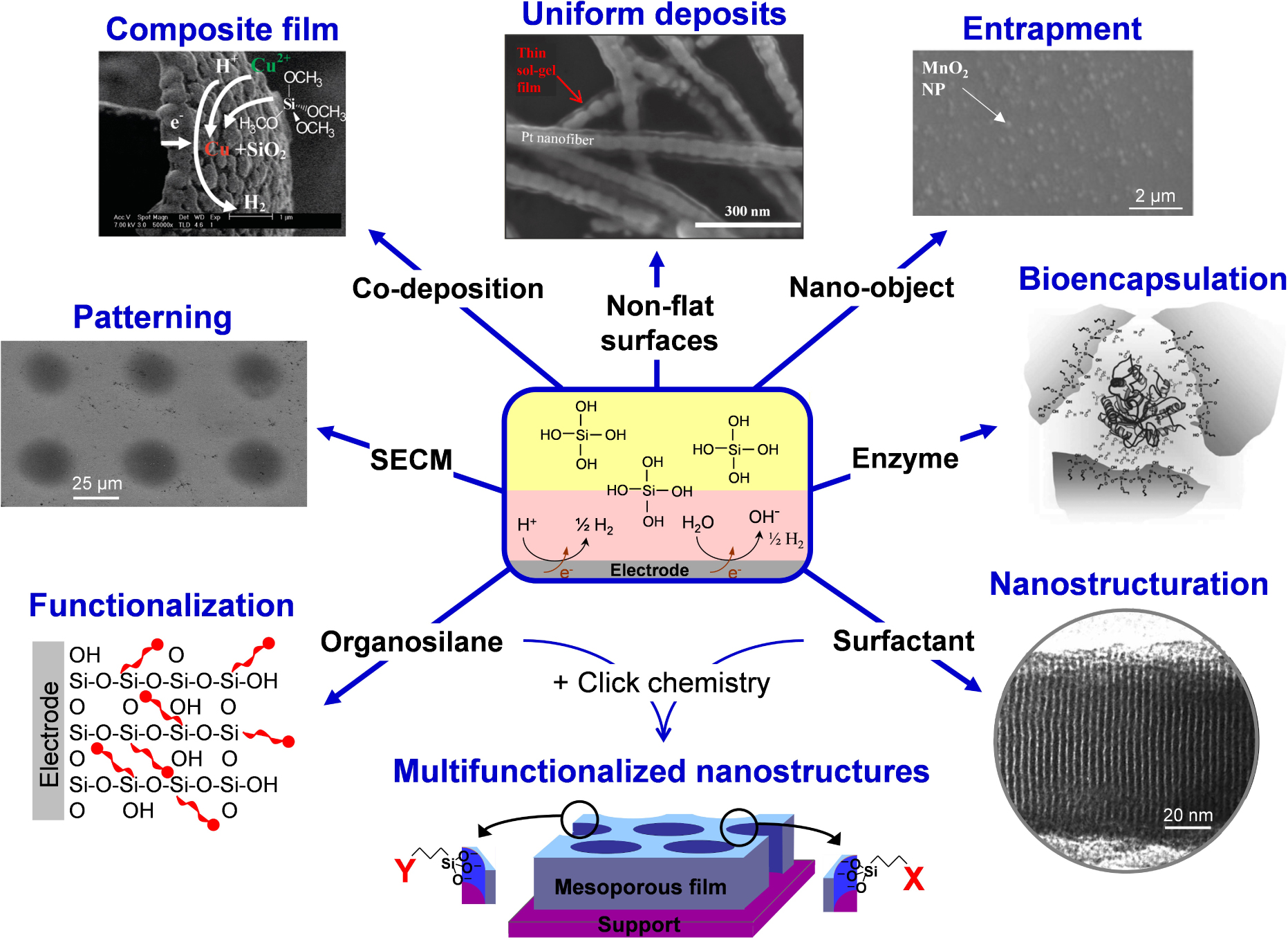
Illustration of the various possibilities offered by the electro-assisted deposition of silica films.
The parameters affecting the growth process are mainly (1) the nature of the precursor and its concentration, (2) the value of the applied cathodic potential, (3) the deposition time, and to lesser extent the temperature and supporting electrolyte [133, 147, 149, 156, 157, 158, 159, 160]. In particular, the film thickness (i.e., the amount of deposited material) has a strong dependence on the applied potential, being much larger when operating at more cathodic potentials because they induce higher interfacial pH values (due to more OH− species generated). The time of electrolysis is also an important parameter governing the film growth as one could expect thicker deposits at longer electrodeposition times. However, if the film thickness is indeed increasing linearly in the sub-micrometer range with the time of applying the cathodic potential in a first stage, it rises up exponentially to form much thicker (>1 μm) and rougher porous deposits in a second stage. As illustrated in Figure 2, these two steps and their consequences on film thickness and roughness can be evidenced from quartz crystal microbalance measurements (Figure 2A), from thickness variation as a function of deposition time (Figure 2B), and by atomic force microscopy imaging (Figure 2C) [149]. The second faster gelation step leading to the presence of thicker deposits in the form of particulate aggregates is due to the fact that the polycondensation catalysts (OH− species) are generated at the electrode/solution interface in a thickness of hundreds of micrometers (corresponding to the diffusion layer originating from the application of the cathodic potential). A quantitative description of the kinetics associated with the growth of sol–gel films generated by electrochemically assisted deposition can be derived from numerical simulations [161]. Finally, varying the concentration of precursors enables to tune the film growth rate [159, 160] and the use of very dilute sol solutions leads to the formation of ultra-thin (<10 nm) silica films [156]. The replacement of some tetraalkoxysilane precursors by organo-trialkoxysilane reagents also leads to thinner films due to slower hydrolysis/condensation reactions [157]. For more details on the fundamentals of sol–gel electrochemical deposition, the reader is directed to an excellent book chapter on the topic [160].
The concept of electrochemically assisted sol–gel deposition opens new ways for electrode surface modification, as illustrated in Figure 3.
- Firstly, it offers the advantage of depositing layers of uniform thickness over the whole surface of non-flat supports (such as metal or carbon nanofibers [162, 163, 164], streaked metal surfaces [133, 165], ordered macroporous metals [156, 166]) or micro- and nanoscale objects (carbon nanotubes [167]), the electrode geometry and size having some impact on film formation and properties [168]. It can cover selectively specific portions of heterogeneous supports, either via electrodeposition only onto the conducting portions of the support (printed circuit, gold mesh, silver grid printed on polyethylene terephthalate) [169, 170] or by taking advantage of different overpotentials for the generation of OH− catalyst (Pt nanoparticles on carbon or Pt–Au bimetallic macroporous electrodes), enabling selective sol–gel electro-assisted deposition onto Pt [166, 171], whereas the films obtained by dip-coating covered the whole surface of the Pt/C or Pt/Au supports. Of related interest is the formation of free-standing porous silica microstructures by exploiting adhesion differences of the deposit on hydrophilic and hydrophobic surfaces, via the selective removal of silica from the more hydrophobic areas of the substrate (thiol-modified Au) while keeping those firmly attached to the hydrophilic ones (indium-tin oxide) [172].
- Secondly, patterning is achievable by local scale electro-assisted deposition of sol–gel dots using a micrometric size electrode and a scanning electrochemical microscope in order to approach the microelectrode closely above the surface of a solid support enabling to generate locally the interfacial pH change to induce gelation and film formation under the microelectrode [173, 174, 175]. Getting patterns of locally deposited sol–gel material can be also performed by electrochemistry at arrays of liquid–liquid micro- or nanointerfaces, yet via a slightly different mechanism involving the electro-assisted transfer of cetyltrimethylammonium surfactant from an organic phase to an aqueous phase containing the tetraalkoxysilane precursor, leading to film formation at such soft interfaces [176, 177, 178].
- Thirdly, composite films can be obtained by co-deposition of metal and sol–gel interpenetrated layers by adding a metal precursor in the starting sol, which is reduced simultaneously to silica gel formation (examples are available for Cu [179], Zn [180, 181], Ni [182] and Au [183]). Co-electrodeposition of silica and other metal oxides is also possible (e.g., TiO2 [153] or MnO2 [184]). Sometimes, the silica part of the composite is dissolved to get porous metals or metal oxides [182, 183, 184]. Conductive polymer–silica hybrid thin films can be obtained by combining electropolymerization and sol–gel electrodeposition from a medium containing together the organic polymer precursor (e.g., pyrrole) and a tetraalkoxysilane. It involves performing the anodic polymerization of the conductive polymer and the cathodic sol–gel electrodeposition successively (separating both processes enables a better control of deposition parameters and thus an easy manipulation of film composition and thickness) [185, 186]. Other kind of organic–inorganic hybrid films based on polymer–silica composites can be indirectly electrodeposited via interfacial pH change, as illustrated for organotrialkoxysilane-chitosan hybrid gels [187, 188].
- Fourthly, nano-object can be physically entrapped within sol–gel films in the course of their electrochemically induced growth, offering a way to immobilize durably some particles that are somewhat difficult to accommodate onto electrode surfaces in a mechanically stable form. Examples are available for clays [189, 190], metal or metal oxide nanoparticles [191, 192, 193], carbon nanomaterials [167, 170, 194, 195], giving rise to composite films applicable in various fields (sensing, electrocatalysis, protective coatings). An interesting approach is the controlled growth of sol–gel matrices with different thickness and functional groups as nanoparticle imprinted thin films on electrodes that are likely to recognize and detect selectively gold nanoparticles based on their dimensions [196]. The electrochemical generation of sol–gel films can be also performed in the presence of polymeric nanobeads, leading to their entrapment in the inorganic coatings, and their subsequent dissolution leads to pore formation and enhanced mass transport of the resulting porous films [197].
- Fifthly, adding molecules or biomolecules and even microorganisms in the starting sol enables in principle their entrapment in the film during the formation of the silica framework. This is indeed successful for redox proteins, as pioneeringly demonstrated for haemoglobin and glucose oxidase in electrodeposited silica films [198, 199, 200], taking advantage of the room temperature sol–gel processing (which appears as a robust and versatile way for the effective immobilization of biologicals [201, 202, 203]). The use of additives in the starting sol may help at improving the biocomposite film properties, such as polymers (i.e., polyelectrolytes to favour bioencapsulation/biocompatibility) [204], noble metal nanoparticles (to enhance conductivity) [200, 205] or biodegradable polymeric nanoparticles [206]. The immobilization of larger biological materials (e.g., bacteria) in electrogenerated sol–gel films is also possible, leading to the production of electroactive artificial biofilms [207, 208]. On the other hand, if examples are available for entrapping smaller molecular species in electrodeposited sol–gel films (e.g., methyl parathion or dopamine via molecular imprinting during electro-assisted deposition [209, 210]), they are usually not durably immobilized and are quickly lost in solution, requiring suitable strategies to attach them more firmly (as pointed out for NADH, for example) [211]. More detail is given in Section 3.
- Sixthly, to the electrosynthesis medium can be doped with organosilane reagents that are likely to co-condense with the tetraalkoxysilane precursors to form an organic–inorganic hybrid film with the organic components being covalently bonded to the silica framework [147, 149, 165]. This enables to avoid leaching of the organo-functional groups, as common for other types of sol–gel materials [212, 213, 214, 215], thanks to the non-hydrolysable metal–carbon bond in metal alkoxides [216]. The organo-functional groups are selected for their particular properties, promoting tailor made features to the hybrid films, such as preconcentration ability [80, 165, 217], molecular recognition [209, 218], ion exchange [219, 220], electrocatalysis [221, 222], selective permeation [61, 157, 223, 224], superhydrophobicity [225, 226, 227] or protection against corrosion [227, 228, 229].
- Seventhly, combining the electro-assisted sol–gel formation with the inclusion of a surfactant template enables to get highly ordered and nanostructured deposits (see, e.g., pioneering works for vertically aligned mesoporous silica films [230] or lamellar structured zinc oxide films [136]). In this case, the applied potential does not only contribute to enforce the pH change at the electrode–solution interface but it also contributes to some organization of the surfactant species onto the electrode surface (adsorbed micelles or hemi-micelles formed as a result of potential-controlled surface aggregation of surfactants at electrode surfaces [231]). This combination of electrochemical modulation of the sol–gel process and electrochemical interfacial surfactant templating leads to the growth of nanostructured oxides with preferential pore orientation [135, 136, 230, 232]. The development of this rapidly expanding field is presented in detail in Section 4. Note that mesoporous metals can be formed by direct electrodeposition through organized surfactant phases on electrodes [233, 234], or mesoporous metal oxides films from electrochemically induced precipitation in the presence of ionic surfactants [136, 235, 236, 237, 238], but this is conceptually distinct from the electro-induced self-assembly of sol–gel films by generating the polycondensation catalyst [230]. Of related interest is the electrogeneration of porous silica films by inclusion of β-cyclodextrin [239], taking advantage of the templating ability of cyclodextrin assemblies [240] and the potential-controlled adsorption of such stacks onto an electrode surface [241]. Dendrimer pillars forming at solid–liquid interfaces [242, 243] can be also exploited as template for the electrogeneration of nanoporous silica [244].
- Finally, one can combine nanostructuration and functionalization. Because post-synthesis grafting may lead to pore blocking, the co-condensation approach is often preferred for the electro-assisted self-assembly of functionalized mesoporous silica films [137]. It is however restricted to rather simple organo-functional groups (e.g., aminopropyl) and quite low organic group contents (i.e., 10%) [245]. An elegant way to overcome this limitation is to combine the electro-assisted self-assembly of azide- and/or thiol-functionalized films with click chemistry, a method likely to generate nanostructured silica films bearing a richness of organo-functional groups [137, 246] and even multifunctional nanostructures by exploiting two different click reactions [247]. This opens the door to a series of advanced applications and most recent developments are presented and discussed in Section 4.3.
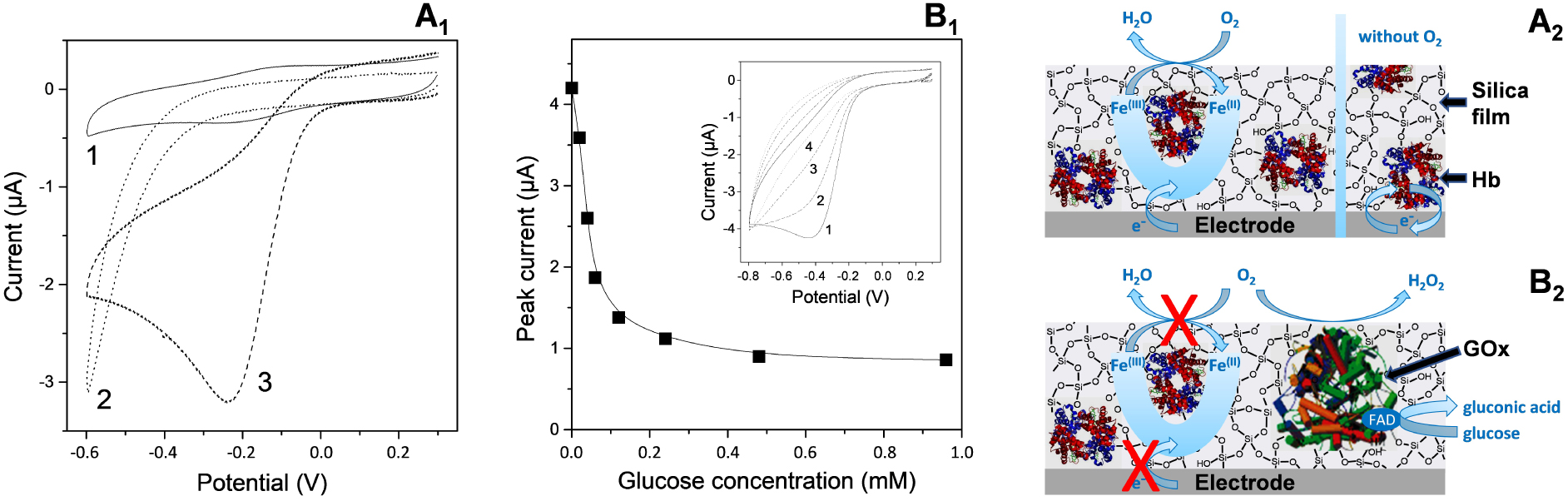
Illustration of sol–gel bioencapsulation. (A) Comparison of cyclic voltammograms (CV) recorded using GCE with electrodeposited silica gel entrapping Hb (1,3) or not (2), as obtained in anaerobic (1) or aerobic (2,3) conditions. Electrodeposition was performed from a sol solution containing 227 mM tetraethoxysilane (TEOS) and 20 μM Hb, by applying a potential of −1.2 V for 10 s. (A1 = data; A2 = illustrative scheme). (B) Variation of the cathodic peak currents measured at GCE with electrodeposited silica gel entrapping Hb and GOx, in an air-saturated phosphate buffer solution (pH 6.5) to which increasing amounts of glucose have been added, as a function of glucose concentration. Inset represents CV curves recorded in the absence (1) and in the presence of glucose at 0.02 mM (2), 0.04 mM (3), and 0.06 mM (4). The biocomposite film was prepared as in (A) but from a TEOS-based sol containing 20 μM Hb and 50 μM GOx in mixture. (B1 = data; B2 = illustrative scheme). Reprinted from Nadzhafova et al. [198], Copyright (2007), with permission from Elsevier Science. Masquer
Illustration of sol–gel bioencapsulation. (A) Comparison of cyclic voltammograms (CV) recorded using GCE with electrodeposited silica gel entrapping Hb (1,3) or not (2), as obtained in anaerobic (1) or aerobic (2,3) conditions. Electrodeposition was performed from a sol solution containing ... Lire la suite
3. Electrochemically induced sol–gel bioencapsulation and extension to the entrapment of other materials
Thanks to their synthesis at room temperature and under mild conditions, sol–gel oxides are good hosts for the encapsulation and stabilization of biochemicals such as enzymes, antibodies, living cells and bacteria [248]. Sol–gel bioencapsulation, by keeping the biological activity of the immobilized biorecognition elements [201, 202, 203], leads to bioceramic materials that are attractive for the development of biosensors [249, 250, 251]. For bioelectrochemical applications, the bioceramic materials needs to be in contact with a conductive surface, which is usually achieved by dispersing biocomposite particles into conductive matrices or by coating bioceramic films formed by evaporation on electrodes [40, 43, 252], which is however not appropriate for uniform deposition on surfaces with complex morphologies. As briefly aforementioned, sol–gel bioencapsulation can be electrochemically induced to form biocomposite thin films, even on non-flat surfaces [156, 204, 205, 253], leading to a novel one-step electrochemical approach of biomolecule immobilization for bioelectrochemical applications (beside electrodeposited polymers, for instance) [254]. Historical aspects and major advances made in this field are summarized hereafter, ending with few words on the electro-assisted sol–gel encapsulation of non-biological materials.
3.1. Feasibility of electrochemically induced sol–gel bioencapsulation
The first examples of electrochemically induced sol–gel bioencapsulation were reported in 2007 by two independent groups (Nadzhafova et al. [198] and Jia et al. [199]), describing the one-step immobilization of glucose oxidase (GOx) and/or haemoglobin (Hb) in an electrogenerated silica film. Once immobilized, the biomolecules kept their biological activity, as pointed out for GOx via the detection of glucose [198, 199], and the redox proteins were electrochemically accessible in spite of the insulating nature of the silica host, as demonstrated for Hb [198, 200]. This is attributed to the biocompatibility of sol–gel matrices [255, 256], which can even lead to enhanced stability when an enzyme is encapsulated in silica [257]. Figure 4 illustrates the proof-of-concept. In part A of the figure, one can see the typical reversible voltammetric signal of a glassy carbon electrode (GCE) covered with an electrogenerated silica film with encapsulated haemoglobin in a deaerated solution (curve 1) and its electrocatalytic response in the presence of oxygen (curve 3), with control experiment showing no response to oxygen when using a haemoglobin-free silica film on GCE (curve 2). In case of co-immobilized Hb and GOx, the biocomposite film becomes sensitive to the addition of increasing concentrations of glucose in the medium, resulting in the progressive decrease of the electrocatalytic signal (Figure 4B) due to oxygen consumption during the enzymatic bioconversion of glucose [198]. This demonstrates both the electrochemical and biochemical activities of the encapsulated biomolecules. For very open silica films (i.e., as those formed at very cathodic potentials for which the generation of hydrogen bubbles induces the formation of a highly porous silica matrix), the bioelectrochemical response of encapsulated glucose oxidase may drop. This is due to some enzyme loss from the silica layer but it remains better than the poor stability of enzymes physically adsorbed onto electrode surfaces [199]. On the other hand, GOx nanoconfinement in well-structured silica material such as inverse opal matrix revealed enhanced enzyme activity relative to that of GOx in aqueous solution [258]. Note that bioceramic thin films can be also prepared by spin-coating or dip-coating on electrodes and used for electrochemical biosensing [252]. This approach was restricted to flat surfaces only and usually required multi-step preparation procedures to get multilayer configurations to ensure stability, contrary to the one-step electro-assisted sol–gel deposition of biocomposite films, which is applicable to uniform film formation on microstructured electrodes as well [156, 204, 253].
3.2. Generalization, importance of additives
The first attempts to extend the above electro-induced sol–gel bioencapsulation method to other systems, simply by replacing Hb and GOx by other enzymes in the sol solution, failed as illustrated in Figure 5A for d-sorbitol dehydrogenase (DSDH) for which no response to d-sorbitol is observed [259]. The reason is the lack of favorable interactions between the negatively charged silica network and the anionic groups on the DSDH surface. An effective way to circumvent this problem is to add a cationic polyelectrolyte in the synthesis medium (i.e., poly(dimethyldiallylammonium chloride), PDMDAAC, or protonated polyethyleneimine, PEI), acting as stabilizer by favoring electrostatic interactions during the biocomposite formation [259]. In this case, the encapsulated DSDH recovered its biological activity and the biocomposite film is operating in both anodic and cathodic modes in the presence of d-sorbitol and d-fructose, respectively (Figure 5B). The method is compatible with the co-immobilization of several biomolecules (e.g., DSDH and diaphorase [259] or glucose dehydrogenase and diaphorase [260]) and has been used so far for the entrapment of various other enzymes such as lipase [253], laccase [261], polyol dehydrogenases [262], or membrane-bounded (S)-mandelate dehydrogenase [163]. The biopolymer chitosan has been also used as stabilizer [261, 263] and the co-encapsulation of gold nanoparticles contributed to further enhancement of the bioelectrode response [260, 261, 264], thanks to the preservation of encapsulated enzymes in silicate composite films with additives (polymers, smart nanomaterials) as well as their contribution to ensure porosity for reactants [251, 265]. Finally, one has to mention alternative electrodeposition methods, such as the generation of free standing silica-based biocomposite films by electrochemically induced ion transfer at liquid/liquid interfaces [266] or with the help of an electrode/organic-phase/aqueous-electrolyte three-phase junction [267].
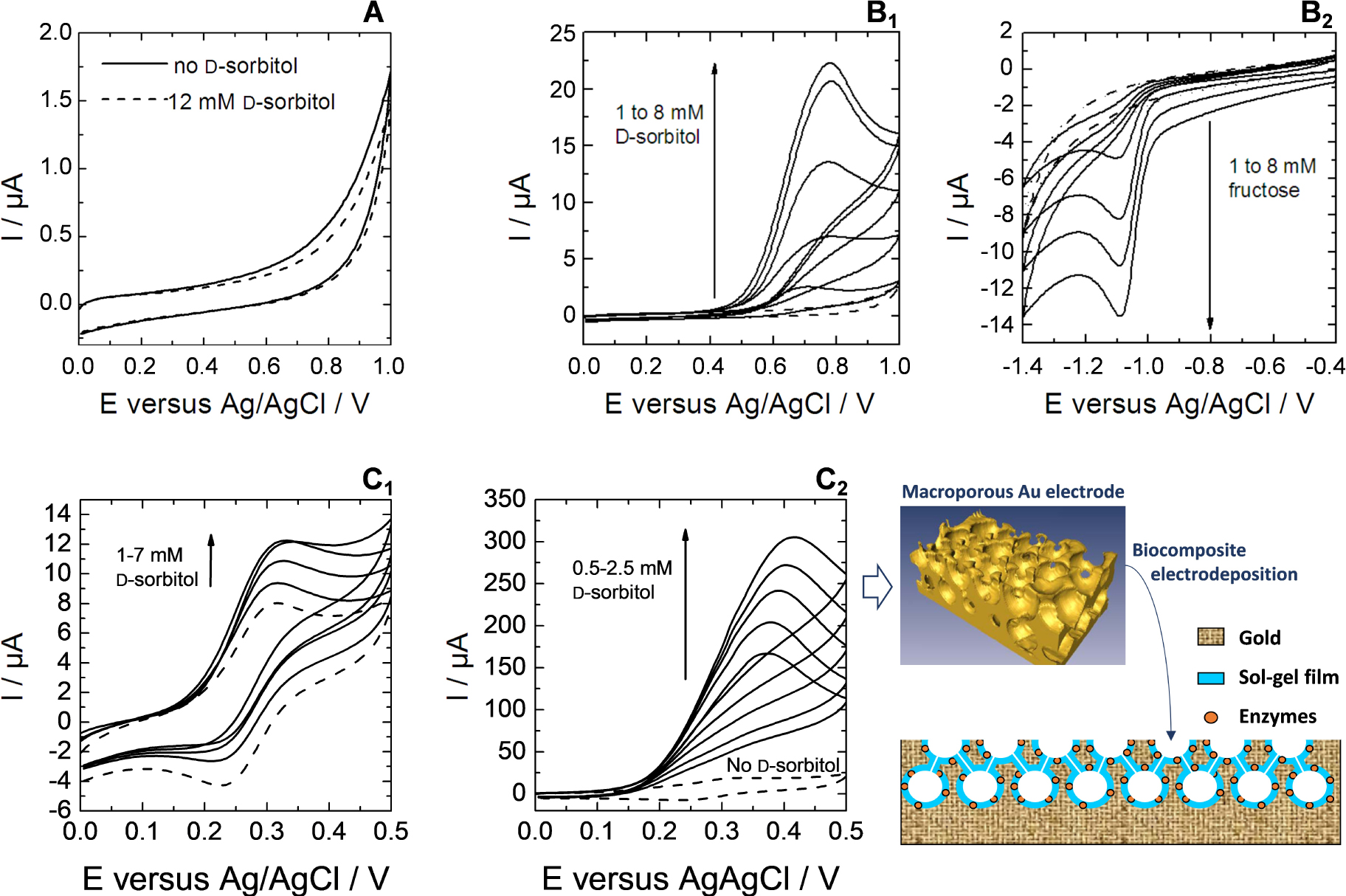
(A, B) Electrochemical responses to d-sorbitol (A, B1) and to fructose (B2) measured at GCE modified by DSDH with using sol–gel electro-assisted deposition done at −1.3 V for 60 s from a sol containing 0.17 M TEOS, 3.3 mg⋅cm−3 DSDH, respectively without (A) and with 6.7 wt% PDMDAAC (B). (A) Responses in the absence of d-sorbitol (dashed line) and in the presence of 12 mM d-sorbitol (solid lines). (B) Responses in the absence of enzymatic substrate (dashed lines) and in the presence of d-sorbitol (B1) or d-fructose (B2) from 1 to 8 mM (solid lines). CVs were recorded either in tris-HCl buffer (pH 9) containing 1 mM NAD+ (B1) or in 0.1 M phosphate buffer (pH 6.5) containing 1 mM NADH (B2). (C) CV responses to d-sorbitol of flat and macroporous gold electrodes modified by silica thin films with co-encapsulated DSDH and diaphorase electrodeposited at −1.1 V for 30 s from a sol containing 6.7 wt% PDMDAAC, 0.17 M TEOS, 3.3 mg⋅cm−3 DSDH and 0.83 mg⋅cm−3 diaphorase. (C1) Responses of a flat gold electrode in the absence (dashed line) and in the presence of increasing concentrations of d-sorbitol from 1 to 7 mM (solid lines). (C2) Responses of a gold macroporous electrode (three half-layers, see scheme in right bottom) in the absence (dashed line) and in the presence of increasing concentrations of d-sorbitol from 0.5 to 2.5 mM (solid lines). All CVs were performed in tris-HCl buffer at pH 9 in the presence of 1 mM NAD+ and 0.1 mM ferrocenedimethanol in solution. Reprinted from Wang et al. [259], Copyright (2011), with permission from Elsevier Science. Masquer
(A, B) Electrochemical responses to d-sorbitol (A, B1) and to fructose (B2) measured at GCE modified by DSDH with using sol–gel electro-assisted deposition done at −1.3 V for 60 s from a sol containing 0.17 M TEOS, 3.3 mg⋅cm−3 DSDH, respectively without ... Lire la suite
Interestingly, the biocomposite films electrogenerated onto large area electrode supports (such as electrospun platinum nanofibers [162], ordered gold macroporous inverse opal structures [156], dealloyed nanoporous gold [253], or carbon nanotubes layers [268]) led to dramatic improvement of the catalytic efficiency of the bioelectrode. This is illustrated in Figure 5C, showing clearly a much larger bioelectrocatalytic response (by ca. two orders of magnitude) when passing from a silica film with co-encapsulated DSDH and diaphorase electrodeposited on flat gold electrode (Figure 5C1) to the same biocomposite layer formed onto the internal surfaces of a macroporous gold electrode (Figure 5C2). Such advantage is attributed to the ability of the electrochemically assisted deposition process to form thin films of uniform thickness over the entire large area internal surface of the macroporous gold electrode without any pore clogging effect [156]. This is basically promising for the development of bioreactors directed to the quantitative bioconversion of the enzymatic substrates, on the condition of being able to immobilize all the necessary components (i.e., the biomolecule and its cofactor, the redox mediator) to get reagent-free bioelectrocatalytic devices (the data depicted in Figure 5C were obtained from biocomposite films entrapping only the enzymes, while the NAD+ cofactor and ferrocenedimethanol mediator were added to the solution [259]).
3.3. Co-immobilization of mediators and cofactors
In addition to biocompatibility, another interest of silica-based sol–gel materials for bioelectrochemistry purposes is the ease of chemical modification, either by physical entrapment of various electroactive and/or catalytic reagents and additives or via covalent grafting thanks to a rich organosilane chemistry, which has been largely exploited in the field of electrochemical biosensors [251, 269]. In view of developing bioelectrocatalytic reactors, this has to be extended to large area porous electrodes and the electrochemically assisted deposition may help on that matter. In particular, we have investigated the field of NAD(H)-dependent dehydrogenases operating in both oxidation and reduction modes. One already knows from above that enzyme immobilization can be achieved by physical entrapment (Figure 5). For the confinement of the necessary mediators (useful to regenerate the NAD+ and NADH cofactors [270] because their direct electrochemical reduction or oxidation may lead to enzymatically inactive or adventitious side products [271]), several strategies are available. When wishing to work in the anodic side, numerous redox polymers can be (electro)deposited—or molecular redox mediators (electro)grafted—onto electrode surfaces for electrocatalytic oxidation of NADH and thus NAD+ regeneration [272, 273]. In our group, we have explored different systems compatible with electro-induced sol–gel bioencapsulation. They encompass the use of a ferrocene-functionalized PEI polymer acting both as a polyelectrolyte stabilizing the enzyme in the silica matrix and as a mediator for NADH oxidation [211], the one-step co-entrapment of the enzyme together with carbon nanotubes wrapped with an osmium-complex modified polymer electrocatalyst [274] or modified with adsorbed vitamin K3 [260], and the electropolymerization of poly(methylene green) mediator onto the electrode surface prior to the electrodeposition of the biocomposite gel [275]. When wishing to work in the cathodic side, fewer redox mediators are available for the efficient non-enzymatic regeneration of NADH from NAD+ reduction [276, 277]. The most effective ones are rhodium complexes, such as (2,2′-bipyridyl) (pentamethylcyclopentadienyl)-Rhodium ([Cp*Rh(bpy)H2O]2+ or [Cp*Rh(bpy)Cl]+) [278, 279]. Their covalent immobilization in an active form onto an electrode surface is however challenging as it is subject to inactivation in the presence of enzymes [280] or organic ligands [281] (i.e., likely to affect the coordination chemistry of Rh centers). We found a way to solve this problem by combining diazonium electrografting, click chemistry and metal complexation, to have [Cp*Rh(bpy)Cl]+ species covalently bonded to carbon electrodes (Figure 6A), giving rise to effective electrocatalytic reduction of NAD+ (Figure 6B) [282]. Then, the DSDH enzyme can be co-immobilized, by either covalent attachment to the electrode surface [283] or entrapment in a silica layer overcoated onto the porous carbon electrode bearing the Rh mediator [284]. In both cases the enzyme and [Cp*Rh(bpy)Cl]+ species were physically separated from each other, leading to effective bioelectrocatalytic responses to d-fructose when DSDH was co-immobilized with the Rh mediator while no response was observed in the control experiment without enzyme (see respectively curves (a) and (b) in Figure 6C).
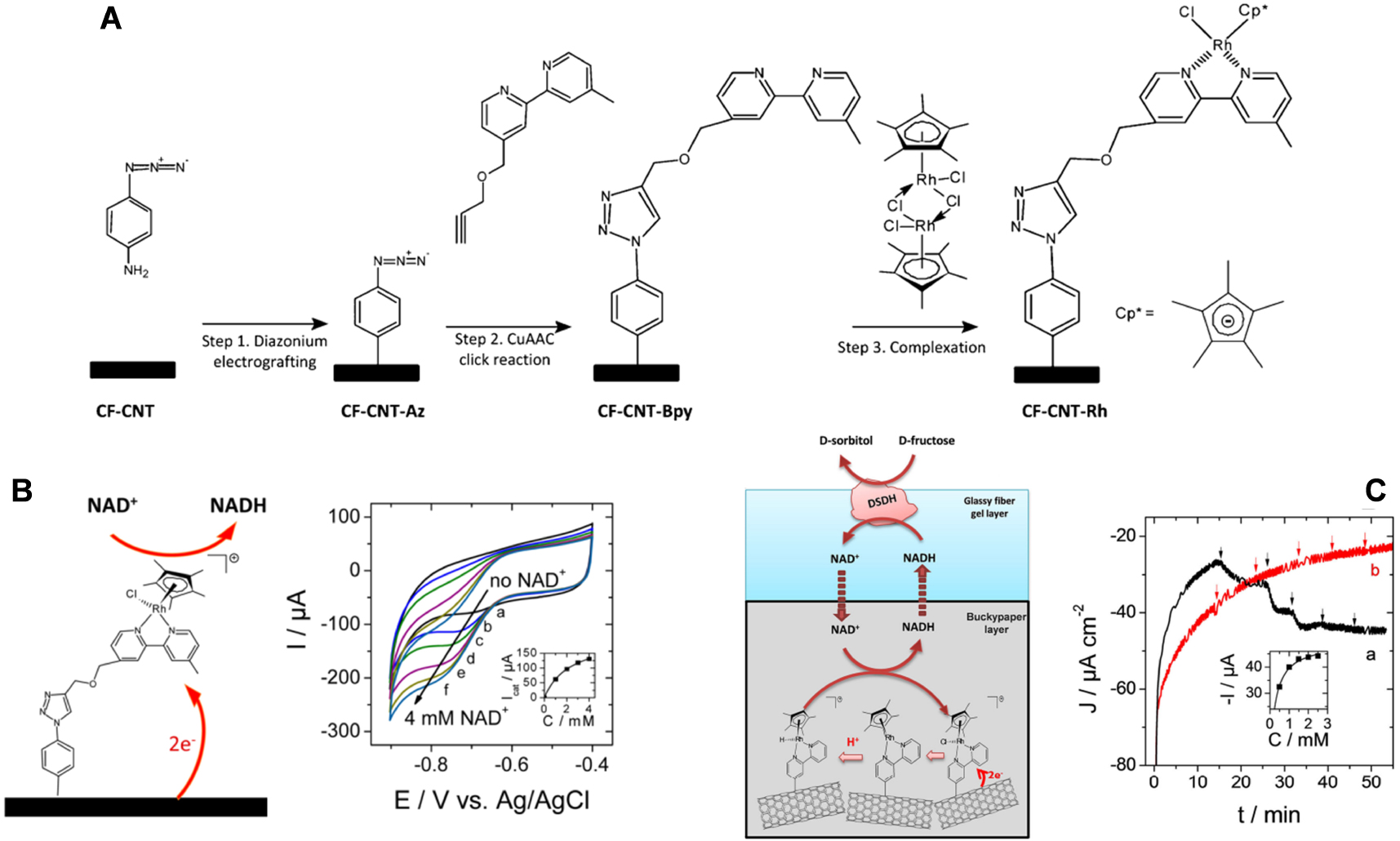
(A) Scheme illustrating the synthetic route followed for the functionalization of a carbon electrode surface with [Cp*RhIII(bpy)Cl]+. (B) NADH regeneration mediated by [Cp*RhIII(bpy)Cl]+ covalently bonded to a carbon felt–carbon nanotubes (CF-CNT-Rh) electrode. CV curves recorded at a potential scan rate of 5 mV⋅s−1 using the CF-CNT-Rh electrode in 50 mM PBS buffer (pH 6.5), under nitrogen, to gradually added NAD+: (a) 0 mM, (b) 0.5 mM, (c) 1 mM, (d) 2 mM, (e) 3 mM, and (f) 4 mM. Reprinted with permission from Zhang et al. [282], Copyright (2017) American Chemical Society. (C) Configuration and electroenzymatic process involved in the bioelectrochemical system with DSDH immobilized in a silica layer overcoated onto a bucky paper functionalized with the rhodium complex. Amperometric response to additions of 0.5 mM d-fructose (each addition is marked with an arrow) recorded using a 1 cm2 electrode (a) in the presence of DSDH in the silica layer and (b) in the absence of enzyme in the composite electrode. The measurements were performed under nitrogen at −0.72 V vs. Ag/AgCl in 50 mM PBS (pH 6.5) with 1 mM NADH in solution. From Zhang et al. [284], reproduced with permission of WILEY-VCH Verlag GmbH. Masquer
(A) Scheme illustrating the synthetic route followed for the functionalization of a carbon electrode surface with [Cp*RhIII(bpy)Cl]+. (B) NADH regeneration mediated by [Cp*RhIII(bpy)Cl]+ covalently bonded to a carbon felt–carbon nanotubes (CF-CNT-Rh) electrode. CV curves recorded at a potential scan rate ... Lire la suite
The remaining challenge is the co-immobilization of the NAD+/NADH cofactor. Indeed, its simple entrapment in the sol–gel matrix led to rapid leaching in solution and thus poor operational stability of the bioelectrode in the absence of additional protective silica overlayer [285]. Covalent bonding would lead to more durable immobilization, as shown for biosensors based on NADH bonded to water-soluble polymers [286, 287], but special care should be taken to the regioselectivity of the coupling reaction in order to maintain the enzymatic activity of the cofactor [288]. We have been able to reach the goal of durable cofactor immobilization in sol–gel bio-composite thin films by using a suitable organosilane, 3-glycidopropyltrimethoxysilane (GPS), which is likely to bind NAD+ (through the C-6 amino group rather than the N-1 ring nitrogen via Dimroth rearrangement [289]) in the course of the sol–gel process [211]. In such a way, when co-immobilized with DSDH and diaphorase (DI) enzymes and ferrocene-functionalized PEI (PEI-Fc) as mediator and polyelectrolyte, the resulting biocomposite film on GCE (Figure 7A) can be used as a reagent-less bioelectrode for the conversion of d-sorbitol (as pointed out by CV, see Figure 7B). In this case, long-term operational stability is achieved (several hours, see blue line in Figure 7C), whereas the simple entrapment of the cofactor in the film led to very fast vanishing of the bioelectrochemical response (see red line in Figure 7C) due to its fast leaching in solution [211]. This approach can be extended to other electrode supports and other immobilized mediators, such as poly(methylene green) or Os-complex modified polymer on carbon nanotubes, which were covered with an electrodeposited sol–gel silica-GPS-NAD-DSDH and applied to the reagent-less biotransformation of d-sorbitol, with good stability upon continuous use in hydrodynamic conditions [274, 290].
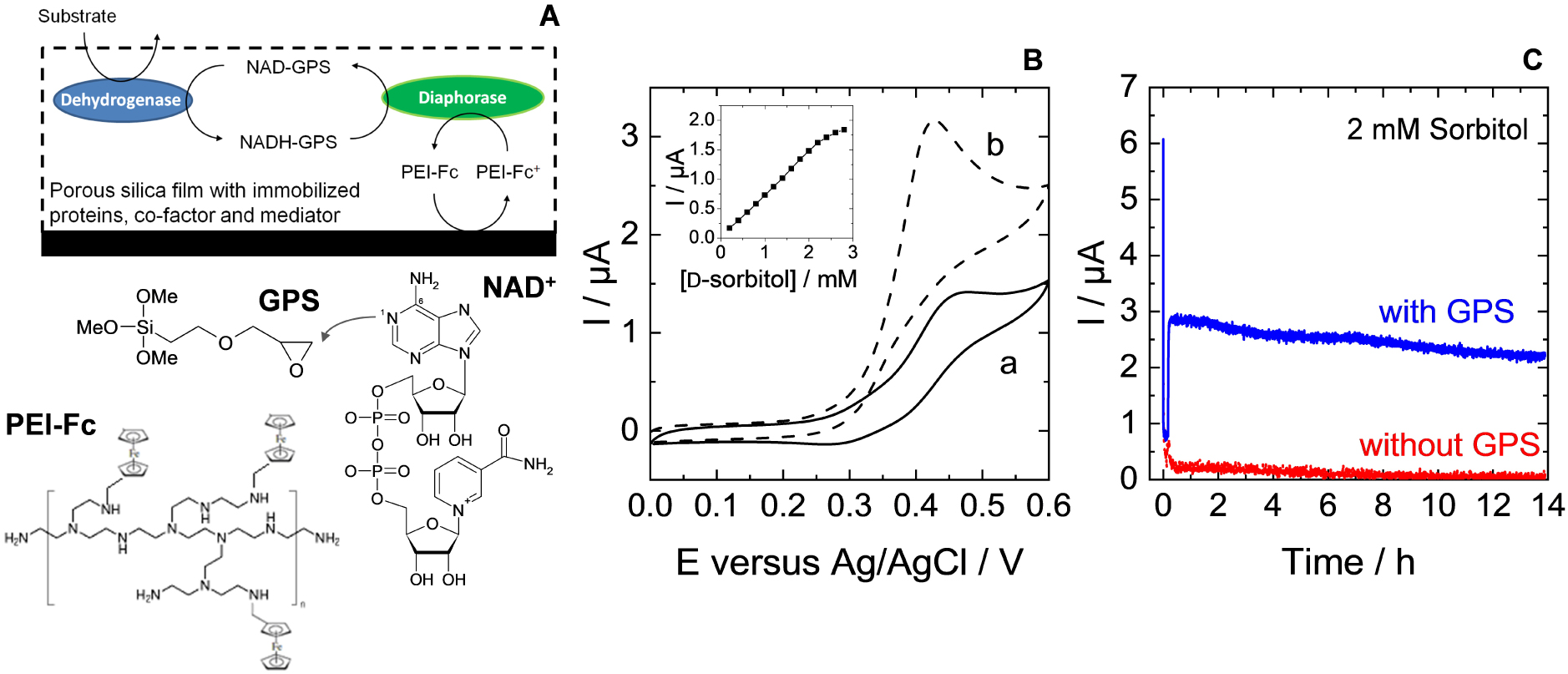
(A) Illustration of the electrochemical pathway used with the immobilized cofactor NAD+ for the detection of the dehydrogenase enzymatic substrate. (B) Cyclic voltammograms recorded with GCE/(TEOS + GPS)/(DSDH + DI)/NAD-GPS/PEI-Fc containing all elements (cofactor, mediator and proteins) in the absence of d-sorbitol and in the presence of 2.8 mM d-sorbitol. Inset plot shows the variation of peak currents sampled at 0.4 V by successive additions of 0.2 mM d-sorbitol. (C) Amperometric responses obtained at an applied potential of +0.4 V using either GCE/(TEOS + GPS)/(DSDH + DI)/NAD-GPS/PEI-Fc (i.e., in the presence of GPS, blue curve) or GCE/(TEOS)/(DSDH + DI)/NAD+/PEIFc (i.e., in the absence of GPS, red curve). Measurements have been performed for 14 h oxidation under convective conditions in 0.1 M Tris–HCl buffer (pH 9) containing 2 mM d-sorbitol. Reprinted from Wang et al. [211], Copyright (2012), with permission from Elsevier Science. Masquer
(A) Illustration of the electrochemical pathway used with the immobilized cofactor NAD+ for the detection of the dehydrogenase enzymatic substrate. (B) Cyclic voltammograms recorded with GCE/(TEOS + GPS)/(DSDH + DI)/NAD-GPS/PEI-Fc containing all elements (cofactor, mediator and proteins) in the absence ... Lire la suite
3.4. Development of bioelectrocatalytic reactors
Taking advantage of all the above features of electrochemically assisted sol–gel bioencapsulation in terms of uniformly coating large area porous electrodes with biocomposite layers and effective immobilization of all the necessary ingredients (enzymes, cofactors, mediators), we were able to design bioelectrocatalytic flow reactors. For example, a lab-scale flow bioreactor comprising a porous carbon felt electrode (16 cm2 geometric area) with CNT wrapped by poly(methylene green) (mediator) and covered with sol–gel layer comprising DSDH enzyme resulted in the quantitative transformation of d-sorbitol into d-fructose in the mM concentration range at the rather low overpotential of +0.1 V (vs. Ag/AgCl pseudo reference) [275]. If combining such bioanode to an oxygen-gas diffusion cathode, the flow bioreactor was likely to operate in an energy-producing mode as illustrated in Figure 8. It led to a lower biotransformation yield (60%), but reached an energy power output up to 14.6 μW⋅cm−3 and a conversion rate of 1.18 mg⋅day−1 in conditions of bioconversion with continuous substrate feeding [275]. Such bioreactors with enzymes encapsulated in electrodeposited silica-based materials were found to operate continuously in a stable way for a week, positioning the electro-assisted sol–gel bioencapsulation at a good place among the one-step electrochemical approaches of enzyme immobilization for bioelectrochemical applications [254]. Note that even better performance can be reached for enzymes immobilized in conducting polymers (as exemplified for the production of H2O2 in a GOx-based biocatalytic flow reactor [291]). Recent efforts are directed to the development of hydrogenase-based hybrid flow bioreactors operating in the cathodic mode with good Faradic efficiency and high turnover numbers [292, 293].
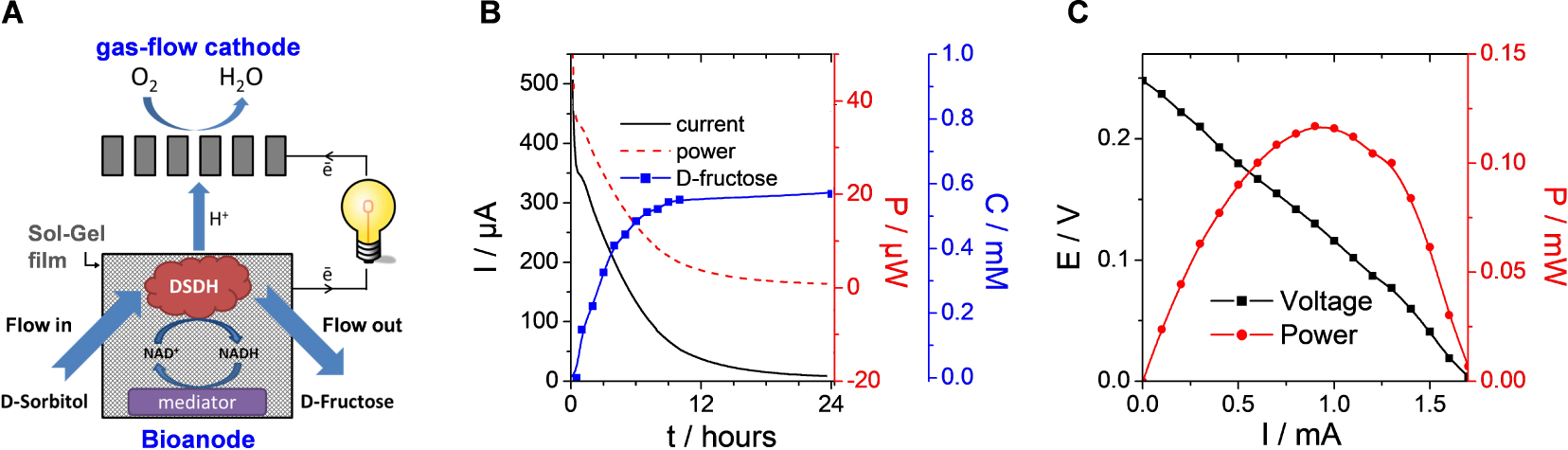
(A) Illustration of the flow bioreactor composed of a gas-flow cathode and a bioanode made of carbon felt (16 cm2) modified with carbon nanotubes, wrapped with poly(methylene green) mediator, and subsequently subjected to electrodeposition of a biocomposite layer from a sol solution containing TEOS, GPS, PEI and DSDH (the enzyme concentration in the bioanode was 0.375 mg⋅cm−3). (B) Conversion of d-sorbitol (1 mM initial concentration) to d-Fructose in the bioreactor with simultaneous energy production. The 100 mL flowing electrolyte was composed of 0.1 M Tris-HCl buffer (pH 9) and 0.1 mM NAD+. (C) Polarization curve of the bioreactor when flowing the of d-sorbitol solution. Reprinted from Mazurenko et al. [275], Copyright (2016), with permission from Elsevier Science. Masquer
(A) Illustration of the flow bioreactor composed of a gas-flow cathode and a bioanode made of carbon felt (16 cm2) modified with carbon nanotubes, wrapped with poly(methylene green) mediator, and subsequently subjected to electrodeposition of a biocomposite layer from a sol ... Lire la suite
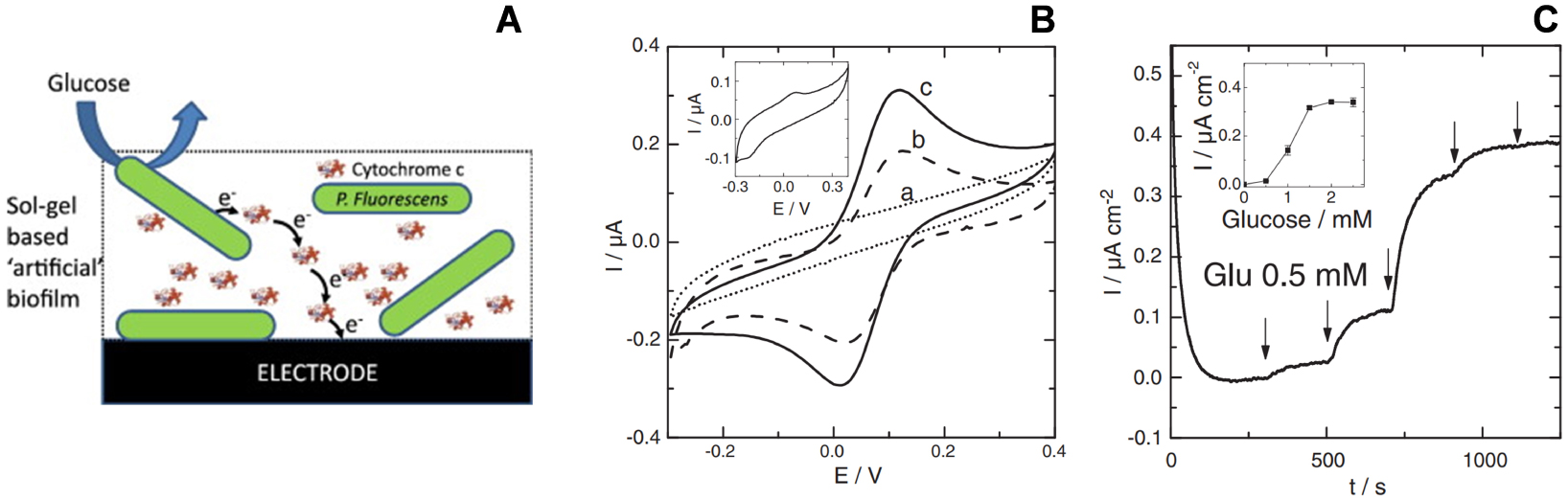
(A) Illustration of an electroactive artificial biofilm made of Pseudomonas fluorescens bacteria and cytochrome c co-immobilized in a silica sol–gel film formed by electrochemically assisted deposition on glassy carbon electrode (GCE). (B) Cyclic voltammograms for (a) bare GCE, (b) cytochrome c in a sol–gel film on GCE, (c) cytochrome c and Pseudomonas fluorescens in a sol–gel film on GCE. Inset: Cyclic voltammetric response of P. fluorescens alone in the sol–gel film. (C) Amperometric current responses measured upon the addition of glucose using GCE modified with the artificial biofilm depicted in (A). The measurements were performed under stirring in phosphate buffer (pH 7) at room temperature at an applied potential of +0.15 V vs. Ag/AgCl reference electrode. Reprinted from Ghach et al. [208], Copyright (2014), with permission from Elsevier Science. Masquer
(A) Illustration of an electroactive artificial biofilm made of Pseudomonas fluorescens bacteria and cytochrome c co-immobilized in a silica sol–gel film formed by electrochemically assisted deposition on glassy carbon electrode (GCE). (B) Cyclic voltammograms for (a) bare GCE, (b) ... Lire la suite
3.5. Encapsulation of bacteria: towards electroactive artificial biofilms
The efficient electrochemical communication between living cells and electrode surfaces is a challenge of fundamental interdisciplinary research and the immobilization of such biological/living material in an (electro)active form onto a suitable conductive support is a key prerequisite to their effective application in (bio)electrochemistry [294]. The electrochemically assisted sol–gel deposition may contribute to the field, by providing a way to encapsulate bacteria in thin silica films in order to make them more practical for use in bioelectronics devices. This was first demonstrated by forming bacteria-silica composite films, either by sol–gel electrodeposition over bacteria immobilised on ITO electrode or in one-step where the sol and the bacteria were deposited at the same time [207]. In both cases, rehalose, poly(ethylene glycol) and chitosan were added to the sol to improve cell viability. With optimum conditions, 95% cell viability was observed after one month. The proof-of-concept of sol–gel based “artificial” biofilms on electrodes was then developed using cytochrome c as a biological redox shuttle between Pseudomonas fluorescens bacteria (both entrapped in a sol–gel film, Figure 9A) and a glassy carbon electrode surface [208]. The electrochemical addressability of the bacteria in the film was evidenced from the increase in current response of cytochrome c in the presence of P. fluorescens (Figure 9B). The feasibility of bacteria/cytochrome c electron transfer in the sol–gel layer was also proven by detecting the microbial communication with the electrode upon successive additions of glucose as electron donor for respiration (Figure 9C). As shown for G. sulfurreducens cells immobilized in composite materials associating silica gel and carbon felt fibers, such ready-to-use artificial bioelectrodes are promising for the versatile time- and cost-saving development of microbial electrochemical systems [295].
3.6. Extension to the entrapment of other (non-biological) materials
Using similar approaches as for bacteria entrapment by electro-assisted sol–gel deposition, hard inorganic particles have been encapsulated into electrogenerated silica films, such as clays [189] or organoclays [190], metal oxides [193], gold nanoparticles [191, 192, 196, 296], graphene oxide [194, 195], or carbon nanotubes [170, 274, 290, 297, 298]. Again, two strategies were employed, either the electrodeposition of the silica layer onto preformed assemblies of these nano-objects onto the electrode surface [189, 190, 193, 194, 196, 274, 290, 297, 298], or the one-step co-electrodeposition of the composite film from a sol solution containing the nano-objects in suspension [170, 191, 192, 195, 296]. In the particular case of mixed polypyrrole-silica molecular imprinted polymer (PPy@sol–gel MIP) films [297, 298], one does not know what was the driving force of the composite electrodeposition process (PPy electropolymerization or sol–gel electro-assisted precipitation), even if one knows that molecularly imprinted silica films can be generated by electro-assisted sol–gel deposition [210]. Several applications have been developed by exploiting the particular properties of the nanomaterials entrapped in the silica layer and their durable immobilization on the electrode surface. They encompass electrochemical sensors (for metal ions [189], biologically relevant molecules [191], dugs or pharmaceuticals [190, 297, 298], or even the nanoparticles themselves [196]), electrocatalysis [191, 193], electrochemiluminescence [192], corrosion protection [195], or antireflection and non-linear optics [170].
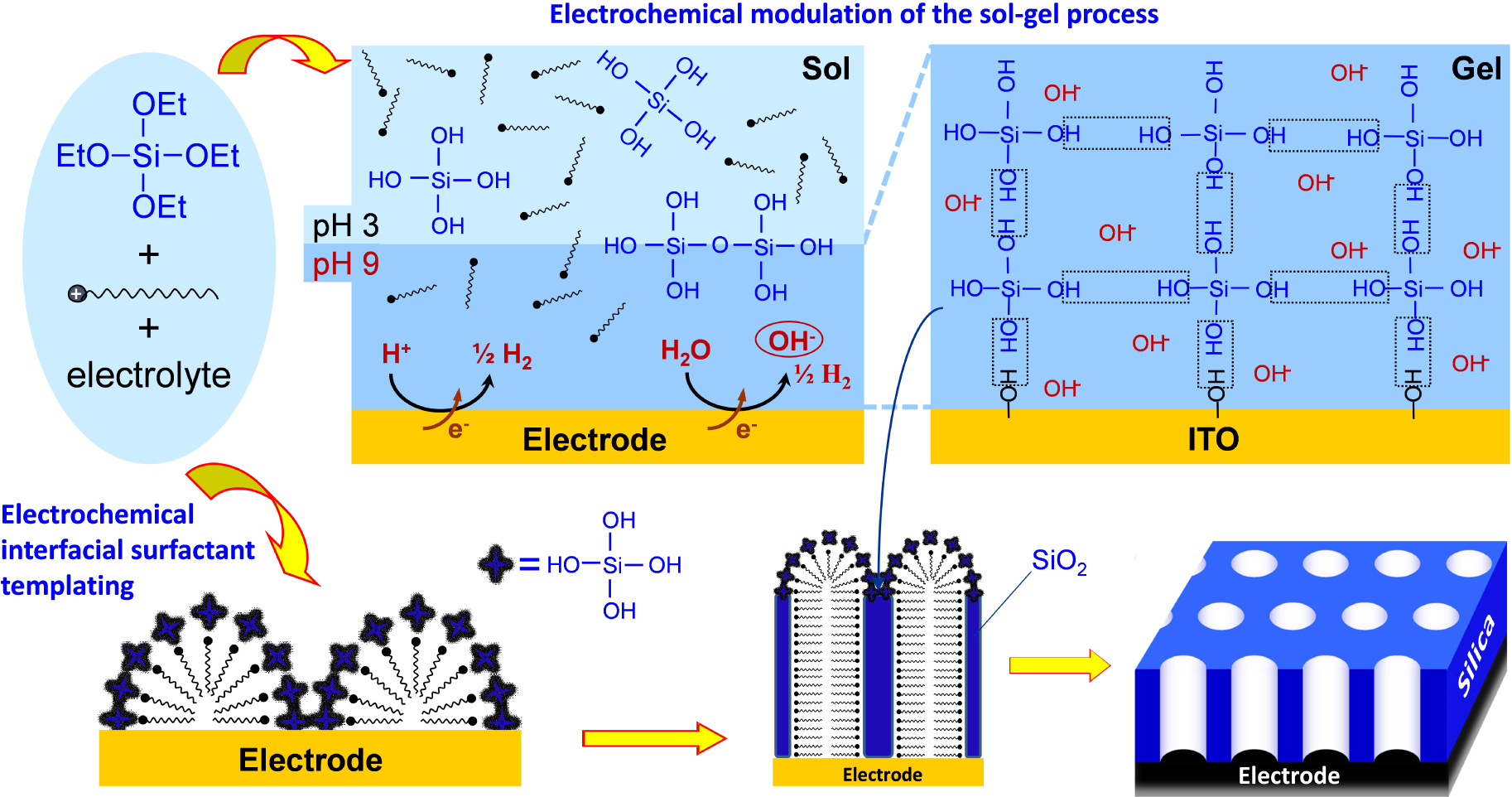
Schematic illustration of the electrochemically assisted self-assembly of a vertically aligned mesoporous silica film onto an electrode surface from a sol solution containing Si(OEt)4 precursors, a cationic surfactant and a supporting electrolyte. Reprinted with permission from Mousty and Walcarius [135], Copyright (2015) Springer.
4. Nanostructuration of electrode surfaces by electrochemically assisted deposition in the presence of surfactant templates
An important feature regarding the application of isolating thin films on electrodes is their propensity to allow efficient accessibility to the underlying conductive surface and fast transport processes through the film. For silica-based materials, this can be achieved with the aid of widely open inorganic frameworks (such as silica aerogels [299, 300]) or three-dimensional nanostructured architectures (as ordered mesoporous silica films generated by evaporation-induced self-assembly on electrodes [59, 301]. The porosity can be also created in electrogenerated silica films. The straightforward way is to work at very cathodic potentials for sol–gel electrodeposition in order to exploit the hydrogen bubbles produced during water reduction as a dynamic template in forming the silica matrix [302, 303] (as otherwise applied to the production of porous metallic foams [304, 305]). Three-dimensional macroporous and mesoporous silica films can be also obtained from the electro-assisted deposition through assemblies of polymeric particles (mainly polystyrene (nano)beads [197, 224, 306]) or dendrimers [224, 244]), among some other related approaches [47]. An elegant way to control both the film structure at the nanoscale and mesopore orientation is via the combination of electrochemical interfacial surfactant templating and electrochemical modulation of the sol–gel process, as show hereafter through the concept of electrochemically assisted self-assembly (EASA).
4.1. The electrochemically assisted self-assembly (EASA) concept: an efficient route to ordered and vertically oriented mesoporous silica films
When performing the electrochemically assisted deposition of sol–gel films using tetraethoxysilane (TEOS) in the presence of a cationic surfactant (cetyltrimethyl-ammonium bromide, CTAB) in the starting sol, the applied potential is not only responsible for the supply of the polycondensation catalysts (OH− species), but it also contributes to arrange the surfactant molecules in the form of transient hemi-micelles, thus contributing to the vertical growth of silica walls around this template [135, 137, 232], as schematically depicted in Figure 10. The EASA concept was discovered by our group in 2007 [230], and then developed and optimized by thoroughly investigating the various parameters (TEOS and CTAB concentrations, sol aging, electrolysis mode, deposition time) likely to affect the film structure, morphology and thickness [307]. It indeed enables to get quickly a continuous and uniform film covering the whole electrode surface (Figure 11A), which is made of regularly organized mesopore channels in a hexagonal geometry (Figure 11B1), all perfectly oriented perpendicular to the plane of the membrane (Figure 11B2). The hexagonal ordering and the vertically oriented mesostructure are evidenced by both electron diffraction (see inset in Figure 11B1) and grazing-incidence X-ray diffraction (GIXD) showing the presence of diffraction spots in the equatorial plane (Figure 11C). After surfactant removal (by either calcination or solvent extraction [308]), the ordered and oriented mesostructure is maintained and the porosity released, without altering the pore diameter or lattice parameter [307]. The electrodeposition can be performed potentiostatically (by applying a suitable cathodic potential) or in galvanostatic conditions (by applying a negative current). Increasing the TEOS concentration and/or lengthening the electrolysis time and/or applying a more cathodic potential (or a higher current density in the galvanostatic mode), contributed to accelerate the film growth, and thus increasing its thickness, but also resulted in the formation of a side-product (particulate aggregates) over the uniform mesoporous film (see Figure 11D1). This is also visible on the GIXD pattern (Figure 11D2) by the presence of out-of-plane rings (in addition to the in-plane spots), indicating additional materials with wormlike mesostructures. By carefully controlling the electrodeposition conditions (i.e., deposition time < 15 s; [TEOS] < 125 mM; CTAB/TEOS ratio < 0.32), however, one can avoid the precipitation of such particles. So, one does obtain uniform and aggregate-free films on various solid supports (gold, platinum, glassy carbon, ITO and copper [230, 307]), including on supports of various size and shape (microfibers [168], exfoliated graphite [309] or macroporous 3D graphene [310]), and even at the local scale (with an ultramicroelectrode generating locally the hydroxyl catalyst [174]). Applying EASA through polystyrene bead assemblies led to hierarchical macro-mesoporous films with the vertical orientation maintained throughout the whole membrane [311]. Other methods were then described to form vertically aligned hexagonal mesostructures (Stöber-solution growth [312] or oil-induced co-assembly [313]), but comparative studies by independent groups [314, 315] suggested faster and easier access to highly ordered films on electrodes using the EASA process, positioning it among the most versatile ways to ordered mesoporous films with preferential orientation control [316].

(A) FE-SEM micrograph obtained for electrodeposited surfactant-templated mesoporous silica film on ITO (cross-section view + enlargement), as generated for short time (10 s); (B) High-magnification TEM micrographs obtained for a surfactant-templated mesoporous silica film electrogenerated on glassy carbon: top view (B1) and cross-sectional view (B2); the inset in B1 shows the electron diffraction pattern of the mesostructured film; (C) Grazing-Incidence X-Ray Diffraction (GIXD) pattern of the film obtained as in (A); (D) FE-SEM micrograph (D1, top view + magnified cross-section in inset) and GIXD pattern (D2) observed for a film prepared similarly as in (A) but at longer electrodeposition time (30 s). Reproduced with permission from Walcarius et al. [230], Copyright (2007) Nature Publishing Group, and from Goux et al. [307], Copyright (2009) American Chemical Society. Masquer
(A) FE-SEM micrograph obtained for electrodeposited surfactant-templated mesoporous silica film on ITO (cross-section view + enlargement), as generated for short time (10 s); (B) High-magnification TEM micrographs obtained for a surfactant-templated mesoporous silica film electrogenerated on glassy carbon: top view ... Lire la suite
The film thickness can be basically tuned by adjusting the sol composition and the deposition parameters that are likely to control the film growth rate. However, too low TEOS or CTAB concentrations and too short deposition times prevented film formation [307], thus limiting the access to ultra-thin films, while too high TEOS or CTAB concentrations and too long electrodeposition led to the precipitation of particulate aggregates on top of the ordered and oriented mesoporous coatings (Figure 11D1). Some strategies were recently proposed to circumvent these limitations, such as the soft wet etching to decrease film thickness [317], and the alternative current-assisted deposition or hydrodynamic electrochemistry approaches to get aggregate-free films beyond microns in thickness [318, 319]. Applying EASA in sequential deposition mode enabled to form multilayered mesoporous silica films up to 400 nm in thickness without huge aggregates on top of them [320]. Most investigations made so far with EASA films were based on samples prepared using either CTAB or OTAB (octadecyltrimethylammonium bromide) as surfactant, leading to materials with pore sizes in the 2–3 nm range [128, 137, 321]. Actually, the range of accessible mesopore diameters can be extended from 1.3 to 4.4 nm by adjusting the alkyl chain length of the alkylammonium template (from C12 to C22) [128, 322]. Other cationic compounds such as alkyl-imidazolium-based ionic liquids have been used to generate mesoporous silica films with 2.5–3.5 nm wide oriented nanochannels [323, 324], the driving force of the synthesis being always the cooperative self-assembly induced by favourable electrostatic interactions between the positively charged surfactant and both the negatively charged silica precursors and the negatively charged support [325]. Such electrostatic attraction was otherwise exploited for the EASA growth of oriented mesoporous metal–organic framework thin films upon electro-assisted pH increase [326]. Finally, an important point for further practical applications is the good adhesion of the mesoporous layer onto the underlying electrode surface. If it is straightforward for silica films on hydroxide-containing electrode materials (such as ITO or FTO) thanks to their covalent bonding [307, 327], or on reduced graphene oxide thanks to strong interactions with surface oxygen-containing groups and hydrophobic π-conjugated structure [325, 328], their durable attachment to other electrodes may require suitable pre-treatments. They encompass notably the electrochemical activation of glassy carbon surfaces [329, 330], the electrografting of 3-aminopropyl-triethoxysilane (APTES) on carbon electrodes [331, 332], and the chemisorption of 3-mercaptopropyl-trimethoxysilane (MPTMS) on gold electrodes [333, 334], APTES and MPTMS acting as a “molecular glue” between the electrode surface and the electrodeposited silica layer.
4.2. Size and charge selectivity at the molecular level
Figure 12 illustrates the typical voltammetric responses obtained for redox probes of different charge (the cationic
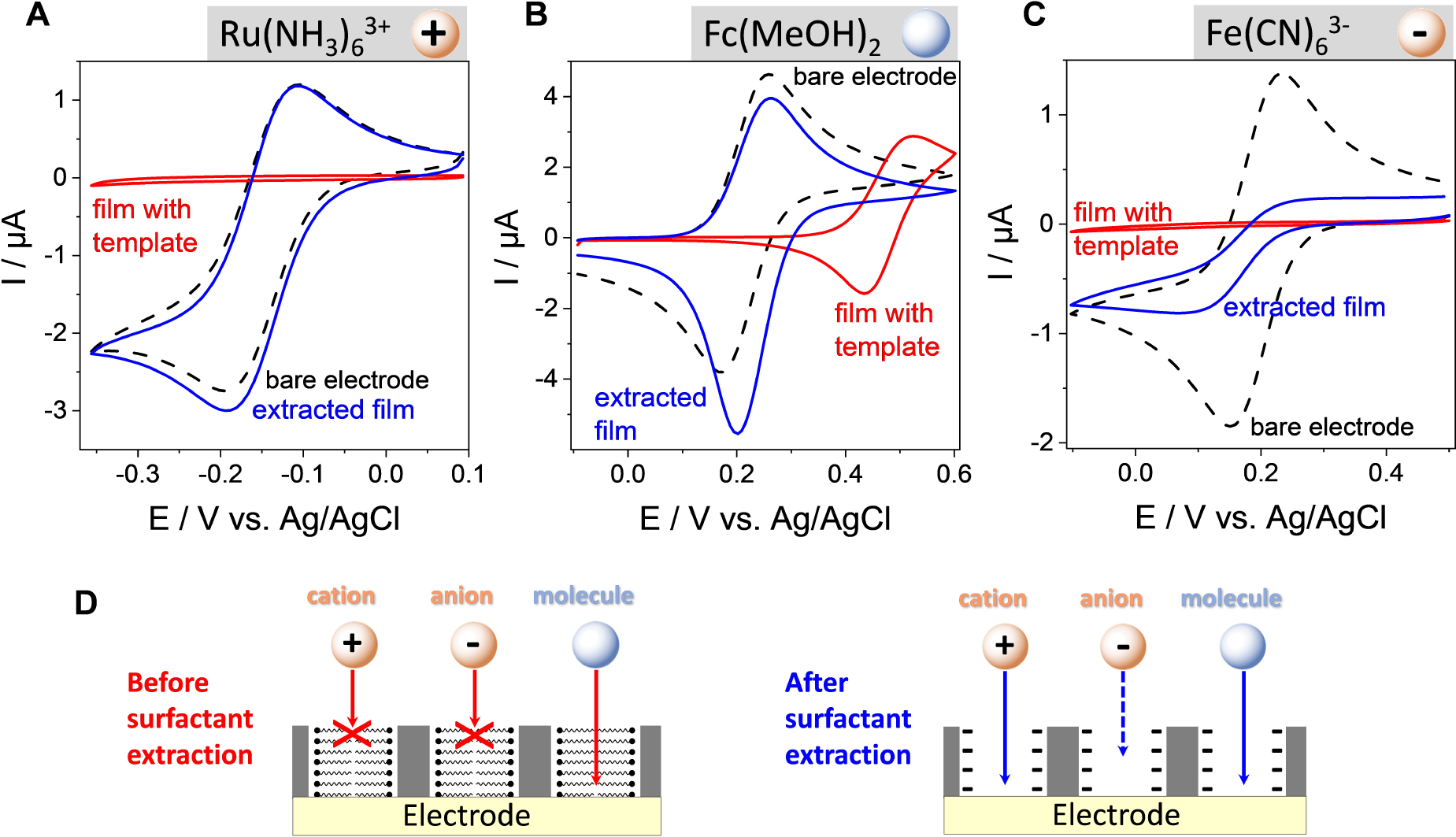
(A–C) CV curves recorded at a bare ITO electrode (curves in black) and at ITO covered with an oriented mesoporous silica film (pore ∅ = 2.0 nm) electrode, respectively before (red curves) and after (blue curves) surfactant removal using (A)
(A–C) CV curves recorded at a bare ITO electrode (curves in black) and at ITO covered with an oriented mesoporous silica film (pore ∅ = 2.0 nm) electrode, respectively before (red curves) and after (blue curves) surfactant removal using (A) ... Lire la suite
The rejection of anions is even more effective, almost totally blocking, if operating in a much diluted electrolyte medium (see black curve in Figure 13A). Actually, varying the ionic strength of the solution is a convenient way to tune the permselective properties of the film towards anions by playing with the thickness of the electrical double layer (EDL) [335]. The EDL overlaps over the whole mesopore channels in diluted electrolyte media, whereas high ionic strengths induce EDL thickness becoming smaller than the pore radius, thus making possible the free diffusion of anions through the channels (see illustrative schemes in Figure 13A) resulting correspondingly in large voltammetric signals (see blue curve in Figure 13A). The charge selectivity can be reversed by shifting pH towards lower values [336], and such effect of pH-modulated ion transport and amplified redox response is illustrated in Figure 13B for a bulky polyanionic probe (polyoxometalate, POM). As shown, a more acidic medium will induce the protonation of surface silanol groups and contribute to make the nanochannels positively charged, thus enhancing the ingress of the negatively charged POM species through the membrane and their electrochemical response on the underlying electrode (compare blue and black curves in Figure 13B). Again, these processes are influenced by the ionic strength of the medium, but also by the membrane pore size and POM probe charge [337]. Besides electrostatic effects, permselectivity can be also determined by lipophilic effects or external forces (convection, migration), and detailed discussion on the permselective behavior of silica nanochannel membranes (including those prepared by the Stöber growth method [312]) is available elsewhere [51].
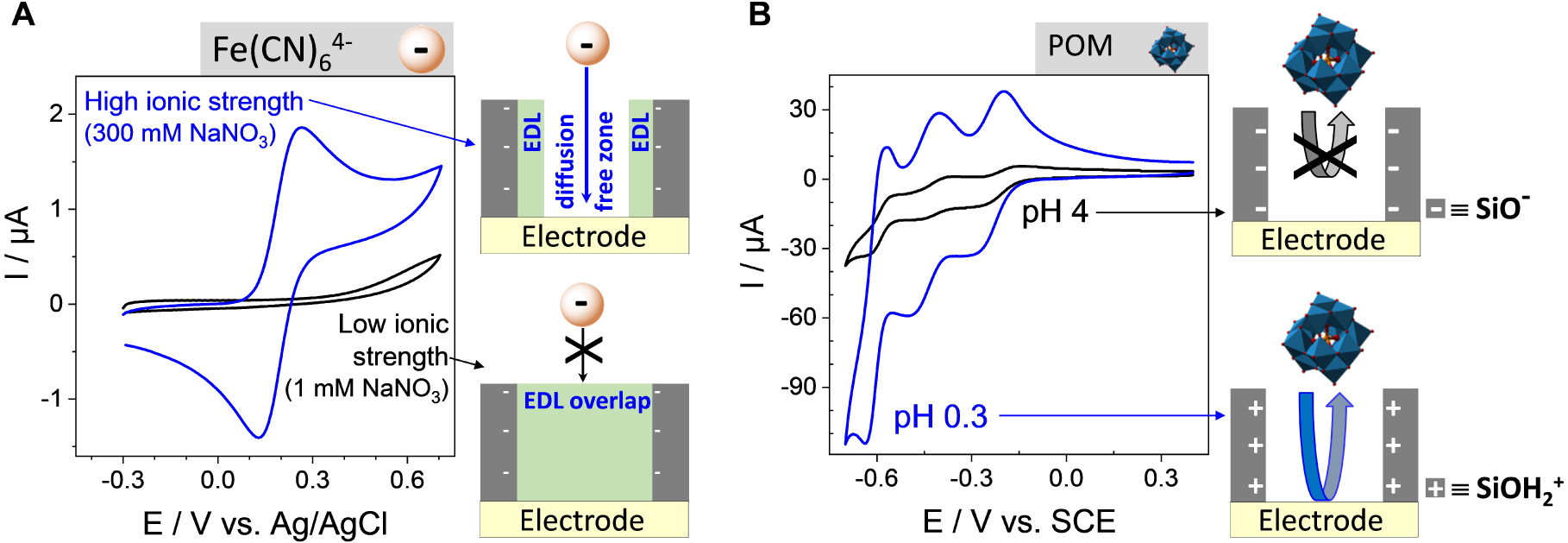
Effect ionic strength and pH on charge selectivity at ITO electrode covered with a surfactant-extracted mesoporous silica film (pore ∅ = 2.0 nm). (A) CV curves recorded at the film electrode using
Effect ionic strength and pH on charge selectivity at ITO electrode covered with a surfactant-extracted mesoporous silica film (pore ∅ = 2.0 nm). (A) CV curves recorded at the film electrode using
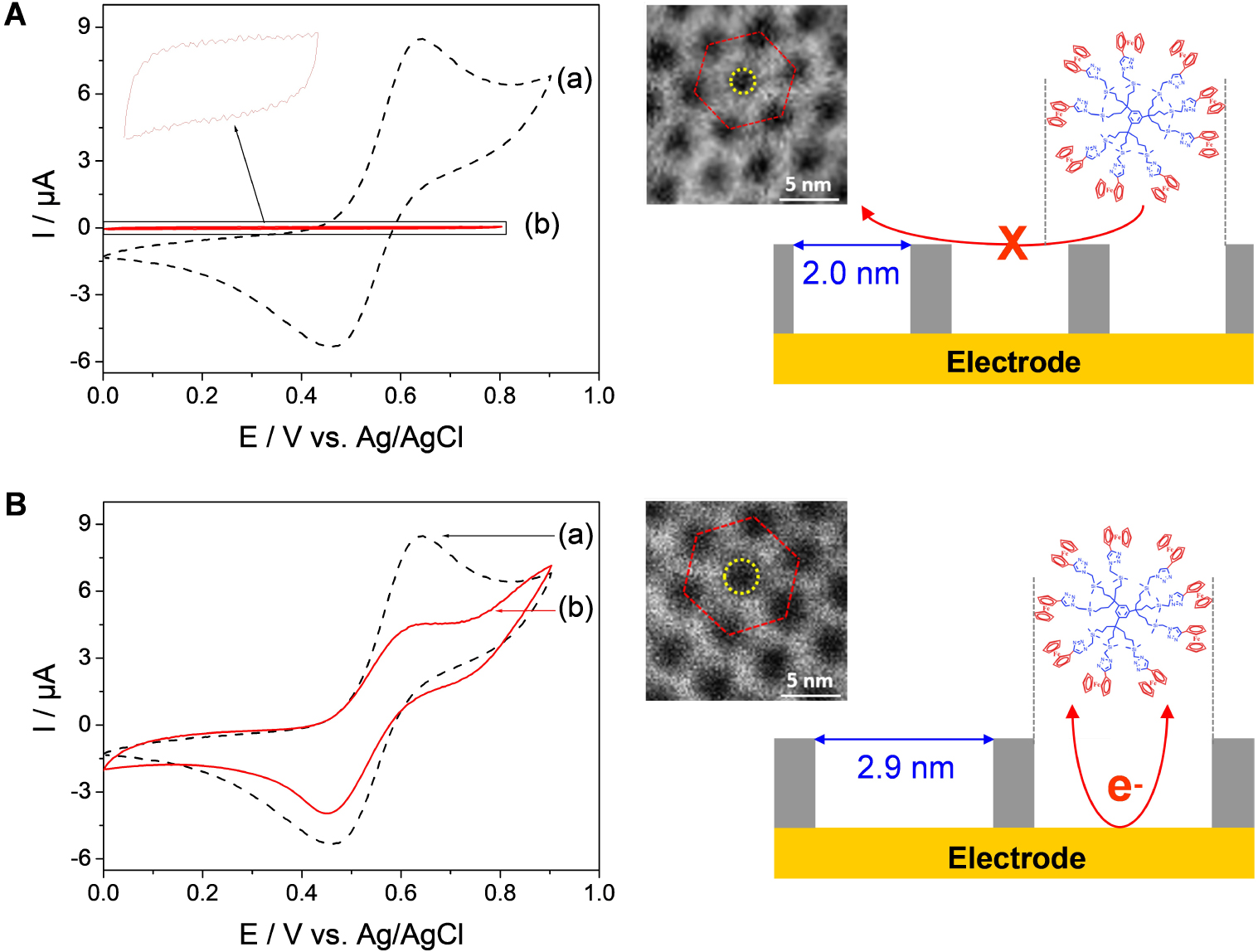
CV curves recorded in 0.12 mM 9-ferrocenyl-terminated dendrimer (in THF + 0.2 M TBABF4) using (a) bare ITO and (b) ITO electrodes modified with (A) CTAB-based and (B) OTAB-based mesoporous silica films after surfactant removal. On the right part, both high resolution STEM of the films (the sides of the red hexagon represent the d-spacing and the yellow circle one mesopore aperture) and simplified schemes of the systems have been added. Reprinted with permission from Vilà et al. [321], Copyright (2016) American Chemical Society. Masquer
CV curves recorded in 0.12 mM 9-ferrocenyl-terminated dendrimer (in THF + 0.2 M TBABF4) using (a) bare ITO and (b) ITO electrodes modified with (A) CTAB-based and (B) OTAB-based mesoporous silica films after surfactant removal. On the right part, both high resolution STEM ... Lire la suite
Another important characteristic of electrodes coated with vertically oriented mesoporous silica films with monodisperse pore diameters in the nanometer range is their ability to differentiate redox probes based on their size. Indeed, if small probes (as above) are likely to cross the membrane and give well-defined voltammetric signals (Figure 12), bigger molecules or biomolecules (such as haemoglobin, for instance) are not electrochemically accessible as they cannot reach the underlying electrode surface [338]. This imparts size selectivity to the films, at the molecular level, as better demonstrated by using an electroactive nanoferrocenyl dendrimer (with dimensions of 1.9 × 2.4 × 2.7 nm3) and two oriented mesoporous films with distinct but close pore diameters (i.e., 2.0 and 2.9 nm) [321]. As shown in Figure 14, the dendrimer gives rise to well-defined reversible voltammetric signals at a bare ITO electrode (see dashed black curves in Figure 14) but they are totally suppressed when the ITO surface is covered with a mesoporous film of 2.0 nm aperture (see red curve in Figure 14A) and partly restored for nanochannels with 2.9 nm diameter (see red curve in Figure 14B). In this latter case, the lower peak current intensities compared to bare ITO are due to some diffusional limitations for such large probe in the silica nanochannels [321]. This demonstrates unambiguously the molecular sieving properties of these oriented honeycomb structured membranes with tunable pore size. When removed from their support, this kind of thin membranes with adjustable-size vertical mesochannels are expected to be good candidates for, e.g., nanofiltration or nanosize-based separation [339, 340], or nanovalves that enable unidirectional molecular transport [341].
4.3. Functionalization of the vertically aligned nanochannels
4.3.1. Growth of nanofilaments through the mesopore channels
In the same way that we commonly use track-etched polycarbonate membranes or anodic aluminum oxide templates for designing 1D nanostructures arrays [342, 343, 344, 345], one could think about using our oriented mesoporous silica films as a mold for the electrochemical growth of nanofilaments, with the wish to reach smaller dimensions due to the nanometer-scale diameters of the vertically aligned nanochannels. Even if metal ions can be electrochemically reduced on electrodes coated with mesoporous silica films (as pointed out for copper or silver detection by anodic stripping voltammetry, for instance [245, 346]), the electrodeposition of ultra-small size metallic nanowires with macroscopic continuity remains challenging [347, 348]. A recent interesting study reports the interest of electrodeposition from a supercritical fluid to produce tin nanowires through nanoporous membranes, but too small pore diameters (<7 nm) do not allow the formation of stable nanowires in this template [349]. Actually, when attempting to electrodeposit metallic nanowires through EASA-generated films, they were usually of larger diameter than the silica nanochannels (e.g., 50 nm for palladium wires [350]), suggesting a growth out of the mesoporous membrane. An exception is the growth of individual copper nanowires from the top of the mesoporous film to the underlying electrode support (using a scanning electrochemical microscopy configuration), leading to filaments behaving as atomic contacts with improved stability when protected by the silica nanochannels than those formed on a bare electrode [351]. Nevertheless, 2–3 nm size metallic 1D nanostructures arrays that would have been electrodeposited in a stable form through oriented mesoporous silica films are not yet described. In the following paragraph, we will discuss if it would be possible for “synthetic metals”.
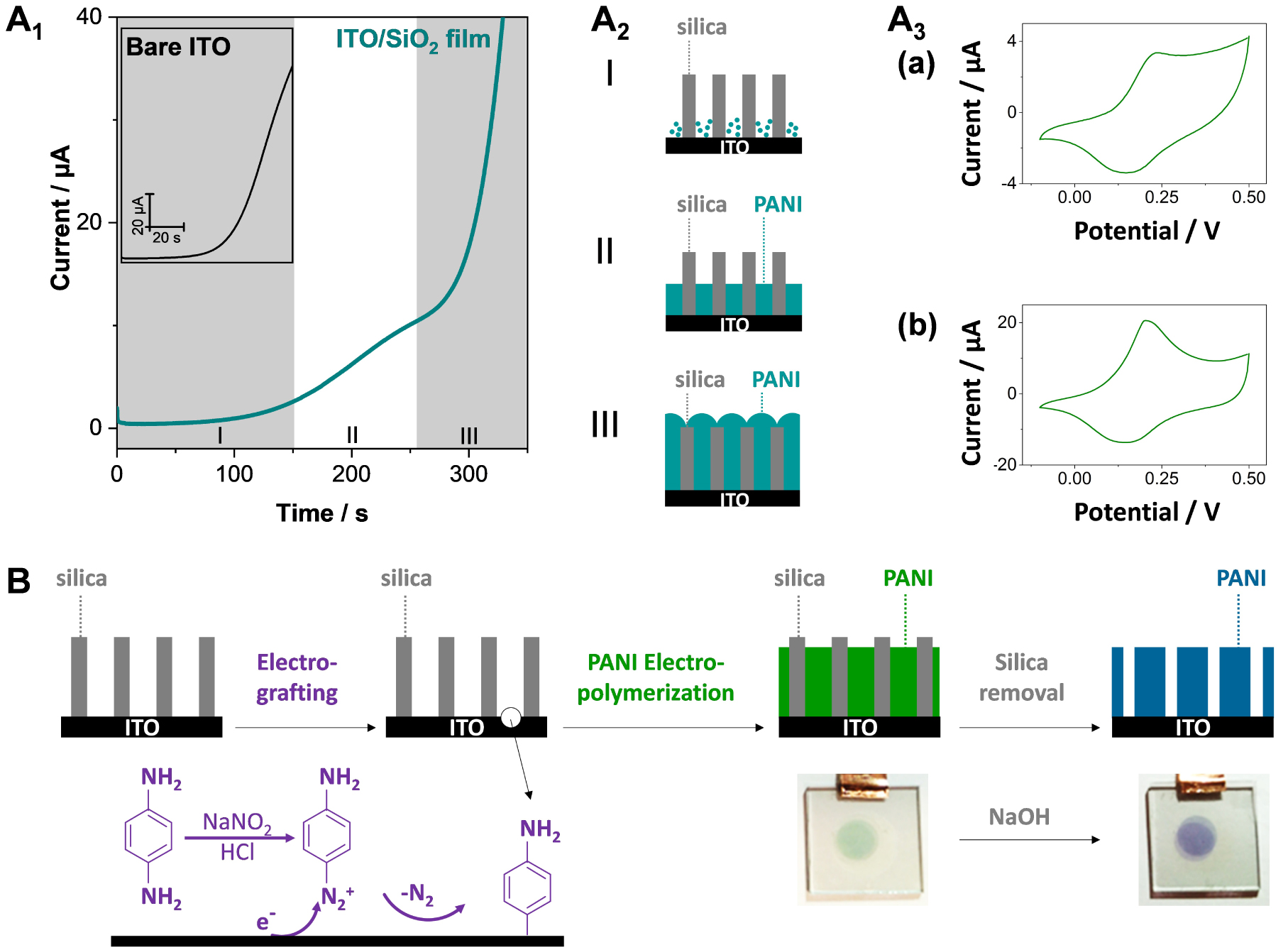
(A1) Potentiostatic electropolymerization of PANI at an ITO electrode modified with a vertically aligned mesoporous silica membrane as the hard template (up to Q = 3 mC), with the electropolymerization at a bare ITO as inset. (I) Induction and nucleation; (II) PANI nanowire formation within the mesochannels; (III) 3D PANI growth. Solution was made of 0.1 M aniline in 0.5 M H2SO4, and applied potential was +0.85 V. (A2) Schematic view of the three electropolymerization stages. (A3) CVs recorded at 50 mV⋅s−1 in 0.5 M H2SO4 after PANI electropolymerization for Q = 1.5 mC (a) and 2.7 mC (b). Reprinted with permission from Gamero-Quijano et al. [352], Copyright (2017) American Chemical Society. (B) Schematic illustration of the generation of PANI nanofilaments by combining electrografting of the monodiazonium salt of p-phenylenediamine and electropolymerization of aniline, with subsequent removal of silica to get the template-free nanofilaments (photos of the deposits are also shown). Reprinted with permission from Ullah et al. [353], Copyright (2022) The Royal Society of Chemistry. Masquer
(A1) Potentiostatic electropolymerization of PANI at an ITO electrode modified with a vertically aligned mesoporous silica membrane as the hard template (up to Q = 3 mC), with the electropolymerization at a bare ITO as inset. (I) Induction and nucleation; ... Lire la suite
Several reports are dealing with the synthesis of conducting polymers by electropolymerization using vertically oriented silica nanochannels as a template, notably for polypyrrole (PPy) [354, 355], polythiophene [356, 357] or poly(3,4-ethylenedioxythiophene) (PEDOT) [358, 359], polyaniline (PANI) [360, 352] and polyquinone [361, 362]. Sometimes the silica membrane is deposited onto a preformed layer of bulk polymer prior to nanowires growth in order to ensure a good adhesion of these latter onto the electrode support [355, 359, 362]. However, the observed diameters are mostly much larger (10–30 nm [355, 356, 363]) than the aperture of the silica nanochannels (2–3 nm), suggesting a growth outside the membrane, even if thinner deposits (ca. 3–5 nm size) are reported in some cases [357, 360, 352]. Of related interest is the electrogeneration of Prussian Blue nanowires, exhibiting an electrochemical behavior suggesting very small size but without direct evidence of the nanofilaments in the nanochannels [364]. A way to control carefully the growth of nanofilaments through oriented silica membranes is demonstrated for PANI by applying electropolymerization in the potentiostatic mode [352]. As shown in Figure 15A1, one can clearly identify three stages in the process: (I) a first nucleation step with low currents, (II) a second step corresponding to a rate-limited growth of 1D PANI nanofilaments in the nanochannels (plateau current), and (III) an exponential current increase corresponding to the 3D growth of the polymer on top of the silica membrane (see a schematic illustration of the three situations in Figure 15A2). On bare ITO, one can only observe the nucleation and 3D growth steps (Figure 15A1, inset). Once formed, PANI deposits are electroactive (see typical CV curves in Figure 15A3, corresponding to the leucoemeraldine-emeraldine redox switching of PANI nanofilaments alone (a) and with bulky deposits on top (b)), with higher peak currents in the latter case as expected from larger amounts of produced polymer. Interestingly, the anodic-to-cathodic peak-to-peak separation for the individual nanofilaments separated from each other by the silica walls was close to 60 mV, a much lower value than that observed for bulk PANI films (ca. 120–130 mV), suggesting a more reversible electrochemical behavior in the former case [352] and higher specific capacitance [365]. Unfortunately, such PANI nanofilaments are not durably attached to the underlying electrode support (ITO in this case) and are lost in solution when dissolving the silica membrane [366]. This problem can be solved by preforming a polymer layer prior to electro-assisted self-assembly of the silica membrane and electropolymerization, as aforementioned [355, 359, 362]. If wishing to have only the polymer nanofilaments covalently attached to an untreated electrode surface, however, it is necessary to develop another strategy. This is achieved by the electrografting of the monomer at the bottom of nanochannels prior to electropolymerization, as illustrated for PANI (Figure 15B). This approach is indeed successful and PANI nanofilaments stay on the electrode support after silica dissolution [366]. Same as above (Figure 15A1), one can control the nanofilaments length by tuning the electropolymerization time, as well as the amount of deposited PANI by adjusting the pore diameters of the silica membrane [353]. These silica-free PANI nanofilaments are all electrochemically accessible (ideal thin layer behavior) contrarily to those confined in the silica membrane for which some diffusional limitations do exist due to resistance to mass transport during doping-dedoping processes [353].
4.3.2. Covalent attachment of organo-functional groups
In order to extend the scope of applications of the oriented mesoporous silica films on electrodes (see Section 4.4), one should be able to get them in the form of organic–inorganic hybrids with organo-functional groups firmly attached to the silica walls. If some reagents (e.g.,
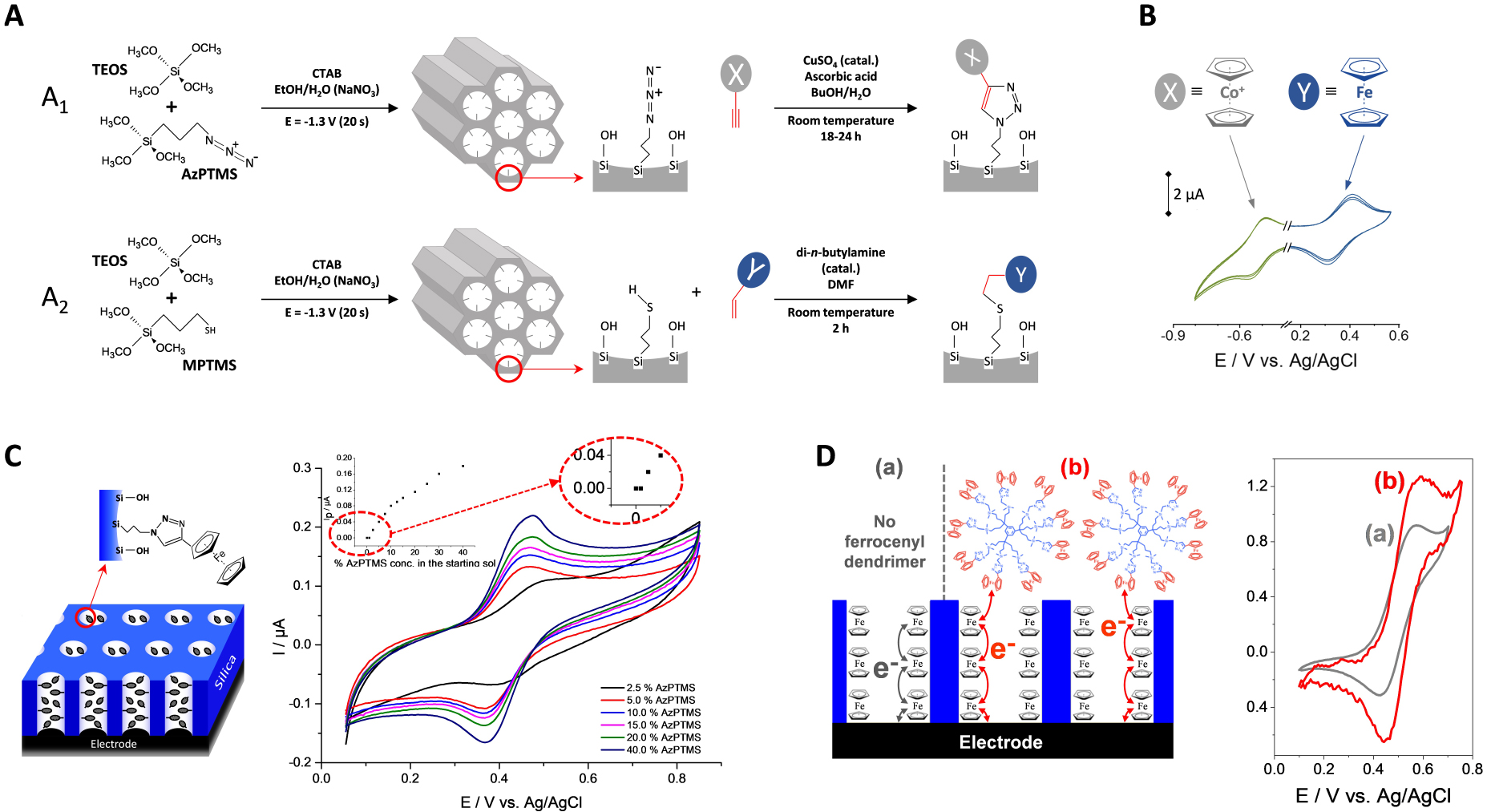
(A) Illustration of the EASA co-condensation process of (A1) AzPTMS and TEOS or (A2) MPTMS and TEOS in the presence of CTAB to get vertically aligned mesoporous silica films respectively functionalized with (A1) azide or (A2) thiol groups, which are further derivatized following (A1) the Huisgen azide–alkyne click coupling or (A2) the thiol–ene reaction. (B) Cyclic voltammograms (4 successive cycles) recorded in DMF (0.1 M TBAPF 6) using an ITO electrode modified with mesoporous silica thin films incorporating azide and thiol groups, as prepared from a sol solution containing AzPTMS/MPTMS/TEOS at the 20/20/60 molar ratio, which were subjected to Huisgen reaction using ethynylcobaltocenium and thiol–ene coupling using vinylferrocene. From Vilà et al. [247], reproduced with permission of WILEY-VCH Verlag GmbH. (C) Cyclic voltammograms recorded in acetonitrile (+0.1 M TBAClO4) at 20 mV⋅s−1 using ITO electrodes covered with ferrocene-functionalized silica films (schematically represented on the left part) as prepared from various AzPTMS/TEOS ratios (100 mM total concentration). From Vilà et al. [246], reproduced with permission of WILEY-VCH Verlag GmbH. (D) Simplified scheme illustrating the electron transfer processes in vertically-oriented ferrocene-functionalized mesoporous silica film in the absence or presence of a ferrocenyl dendrimer in solution (left) and CV curves recorded in THF (+0.2 M TBABF4) without (a) or with 0.12 mM 9-ferrocenyl-terminated dendrimer (b), using an ITO electrode modified with a CTAB-based mesoporous silica film incorporating azide groups (as obtained from a sol prepared with 20% AzPTMS and 80% TEOS) subjected to CuAAC coupling with propargyl-ferrocene, as recorded at a potential scan rate of 5 mV⋅s−1 (right). Reprinted with permission from Vilà et al. [321], Copyright (2016) American Chemical Society. Masquer
(A) Illustration of the EASA co-condensation process of (A1) AzPTMS and TEOS or (A2) MPTMS and TEOS in the presence of CTAB to get vertically aligned mesoporous silica films respectively functionalized with (A1) azide or (A2) thiol groups, which are ... Lire la suite
A more versatile and universal approach is the combination of EASA with click chemistry, as illustrated in Figure 16A. The basic idea is to generate first either azide- or thiol-functionalized mesoporous silica films by co-condensation of TEOS with 3-azidopropyltrimethoxysilane (AzPTMS) or MPTMS, respectively, and then coupling them by either the azide–alkyne Huisgen cycloaddition (Figure 16A1) or the thiol–ene reaction (Figure 16A2) to get basically any type of organic group covalently attached to the silica walls [246, 247]. One can indeed obtain highly ordered and oriented mesoporous silica films from sol solutions containing up to 40% AzPTMS [246] or 30% MPTMS [247] relative to the total silane precursors content without altering the mesostructure. The unexpectedly high azide content compatible with keeping a high level of film structuration/orientation is probably due to the zwitterionic nature of azide groups, acting as stabilizer between the positively charged surfactant molecules and the negatively charged silanolate moieties [246]. In spite of the confined space of the nanochannels, the click coupling is effective in both cases (Huisgen cycloaddition and thiol–ene reaction). Moreover, bi-functionalized films containing both azide and thiol functions can be produced by EASA and derivatized according to the two click reactions, successively in any order thanks to the different catalysts used for each coupling [247]. The organo-functional groups are covalently bonded to the silica nanochannels, randomly and uniformly distributed over the entire internal surface area of the mesopores (contrary to more heterogeneous grafting [371]), at a coverage level that can be adjusted by controlling the amount of organosilanes in the synthesis medium [246, 247]. It is also possible to attach very reactive/unstable species (such as tetrazine [372]) in a smooth way. One can even selectively functionalize one or two silica layers in multi-layered mesoporous films [320]. In principle, one could also think about other reactions, such as the amino-carboxyl or amino-aldehyde bonding’s, which are possible from amine-functionalized mesoporous silica films [373] but one would be limited by the level of functionalization (max 10% APTES to get ordered and oriented films by EASA [245]). Covalent functionalization of such oriented mesoporous films can be performed only at the pore bottom, either by grafting an hydroxylated electrode surface (as ITO) with an organo-alkoxysilane reagent prior to the electrogeneration of the silica film (as pointed out for propyl-phosphonate and cyanopropyl groups [374]) or by electrografting selectively the underlying electrode support after film formation (i.e., via electrochemical reduction of a diazonium salt, as shown for binding ferrocene units [375]). Finally, the use of organosilane precursors bearing more than one alkoxy group (such as tetra-silylated cyclam, for instance) enables the electro-assisted generation of oriented films containing the organo-functional groups inside the silica walls [315].
If redox-active moieties such as ferrocene or cobaltocenium derivatives are used to functionalize the silica walls, they are electrochemically accessible in spite of the insulating nature of the membrane, giving rise to well-defined voltammetric signals (Figure 16B). The charge transfer mechanism is based on electron hopping between adjacent redox species eventually combined to diffusion processes [246, 247, 371, 376], somewhat similar to what happens with redox polymers (polymer backbones with pendant redox centers) [377]. In the present case, the redox-active moieties are firmly attached to a rigid inorganic structure, without the possibility of motion (contrary to the chains mobility in non-reticulated redox polymers for which interplay of electron hopping and bounded diffusion is possible [378]), except perhaps via some flexibility of the binding arm. As expected, the intensity of the voltammetric peak currents is rising up with increasing the content of electroactive units immobilized in the nanochannels (see Figure 16C for the particular case of ferrocene groups clicked onto oriented mesoporous silica films prepared from increased amounts of AzPTMS). At too low concentrations of electroactive centers (i.e., for films prepared from less than 2.5% AzPTMS), the voltammetric response vanishes rapidly because the distance separating the redox units becomes too long to enable effective electron hopping [246], as otherwise reported for other assemblies of redox centers [379]. When investigating the effect of potential scan rates in cyclic voltammetry, one realizes that the charge transfer processes are mainly governed by diffusion (linear dependence of peak currents on the square root of scan rates), except at very low scan rates (<10 mV⋅s−1) where a thin-layer behavior becomes dominating [376]. In the diffusion regime, the rate-determining step is dominated by the percolation of electrons passing from one ferrocene group to another one, especially at low ferrocene contents. At higher ferrocene contents, the free space in the nanochannels becomes smaller and the overall charge transfer process (involving also charge neutralization with counter-ions) can be limited by the transport of the electrolyte anions in a so-confined environment (as an anion is necessary to compensate the positive charge generated during the oxidation of ferrocene into ferricinium). This is notably evidenced with using a large electrolyte anion (i.e., p-toluene sulfonate), contributing to much lower currents due to restricted diffusion compared to a smaller anion (i.e., perchlorate), in neutralizing the ferricinium species generated by the electrochemical oxidation of ferrocene [376]. It is therefore necessary to find the best compromise between a sufficiently high level of functionalization to fasten the charge transfer by electron hopping but not too high to enable easy mass transport during charge compensation by the electrolyte ions.
4.3.3. Long-range charge transfer through insulating hybrid films
As stated above, charge transfer reactions can occur in the insulating silica membrane, either via the presence of conducting polymer nanofilaments (Figure 15A3) or by electron hopping between redox moieties covalently bonded to the silica walls (Figure 16B,C). The question arises now: can we perform long-range charge transfer reactions through the entire thickness (∼100 nm) of the insulating silica film by taking advantage of the vertical orientation of the nanochannels filled with electroactive materials? The answer is fully yes for conducting polymer nanofilaments as they can be quantitatively and reversibly oxidized/reduced, including when the polymer is grown beyond the silica membrane [355, 353]. An additional demonstration is given by Conductive Atomic Force Microscopy (C-AFM) experiments, resulting in high conductance when approaching a Pt-coated C-AFM tip, due to the formation of molecular junctions with the vertical PANI wires in the silica nanochannels [380]. For mesoporous silica films bearing redox units covalently attached to the nanochannels, long-range charge transfer is also possible through the whole membrane, as pointed out using a size-excluded redox probe that can only be electrochemically transformed onto the top of the functionalized film [321]. For example, a vertically aligned ferrocene-functionalized film is likely to oxidize the film-excluded nonaferrocenyl dendrimer (see the increase in peak current in the presence of the dendrimer in Figure 16D). This requires the application of rather low potential scan rates (<10 mV⋅s−1) to be effective, and ca. one ferrocene out of the nine in the dendrimer is electrochemically accessible, as a result of the quite restricted charge/mass transport processes in such films (as discussed in the previous section) [321]. Anyway, it works and it can even be extended to multilayered films containing the redox units in all the superimposed layers, confirming good communication between them [320]. Much faster and reversible charge transfer can be achieved with spin crossover complexes (such as Fe(Htrz)3 (Htrz = 1,2,4,−1H-triazole)) immobilized as a coordination polymer in a mesoporous silica film generated by EASA [381]. In that case, the film growth is based on the ability of Fe(Htrz)3 to act itself as a real template (instead of the surfactant) during the self-assembly of the oriented mesostructure. Finally, the possibility of redox cycling for freely diffusing redox probes in a nanochannel membrane sandwiched between two ITO electrodes, thanks to the facilitated transport offered by the vertical orientation [382], is also of related interest but this is no more a reagent-free system.
4.4. Applications of the vertically oriented mesoporous (organo)silica films
Due to their regular, widely open and vertically oriented pore structure, the mesoporous silica thin films generated by EASA are ensuring fast transport processes from the external solution to the underlying electrode surface, making them very attractive for various applications, as illustrated in Figures 17, 18 and 19. Films with both filled pores (Figure 17), template-free open pores (Figure 18) and functionalized/grafted channels (Figure 19) have been exploited, notably for electroanalytical purposes (Table 1) but not only. The main recent developments are briefly presented and discussed hereafter.
Illustration of some electrochemical applications involving EASA-derived vertically aligned silica nanochannel membranes with filled pores. (A) Hydrophobic-based accumulation/detection and antifouling (possibility to preconcentrate small lipophilic species while rejecting large molecules/interferences and charged ions. One the right, one can see DPV curves of vertically-oriented mesoporous silica film on plasma-treated 3D graphene electrode in response to different added concentrations of uric acid in human serum and the corresponding calibration curve compared to that on bare electrode. Reprinted from Zhu et al. [383], Copyright (2022), with permission from Elsevier Science. (B) Superhydrophobic surfaces and corrosion protection (superhydrophobic film containing a corrosion inhibitor). On the right is a photograph of common liquids beading up on the surfaces of bare aluminum alloy and superhydrophobic benzotriazole-loaded CTAB-filled mesoporous silica film electrodeposited on aluminum alloy substrate and further grafted with dodecyltrimethoxysilane (liquids from left to right: top row, green tea, milk and orange juice; bottom row, coffee, Gatorade and tomato juice). Reprinted from Zhao et al. [384], Copyright (2020), with permission from Elsevier Science. (C) Electrocatalysis with coordination polymer template: Fe(Htrz)3 (Htrz = 1,2,4,−1H-triazole) confined into a mesoporous silica matrix during its formation by EASA on ITO electrode. In the center, FIA responses to three successive injections of 100 μL aliquots of H2O2 (in an aqueous carrier solution of 0.1 M NaNO3 flowing at 1 mL⋅min−1) at 30, 150 and 300 μM; on the right, CV curves recorded at various potential scan rates (in the 0.2–1.2 V⋅s−1 range) in 0.1 M NaNO3 aqueous solution, as depicted on a TEM view of the mesoporous film. Adapted from Ahoulou et al. [385], with permission of WILEY-VCH Verlag GmbH, and [381], Copyright (2019) American Chemical Society. (D) Electropolymerized nanowires of polyaniline (PANI) that are formed in the vertical nanochannels and stood free by dissolution of the silica membrane: electrochromic response and photographs at switching potentials (20 s each) of +0.3 V (colouration) and −0.2 V (bleaching), and cyclic voltammograms recorded at various potential scan rates from 10 to 200 mV⋅s−1 (every 10 mV⋅s−1). Adapted with permission from Ullah et al. [353], Copyright (2022) The Royal Society of Chemistry. Masquer
Illustration of some electrochemical applications involving EASA-derived vertically aligned silica nanochannel membranes with filled pores. (A) Hydrophobic-based accumulation/detection and antifouling (possibility to preconcentrate small lipophilic species while rejecting large molecules/interferences and charged ions. One the right, one can see DPV ... Lire la suite
Electrochemical sensors designed from EASA-based mesoporous silica thin films on electrode
| Analytea | Electrode configurationb | Detection procedurec | Techniqued | Analytical performance | Ref. | |
|---|---|---|---|---|---|---|
| Concentration range | Det. limit | |||||
| Acetaminophen | GCE/rGO/VMSF | Acc.–Det. | DPV | 2 × 10−7–1.0 × 10−4 M | 9.7 × 10−9 M | [386] |
| Norepinephrine | 1 × 10−7–3.0 × 10−5 M | 1.4 × 10−8 M | ||||
| Tryptophan | 8 × 10−7–2.0 × 10−4 M | 3.6 × 10−8 M | ||||
| Ag+ | CF/(VMSF-SH) | Acc.–Det. | DPV | 2.5 × 10−6–2.0 × 10−6 M | - | [168] |
| 4-Aminophenol | ITO/VMSF | Acc.–Det. | DPV | 5 × 10−7–4.0 × 10−4 M | 3.2 × 10−7 M | [386] |
| Ascorbic acid | AuE/(VMSF-GPS- DNAzyme) | Indirect det. | SWV | 1.0 × 10−6–1.2 × 10−4 M | 7.0 × 10−7 M | [387] |
| Cu2+ | 310 ppb–4.5 ppm | 250 ppb | ||||
| tert-Butyl- hydroquinone | GCE/ErGO/VMSF | Acc.–Det. | DPV | 1 × 10−9–5 × 10−7 M & | 2.3 × 10−10 M | [388] |
| 5 × 10−7–1.2 × 10−4 M | ||||||
| Carbendazim | GCE/BN-rGO/VMSF | Acc.–Det. | DPV | 5 × 10−9–7 × 10−6 M | 2.3 × 10−9 M | [389] |
| Catechol | AuE/ErGO/ (VMSF-CTAB) | Acc.–Det. | DPV | 1 × 10−6–2.0 × 10−5 M & | 1.7 × 10−8 M | [390] |
| 2.0 × 10−5–6.0 × 10−5 M | ||||||
| Hydroquinone | AuE/ErGO/VMSF | 1 × 10−6–6.0 × 10−5 M | 1.5 × 10−8 M | |||
| CA15-3 | ITO/(VMSF-GPS-Ab, Ru(bpy) |
Genosensing | ECL | 0.0001–100 U⋅mL−1 | 9 μU⋅mL−1 | [391] |
| DPV | 0.01–100 U⋅mL−1 | 5.4 mU⋅mL−1 | ||||
| CEA | ITO/(VMSF-Ru(bpy) |
Genosensing | ECL | 0.0001–50 ng⋅mL−1 | 0.06 pg⋅mL−1 | [392] |
| CA15-3 | DPV | 0.005–500 U⋅mL−1 | 2.3 mU⋅mL−1 | |||
| C-reactive protein | SPCE/ErGO/ (VMSF-NH2)/GluA-Ab | Genosensing | DPV | 0.01–100 ng⋅mL−1 | 8 pg⋅mL−1 | [393] |
| Chlorpromazine | SPCE/ErGO/VMSF | Acc.–Det. | DPV | 3 × 10−7–2.3 × 10−5 M & | 6.1 × 10−9 M | [394] |
| 10−6–10−5 M (blood) | 1.6 × 10−8 M | |||||
| Clindamycin | PET-ITO/ (VMSF-Ru(bpy) |
Direct det. | ECL | 10 nM–25 μM & | 4 nM | [395] |
| 25–70 μM & | ||||||
| Clozapine | ep-SPCE/VMSF | Acc.–Det. | DPV | 50 nM–20 μM | 2.8 × 10−8 M | [396] |
| Cu2+ | GCE/(clay + VMSF) | Acc.–Det. | SWV | 1 × 10−7–1.0 × 10−6 M | - | [189] |
| Cu2+ | GCE/(VMSF-NH2) | Acc.–Det. | ASDPV | 1 × 10−8–1.0 × 10−6 M | 3 × 10−9 M | [245] |
| Cysteine | ITO/(VMSF-Fc) | Mediated EC | A, FIA | 3 × 10−6–1.0 × 10−4 M | - | [222] |
| Diclofenac | GCE/APTES/ (Amino-AT + VMSF) | Acc.–Det. | SWV | 3 × 10−7–2.0 × 10−5 M | 5.3 × 10−8 M | [190] |
| DNA | AuE/(VMSF-GPS- DNA Ab) | Genosensing | CV | 5 × 10−9–7.0 × 10−7 M | 2.5 × 10−9 M | [395] |
| Dopamine | GCE/VMSF | Acc.–Det. | DPV | 10 nM–1.0 μM & | 0.88 nM | [329] |
| 0.05–1.0 μM (serum) | ||||||
| Dopamine | AuE/(VMSF-GQDs) | Acc.–EC det. | DPV | 200 nM–20 μM & | 120 nM | [397] |
| 20–100 μM | ||||||
| Dopamine | GCE/(VMSF-NMe |
Acc.–Det. | DPV | 2.0 × 10−5–2.26 × 10−4 M | 9 × 10−6 M | [398] |
| Ascorbic acid | 4.9 × 10−5–2.65 × 10−3 M | 1.1 × 10−5 M | ||||
| Doxorubicin | GCE/VMSF | Acc.–Det. | DPV | 5 × 10−10–2.3 × 10−5 M | 2 × 10−10 M | [399] |
| Doxorubicin | GCE/ErGO/VMSF | Acc.–Det. | DPV | 1 × 10−9–2.0 × 10−5 M | 7.7 × 10−10 M | [400] |
| Guanine | ITO/(VMSF-Ru(bpy) |
EC | CV | 2 × 10−7–5.0 × 10−4 M | 5.8 × 10−8 M | [401] |
| Hydrogen peroxide | ITO/(VMSF-PB wires) | Mediated EC | CV | 1 × 10−4–1 × 10−2 M | [364] | |
| Hydrogen peroxide | ITO/(VMSF-Fe(Htrz)3) | Mediated EC | A, FIA | 5 × 10−6–3.0 × 10−4 M | 2 × 10−6 M | [385] |
| Hydrogen peroxide | CFME/(VMSF-PtNS) | EC | A | 5 × 10−4–2.0 × 10−3 M | - | [402] |
| Imidacloprid | GCE/ErGO/VMSF | Acc.–Det. | CV | 1.0–50 μg⋅mL−1 & | 0.3 μg⋅mL−1 | [403] |
| 50–400 μg⋅mL−1 | ||||||
| Insulin | GCE/APTES/ (VMSF-NH2)/ GluA-cDNA-Apt. | Aptasensing | DPV | 1.0 × 10−8–3.5 × 10−7 M | 3.0 × 10−9 M | [404] |
| Isoproturon | GCE/APTES/VMSF | EO amplif. | SWV | 1.0 × 10−5–1.0 × 10−4 M | - | [405] |
| Lidocaine | FTO/(VMSF-Ru(bpy) |
Direct det. | ECL | 1.0 × 10−8–5.0 × 10−5 M | 8 × 10−9 M | [327] |
| Methidazine | 3DG/VMSF | Acc.–EC det. | DPV | 5.0 × 10−8–1 × 10−5 M | 3.0 × 10−8 M | [406] |
| Nitrate | ITO/(VMSF-Fc) | Indirect det. | IC-A | 2.0 × 10−5–1.0 × 10−4 M | 1.6 × 10−5 M | [407] |
| Fluoride | 5 × 10−6–5.0 × 10−5 M | 3 × 10−6 M | ||||
| Chloride | 5 × 10−6–5.0 × 10−5 M | 4 × 10−6 M | ||||
| Bromide | 1.0 × 10−5–7.5 × 10−5 M | 1.0 × 10−5 M | ||||
| Phosphate | 2.0 × 10−5–1.0 × 10−4 M | 1.2 × 10−5 M | ||||
| Sulfate | 1.5 × 10−5–1.0 × 10−4 M | 1.0 × 10−5 M | ||||
| Paraquat | GCE/APTES/VMSF | Acc.–Det. | SWV | 1.0 × 10−8–1.0 × 10−5 M | 4 × 10−9 M | [332] |
| Paraquat | GCE/3DG-CNT/VMSF | Acc.–Det. | DPV | 2 × 10−9–1.0 × 10−8 M & | 1.17 × 10−9 M | [408] |
| 1.0 × 10−8–1.0 × 10−5 M | ||||||
| PSA | ITO/(VMSF-NH2)/ GluA-Ab) | Genosensing | DPV | 10 pg⋅mL−1–1 μg⋅mL−1 | 8.1 pg⋅mL−1 | [409] |
| PSA | ITO/VMSF/GPS-Ab | Genosensing | ECL | 1 pg⋅mL−1–100 ng⋅mL−1 | 0.1 pg⋅mL−1 | [410] |
| PSA | AuE/(VMSF-GPS-PSA Apt.) | Aptasensing | DPV | 1–300 ng⋅mL−1 | 0.28 ng⋅mL−1 | [411] |
| Rutin | GCE/ErGO/VMSF | Acc.–Det. | DPV | 3 × 10−7–2 × 10−6 M & | 2.3 × 10−9 M | [412] |
| 2 × 10−6–4.0 × 10−5 M | ||||||
| Serotonin | ITO/ErGO-CNT/VMSF | Acc.–Det. | DPV | 1 × 10−7–1.0 × 10−5 M | 5.4 × 10−9 M | [413] |
| 1.0 × 10−5–3.0 × 10−5 M | ||||||
| Melatonin | 1 × 10−6–2.0 × 10−5 M | 1.41 × 10−8 M | ||||
| 2.0 × 10−5–1.0 × 10−4 M | ||||||
| 2,4,6-trichloro- phenol | 3DG/VMSF | Acc.–EC det. | DPV | 1.0 × 10−8–1 × 10−7 M & | 2.4 × 10−9 M | [414] |
| 1 × 10−7–1.5 × 10−5 M | ||||||
| Uric acid | pl-3DG/(VMSF-CTAB) | Acc.–Det. | DPV | 100 nM–30 μM & | 2.3 × 10−8 M | [383] |
| 30–150 μM | ||||||
| Uric acid | SPCE/ErGO/(VMSF-NH2) | Acc.–Det. | DPV | 5 × 10−7–1.80 × 10−4 M | 1.29 × 10−7 M | [415] |
aAbbreviations: CA15-3, cancer antigen 15-3; CEA, carcino-embryonic antigen; DNA, deoxyribonucleic acid; PSA, prostate specific antigen. bAbbreviations: GCE, glassy carbon electrode; rGO, reduced graphene oxide; VMSF, vertically-oriented mesoporous silica film; CF, carbon fiber; VMSF-SH, thiol-functionalized VMSF; ITO, indium-tin oxide; AuE, gold electrode; GPS, (3-glycidopropyl) trimethoxysilane; DNAzyme, deoxyribozyme; ErGO, electrochemically reduced graphene oxide; BN-rGO, boron nitride-reduced graphene oxide; CTAB, cetyltrimethylammonium bromide; Ab, antibody; Ru(bpy)
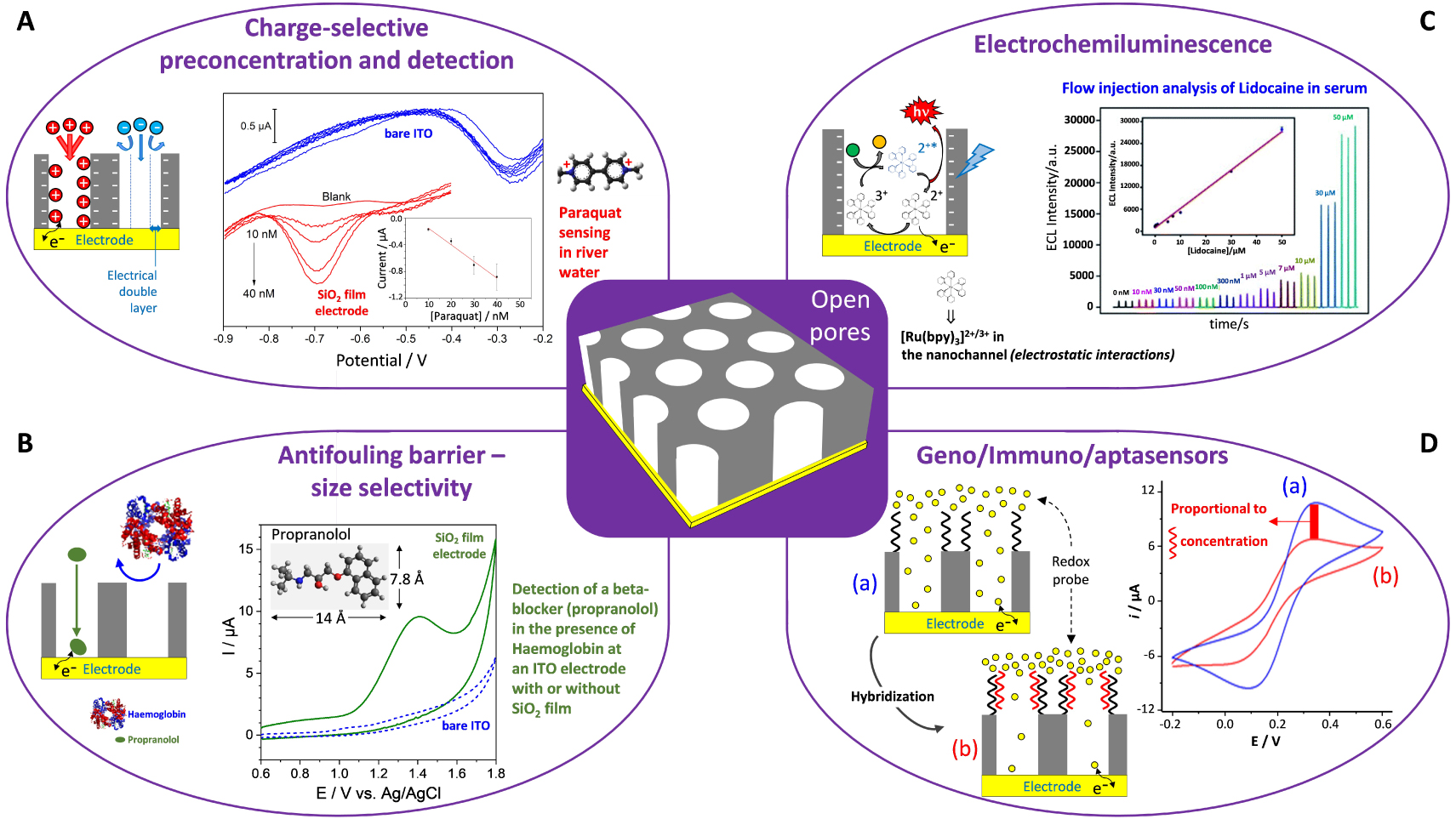
Illustration of some electrochemical applications involving EASA-derived vertically aligned silica nanochannel membranes with open pores. (A) Charge selective preconcentration and detection (possibility to accumulate cations while rejecting anions): paraquat sensing by square wave voltammetry recorded in Meuse River water samples at bare (blue) and modified (red) electrodes, respectively, after spiking with of 0, 10, 20, 30, and 40 nM paraquat (Inset: peak current for paraquat added concentrations of 10, 20, 30, and 40 nM at the modified electrode). Reprinted from Nasir et al. [332], Copyright (2018) American Chemical Society. (B) Antifouling barrier—size selectivity (possibility to detect small species while rejecting the big ones): cyclic voltammetry detection of a beta-blocker (propranolol, at 100 μM) in the presence of Haemoglobin (5 μM) at an ITO electrode with (plain green curve) or without SiO2 film (dashed blue curve). Reprinted from Serrano et al. [338], Copyright (2015), with permission from Elsevier Science. (C) Electrochemiluminescence of transparent FTO electrode coated with a mesoporous silica film bearing
Illustration of some electrochemical applications involving EASA-derived vertically aligned silica nanochannel membranes with open pores. (A) Charge selective preconcentration and detection (possibility to accumulate cations while rejecting anions): paraquat sensing by square wave voltammetry recorded in Meuse River water samples ... Lire la suite
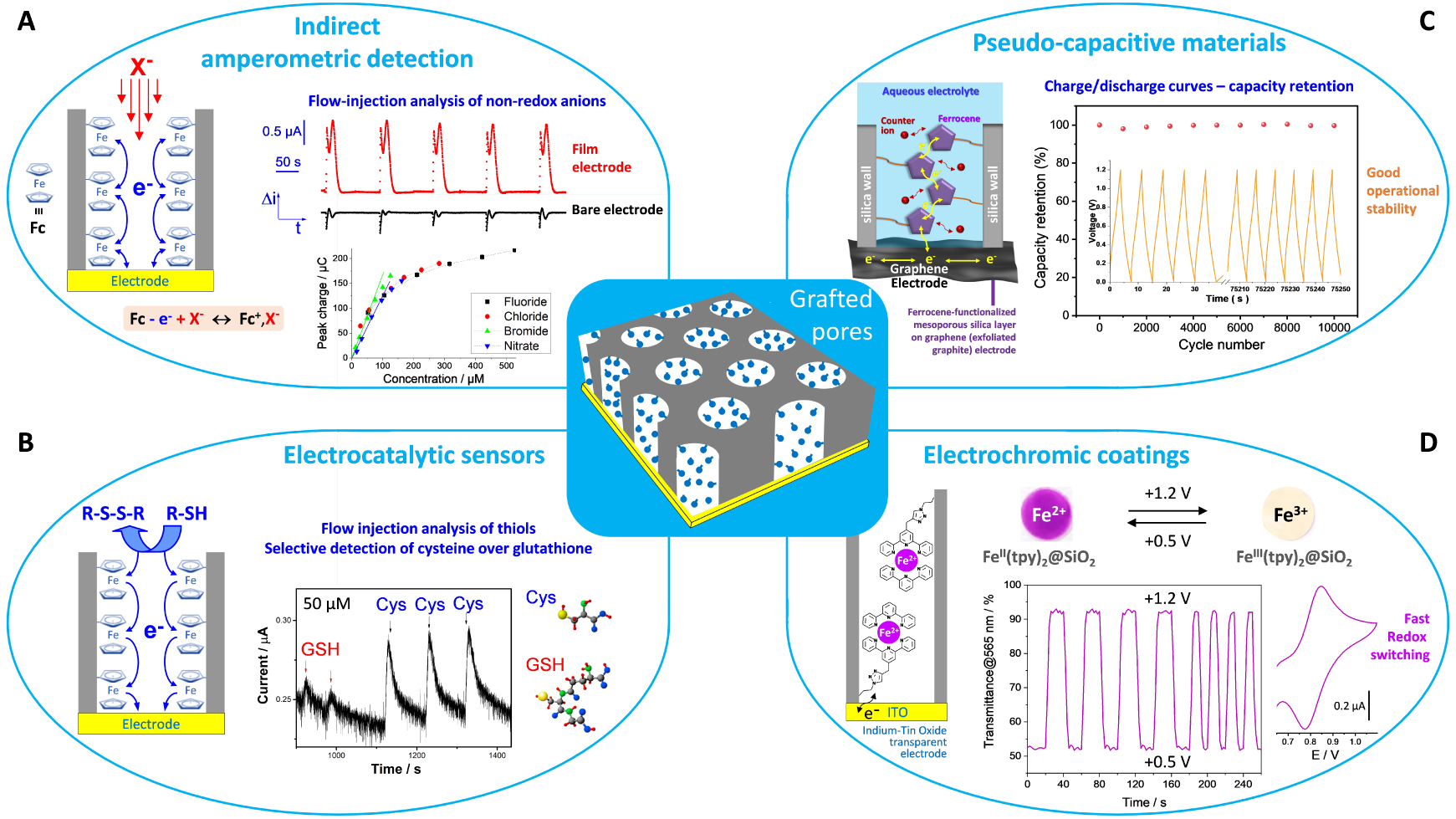
Illustration of some electrochemical applications involving EASA-derived vertically aligned silica nanochannel membranes with grafted pores. (A) Indirect amperometric detection of non-electroactive anions (the oxidation of ferrocene needs an anion to neutralize the generated ferricinium cation). On the top right part, one can see the square wave amperometric detection of 0.1 mM
Illustration of some electrochemical applications involving EASA-derived vertically aligned silica nanochannel membranes with grafted pores. (A) Indirect amperometric detection of non-electroactive anions (the oxidation of ferrocene needs an anion to neutralize the generated ferricinium cation). On the top right part, ... Lire la suite
4.4.1. Preconcentration electroanalysis and permselective coatings
The charge and size selectivity (Figures 12–14) imparting permselectivity properties to these films can be exploited for the accumulation/rejection of target species/interference. Neutral analytes such as biological molecules, drugs or pharmaceuticals [418, 390], on one hand, and pollutants or hazardous species such as pesticides or explosives [419, 420] on the other hand, are likely to preconcentrate into the hydrophobic surfactant phase of the films. This process is quite selective (ionic species and large interferent molecules being excluded), enabling their determination in real media (whole blood, serum, urine, environmental samples, foodstuffs, etc.). An illustrative example is the determination of uric acid in in serum, showing clearly the accumulation effect of the film compared to the bare electrode (Figure 17A), with an anti-interference and antifouling behavior in such a complex matrix [383]. The hydrophobic-based accumulation/detection is usually characterized by calibration curves with two linear dynamic concentration ranges of different sensitivities due to more effective preconcentration from diluted samples [390]. This is also the case for surfactant-extracted films that are likely to accumulate cationic species by favorable electrostatic interactions, as pointed out for paraquat sensing in river water (Figure 18A) [332]. At low concentration (nM to μM range) the voltammetric signal is directly proportional to the square root of potential scan rate (pure adsorption behavior) whereas it becomes more and more diffusion-controlled at higher paraquat concentrations (mM range) [421], explaining the existence of the two dynamic ranges in the calibration [408]. Various organic molecules have been determined by accumulation/detection at EASA-derived mesoporous films, mainly by differential pulse voltammetry (Table 1), including drugs or pharmaceuticals [394, 396, 399, 400, 406, 386], biologically relevant molecules [329, 386, 388, 413], organic dyes [422, 412] and pesticides [389, 403]. Other examples are available for similar films prepared by the Stöber growth method [51], notably based on the modulation of their permeability properties (by the presence or absence of surfactant in the nanochannels) for the direct analysis of both hydrophilic and lipophilic species (as pointed out for antioxidants detection in fruit juice [423]). The question of long-term operational stability was not often considered, which should be rather low for such thin walls made of amorphous silica gel, being evidently not stable at all at high pH [394]. Sometimes, additives like clay particles were added to the film to improve somehow the accumulation efficiency [189, 190]. The covalent bonding of organo-functional groups (e.g., thiol [168], amine [245, 415] or alkylammonium [398]) to the mesoporous silica films is a possibility to enhance further the performance in terms of better sensitivity and selectivity. Another way of improving the preconcentration efficiency is via electro-oligomerization stripping voltammetry, a concept in which the analyte (here isoproturon) is oxidized in the form of oligomeric deposits that are then detected by cathodic stripping square wave voltammetry [405]. An important feature is the protecting role played by the nanochannels of monodisperse diameter to reject interference and (bio)fouling agents. As an example, one can cite the electrochemical detection of the betablocker propranolol in the presence of haemoglobin, which is not possible on bare electrode due to adsorption of the protein. It becomes feasible with the mesoporous silica film acting as a physical barrier preventing the biomolecule from reaching the electrode surface whereas small propranolol species are likely to pass through the film and be oxidized on the underlying ITO electrode (Figure 18B) [338].
4.4.2. Electrocatalytic detection, indirect amperometric detection, and sensors based on hybridization reactions and/or electrochemiluminescence
When the mesoporous silica films are deposited onto nanomaterials-based electrodes (such as reduced graphene oxide or carbon nanotubes, for instance), the analyte preconcentration might be accompanied by electrocatalytic effects. It is notably the case when three-dimensional graphene is used as a support for the films [406, 414]. Nanocatalysts (such as graphene quantum dots [397] or platinum nanoparticles [402]) can be used to modify the silica layer and the resulting nanocomposite be applied to the electrocatalytic detection of various analytes (Table 1). Beside direct electrocatalysis, mediated electrocatalysis is otherwise exploited by incorporating a redox mediator in the nanochannels. Examples are available for silica films simply impregnated with metal complexes (such as
The indirect amperometric detection of non-electroactive anions can be achieved at ferrocene-functionalized mesoporous silica films on the conceptual basis of charge compensation (the oxidation of ferrocene into ferricinium requires the ingress of an anion from the solution to neutralize the so-generated cation), leading to peak currents each time an aliquot of solution containing an anion is injected at the electrode surface (Figure 19A). The flow injection analysis is performed using a pure water mobile phase (to avoid high background currents) and the amperometric signal consists of a first capacitive contribution (due to conductivity change upon electrolyte injection) followed by a faradic one (arising from ferrocene oxidation and charge neutralization by the injected anion). At a bare electrode, one observes only the capacitive contribution. The amperometric response is directly proportional to the anion concentration (Figure 19A, bottom), which tends to level off at larger values because of saturation of the ferrocene sites. The modified electrode can be used as a detector in ion chromatography, enabling the detection of several anions at the same time [407]. Of related interest is the indirect detection of copper and ascorbic acid by exploiting a DNAzyme-based molecular gate [387]. The strategy is the activation of the DNAzyme attached on top of the mesoporous film by the analytes, resulting in a molecular switching able to control the diffusion of a redox probe (
A similar “modulation” principle is applied for the development of geno-, apta- or immunosensors, implying the grafting of an aptamer or an antibody onto the external surface of the mesoporous silica film and using a redox probe in solution. Upon hybridization by the complementary aptamer or antigen, the probe response is decreasing proportionally to the analyte concentration (Figure 18D). This was applied to the detection of cancer [391, 392] and prostate specific [409, 410, 411] antigens, C-reactive protein [393], DNA [416] or insulin [404] (Table 1). The used electrochemical technique was most often differential pulse voltammetry but it could be also electrogenerated chemiluminescence. Actually, mesoporous silica films doped with
4.4.3. Pseudocapacitive hybrid films
As aforementioned, ferrocene-functionalized and oriented mesoporous silica films are characterized by well-defined voltammetric response (Figure 16C) thanks to fast redox kinetics associated to the electron-hopping mechanism [376]. As deposited on ITO electrode, such functionalized films behave as transparent pseudocapacitive materials. They give an almost linear potential response during the galvanostatic charge–discharge processes at various current densities (0.4–20 A⋅cm−3), with an appreciable delivered capacity of 1.10 mC⋅cm−2 (at a current density of 0.4 A⋅cm−3) keeping 48% of its value at higher current density (20 A⋅cm−3) [427]. In order to increase the loading amount of ferrocene groups per surface area, for the sake of increasing capacity, similar films were generated by EASA on a free-standing graphene foam prepared by electro-exfoliation of a graphite electrode [309], exhibiting also well-defined and reproducible charge/discharge curves (Figure 19C). Based on these curves, the capacity can reach 196 mC⋅cm−2 (or capacitance of 326 mF⋅cm−2) at a current density of 2 mA⋅cm−2 [309], with a good operational stability (no noticeable capacity loss after 10,000 cycles, see Figure 19C). They can be also manufactured in the form of graphene-based flexible electrodes [428].
Well-aligned 1D-polyaniline (PANI) nanofilaments as nanoarrays generated by electropolymerization through silica nanochannels membrane (Figure 17D) exhibit improved electrochemical performance compared to bulk PANI deposits [365, 353]. They are promising for application as electrochemical supercapacitors, reaching high specific capacitance (3.00 mF⋅cm−2 at 20 μA⋅cm−2, i.e., an increase by 152% in comparison to PANI directly deposited on ITO substrate) [365]. Similar observations can be made with polypyrrole (PPy) nanowire electrodeposits, for which specific capacitance values 60 times higher than bulk PPy was reported [363], but the nanowires diameter was around 30 nm suggesting that they probably grew also out of the mesoporous membrane. Such nanocomposites based on conducting polymer 1D nanostructures can be considered as prospective electrode materials for the development of supercapacitors [429, 430].
4.4.4. Electrochromic and multi-stimuli smart coatings
As discussed in Section 4.3.1, nanofilaments of conducting polymers (i.e., PANI) can be generated by electropolymerization through the mesoporous films and the resulting vertically aligned nanoarrays can be freed from silicic matrix by dissolution while kept covalently bonded to the transparent ITO electrode surface (Figure 15) [366]. When free, such PANI nanowires exhibit very efficient charge transfer reactions and very fast color change upon reversible switching between the leucoemeraldine and emeraldine forms (Figure 17D), stable for hundreds of cycles, with response time of ca. 0.1 s [353], which is among the fastest one for electrochromic materials based on PANI nanostructures [431]. Similar electrochromic devices based on PEDOT nanowires grown through nanochannels on FTO electrode are reported [432].
Electrochromism can be also induced in the oriented mesoporous films by covalent bonding of metal polypyridyl complexes onto the silica walls, as illustrated for bis(terpyridine) iron(II) species (Figure 19D). Again, one can exploit the rather fast charge transfer reactions via electron hopping to ensure color change by redox switching [417]. The grafting of tris(bipyridine) iron(II) complexes imparts to the hybrid film both electro- and photochemical activity through the manipulation of the oxidation state and spin state of the confined species, being promising for the development of optoelectronic devices [433]. Even rather sensitive/reactive moieties such as tetrazine derivatives can be successfully immobilized in a stable form, while keeping their electrochemical and fluorescence properties, the intensity of which being proportional to the functionalization level [372].
4.4.5. Miscellaneous
In addition to the above uses, the permselective properties and separation ability of mesoporous silica membranes with vertically aligned nanochannels are basically attractive for other applications such as gated molecular transport [62, 375], current rectification [375, 434], ionic and molecular separations [51], electro-osmotic pumping [435], drug release [436], thermoelectric measurements [437], or integration with microfluidics [438]. An extreme situation concerns layers that completely block access to the underlying substrate, such as that encountered for the electrodeposited superhydrophobic coatings [439]. To achieve this goal, the mesoporous silica film containing CTAB in the oriented channels is grafted with long alkyl-chains (using dodecyltrimethoxysilane) on top of the membrane [440], sometimes with an additional corrosion inhibitor (i.e., 1H-benzotriazole) dissolved in the surfactant phase [441, 384]. The resulting films offer good resistance in various electrolyte solutions and their superhydrophobic properties are kept when contacting common liquid products, as demonstrated by contact angle observations (Figure 17B). They also exhibit active corrosion protection of metals and alloys [441, 384]. Of related interest are the self-defensive antibacterial coatings bearing antimicrobial agents in the mesopores and a molecularly engineered functional assembly grafted on top of the film with pH/enzyme dual stimuli-responsiveness and multiple release modes [442].
Unlike blocking/protecting the electrode surface, the exploitation of charge transfer through independent nanochannels in such insulating membrane might be interesting in molecular electronics, optoelectronics or super-dense magnetic memory storage devices [95]. This domain is still in its infancy, but one can cite some pioneering works as protected atomic switches based on single copper nanowires [351], PANI nanofilaments-based molecular junctions in silica nanochannels [380], oriented films with anchored copper phosphonate functional groups with nonlinear optical properties [443], or a dielectric silica layer acting as artificial memristive synapse [444].
4.5. Non-silicate films generated by electro-assisted self-assembly
4.5.1. Oriented mesoporous oxides
The generation of lamellar mesostructures via electrochemical interfacial surfactant templating has been largely developed for ZnO [136, 235, 236, 445, 446, 447, 448] and extended to other metal oxides or hydroxides such as Ni(OH)2 [238, 449], Co(OH)2 [450, 451], SnO2 [452], TiO2 [453] or MnO2 [237]. In those cases, anionic surfactants were mainly used, often with the addition of co-solvents, co-surfactants and cationic ions, the electrodeposition being achievable in both potentiostatic and galvanostatic modes, as well as in cathodic and anodic conditions [445].
The electrodeposition mechanisms have been largely discussed for the production of lamellar structured ZnO films [235, 236, 445, 446, 447, 448]. It was first demonstrated that reduction of nitrate species in a slightly acidic zinc nitrate solution containing dilute (<CMC) sodium dodecyl sulfate (SDS) led to a pH increase at the electrode surface, inducing thereby the precipitation of ZnO in a lamellar structure due to the bilayer arrangement of the SDS surfactant molecules [235, 445]. The driving force of the ordering is the electrostatic interactions between the Zn2+ (or hydrated [Zn(H2O)4]2+) cations and the negatively charged SDS self-assembled onto the electrode surface. Contrary to the silica-based films, the applied potential needed to be less negative (i.e., −0.5 V vs. Ag/AgCl) to get the ordered mesoporosity and avoid metallic zinc deposition. The morphology of the films was different (Figure 20A1), the mesostructure was lamellar (Figure 20A2) instead of hexagonal, and the repeat distance was function of the surfactant concentration or type (Figure 20A3) as a result of different packing during the self-assembly growth [235]. No ordered film can be produced when using CTAB (instead of SDS) because the positively charged head group cannot coordinate zinc cations [445]. Depending on the surfactant concentration and applied potential, the formation of a nanoparticulate ZnO phase can be observed [446, 448]. The electrodeposition can be also performed in the anodic direction. In this case, the starting zinc solution is prepared at high pH (i.e., 13.3) where ZnII species are in the form of anions ([Zn(OH)3]− or [Zn(OH)4]2−) and pH is decreased at the electrode surface by oxidizing ascorbate species added to the medium, leading to the precipitation of ZnO. Surprisingly, the lamellar structures were obtained only with anionic surfactants (like SDS or lauric acid, this time at concentrations above CMC), and not with the positively charged ones (as CTAB) [236]. This indicates that the direct electrostatic interactions between the surfactant and the inorganic precursors are not the main factor affecting the self-assembly process (it is thought that the electrolyte cations play a role in the process) [236]. The morphology of the deposits is still in the form of ZnO plates (Figure 20B1) with some oriented mesopores (Figure 20B2) and a lamellar organization depending on the type of anionic surfactant (with more vertical order with SDS than with lauric acid, compare parts B3 and B4 in Figure 20). Adding alkylammonium to the electrodeposition medium further improved the quality of the mesoporous ordering (Figure 20B5–B8), probably due to the reduction of electrostatic repulsions affecting the ordering and domain size of the bilayer assemblies [445].

(A) Cathodically electrodeposited ZnO films. (A1) SEM image (scale bar: 1 μm) and (A2) TEM view of lamellar-structured ZnO films deposited (at −0.5 V for 10 min at 70 °C) from 0.02 M Zn(NO3)2 solutions containing either 0.25 wt% of SDS (A1) or 0.25 wt% of AOT (A2) anionic surfactants. (A3) Small-angle XRD patterns of ZnO films deposited from 0.02 M Zn(NO3)2 solutions containing (a) 0.02 wt% of SHDS, (b) 0.02 wt% of SDBS, (c) 0.25 wt% of DP, (d) 0.25 wt% of AOT, and (e) 0.25 wt% of SDS, with possible variation of SDS lamellar assemblies that can lead to different repeat distances on the right. Reprinted from Tan et al. [235], Copyright (2005) American Chemical Society. (B) Anodically electrodeposited ZnO films. (B1) SEM image and (B2) TEM view of lamellar-structured ZnO films deposited (at +0.15 V for 30 min at 65 °C) from solutions containing 0.02 M ascorbic acid and 0.01 M Zn(NO3)2 (at a pH of 13.3) and 5 wt% of lauric acid surfactant. (B3, B4) 2D gi-SAXS of ZnO films deposited (B3) with 10 wt% of SDS and (B4) with 5 wt% of lauric acid. (B5–B8) SEM (B5, B6) and TEM (B7, B8) images of ZnO films templated by (B5, B7) 5 wt% of SDS and 1 wt% of CTAB or (B6, B8) 5 wt% of SDS and 1 wt% of TAB. Reprinted from Steinmiller and Choi [236], Copyright (2007) American Chemical Society. (C) Lamellar hybrid nanostructures electrodeposited with the aid of conjugated surfactant molecules. (C1) Morphology: (a,b) SEM images (top (a) and cross-sectional (b) views) of deposited ZnO platelets using PyBA (magnified in the inset for image (a)); (c) TEM image of lamellar sheets within platelets, magnified in the inset; (d) schematic diagram of lamellar ordering composed of inorganic Zn-rich regions and bilayers of PyBA surfactant. Reproduced with permission from Sofos et al. [454], Copyright (2009) Nature Publishing Group. (C2) Structures of the terthiophene-based surfactants (with systematic modifications of their geometry, valency, and flexibility) used for the cathodic electrodeposition of lamellar ZnO films on ITO and PEDOT:PSS-coated electrodes: TAA(T)2 and TBA(T)2 do not lead to ZnO film growth, 3TCA grows isotropic lamellae on both substrates, lamellar coverage from 3TDCA deposition are too weak for a 2D gi-SAXS signal on ITO and isotropic on PEDOT:PSS, 3TBA directs parallel growth on ITO and perpendicular growth on PEDOT:PSS, and 3TDBA deposits lamellae that are preferentially parallel on ITO and isotropic on PEDOT:PSS. Reprinted from Bruns et al. [455], Copyright (2013) American Chemical Society. Masquer
(A) Cathodically electrodeposited ZnO films. (A1) SEM image (scale bar: 1 μm) and (A2) TEM view of lamellar-structured ZnO films deposited (at −0.5 V for 10 min at 70 °C) from 0.02 M Zn(NO3)2 solutions containing either 0.25 wt% of SDS (A1) or ... Lire la suite
Other examples of combining self-assembly and electrodeposition in a single step process are available, notably based on families of surfactants constituted of one or two carboxylate head(s) and conjugated segments/linkers such as pyrene or terthiophene derivatives [454, 456, 455]. For instance, lamellar hybrid films alternating sheets of ZnO and 1-pyrenebutyric acid (PyBA) can be obtained by electro-assisted precipitation of self-assembled Zn(OH)2∕PyBA structures on ITO electrode thanks to favorable zinc-carboxylate interactions and π–π binding of pyrene moieties, and further annealing at 150 °C to transform Zn(OH)2 into ZnO [454]. The morphology is very similar to that obtained for SDS/ZnO films, with near-vertical randomly oriented sheets revealing a lamellar mesostructure having a periodicity of 3.2 nm, with 0.7–1.0 nm thick inorganic walls separated by organic layers of 2.4 nm in thickness (corresponding to PyBA bilayer), as illustrated in part C1 of Figure 20. Adjusting the PyBA concentration in the electrosynthesis medium enables to control between bloomlike and flakelike growth [456]. The vertical orientation is better achieved when covering the ITO surface with poly(3,4-ethylenedioxythiophene:poly(styrenesulfonate) (PEDOT:PSS) defining both hydrophobic and hydrophilic regions. In addition to the substrate, the orientation and long-range alignment of hybrid lamellae are also affected by the molecular structure of the conjugated template, as illustrated for terthiophene-based surfactants (Figure 20C2). Monovalent and/or flexible surfactants gave rise to more anisotropic nanostructures with higher densities, longer persistence lengths, and lower polydispersity, compared to the divalent and/or rigid ones, whereas no ordering at all can be observed with T-shape surfactants [455]. Such hybrid films combining macroscopically oriented n-type semiconductor (ZnO) and p-type semiconducting supramolecular phases (conjugated surfactants) are promising for both photoconductivity and photovoltaic efficiency [454, 455, 457].
Contrary to the oriented mesoporous silica films for which the porosity can be revealed by template extraction, no surfactant removal was reported for the lamellar mesoporous oxides and hydroxides, most probably because their structure cannot be maintained and it would collapse upon removing the organic phase. Nevertheless, besides the above photoconductors alternating n-type and p-type domains, other lamellar organic–inorganic hybrids started to find some applications. The ZnO/SDS films exhibit good optical properties, better than bulk ZnO electrodeposited in the absence of surfactant [447]. They can be loaded with dyes and used as thin films in dye-sensitized solar cell approaching a power conversion efficiency of 2% [458, 459]. Their photocatalytic properties were also exploited for methylene blue photodegradation [460]. Finally, such electrogenerated mesoporous hybrid films are promising for pseudo- or supercapacitor electrodes, as pointed out with Ni-doped Co(OH)2∕ITO nanowires or mesoporous Mn2O3 film [461, 462].
4.5.2. Other mesoporous films electrogenerated in the presence of surfactants
If we leave aside the ordered mesoporous metals or alloys, yet at the origin of the elaboration of oriented mesoporous films by electrochemical interfacial surfactant templating [233, 234], but implying direct metal electrodeposition (different from the concept of indirect electrogeneration of thin films as discussed in this account) and being the subject of existing well-documented reviews [463, 464, 465], there are almost no examples of other mesoporous films electrogenerated in the presence of structure-directing agents. The closest approach is probably the formation of chitosan-based composite films by combining the precipitation of chitosan by electro-induced pH change (easily achievable from an acidic solution at reducing potentials [466]) with the layer-by-layer assembly technique (as shown for alginate–chitosan multilayers [467]) or with other co-deposition or assembly methods [468]. On the other hand, it is surprising that the electropolymerization of conducting polymers has still not been combined with the electrochemical interfacial surfactant templating, especially regarding the need for conducting polymer nanostructures for various applications [469, 470, 471] and the existing soft template-assisted (yet non-electrochemical) polymerization methods [472]. An exception is the electrochemical polymerization performed around the walls of “soap bubble” templates (but the resulting porous conducting polymers were not ordered) [473]. Perhaps one can cite the electrodeposition of enzymes-integrated mesoporous composite films by interfacial templating [474], but they are actually made of amphiphilic phospholipid-templated mesoporous ZnO layers covalently inlaid with the proteins (thus being somehow related to the family of mesoporous oxides presented above). Finally, vertically aligned mesoporous carbon thin films have been also developed from surfactant template assemblies on silver electrodes [475], but they were not electrochemically produced.
5. Conclusions
The electrochemically assisted sol–gel deposition of non-electroactive and non-conductive thin films is a counterintuitive approach overcoming some limitations of the conventional electrodeposition techniques. Uniform silica sol–gel thin films can be produced in a controlled way by electrochemically generated interfacial pH change onto various electrode surfaces, including of irregular topography and heterogeneous nature, as well as at the local scale, or in the form of composites by encapsulation of nano-objects. Using additional organosilanes in the electrosynthesis medium enables to get organic–inorganic hybrid films. The method is compatible with the entrapment of biomolecules without preventing their biological activity, with promising applications in the field of bioelectrocatalytic reactors. Sol–gel electrogeneration in the presence of surfactant template led to the growth of vertically-aligned mesoporous silica nanochannel films via electro-assisted self-assembly. These films exhibit size and charge selectivity at the molecular level, which can be exploited notably for electrochemical sensing (preconcentration electroanalysis, permselective coatings). The oriented nanochannel membranes can used to electropolymerize conducting polymer nanofilaments, durably attached to the electrode surface thanks to electrografting, and exhibiting attractive properties for applications (electrochromism, pseudocapacitors). Combining the electro-assisted self-assembly to click chemistry offers a versatile way to oriented mesoporous films bearing a variety of organo-functional groups that are accessible to external reagents thanks to the vertical alignment of nanochannels of monodisperse diameter. The incorporated redox moieties remain electrochemically accessible, enabling long-range electron transfer, with applications in electroanalysis, electrocatalysis, or electrochemical energy storage. Now that the concepts have been established and considering the numerous possibilities of functionalization, no doubt that further innovative applications will emerge, opening new challenges and prospects in such burgeoning field.
Conflicts of interest
The author has no conflict of interest to declare.