

1 Introduction
The multifunctional features of bis-benzimidazole and its derivatives have received considerable attention as a result of the significant roles they played as ligands in coordination chemistry [1–3], medicinal chemistry as antimicrobial [3–6], antitumor agents [7–9] and for applications, such as dyes and in the field of photography [10–12].
Following successful therapeutical applications of cisplatin as an antitumor drug and of gold-containing auranofin as an antirheumatic drug, a large number of complexes with other metals have been studied and, in several cases, subjected to clinical tests [13–17]. A sizable portion of the effort has been devoted to the interaction of transition metal ion compounds, particularly group-10 metal ions, with cellular targets for developing more potent drugs with improved pharmacological properties and reduced toxicity [18–20].
It is also true for many biosystems that metal ion concentrations must be maintained within a proper range. If the concentration of a given essential metal ion is too low, the organism can suffer from metal ion deficiency. Metal ions can also induce toxicity in organisms, including the human body. Toxicity can arise from excessive quantities of either an essential metal, possibly the result of a metabolic deficiency, or a non-essential metal. Both acute and chronic exposure can be treated by chelation therapy [21–24]. As a part of their biological importance in bioinorganic chemistry, palladium complexes are well known catalysts. They play significant roles in many catalytic industrial bulk processes.
The synthesis of the ligand (L) under conventional heating [25] and of its complexes with the Cu(II) and Cu(I) nitrate were previously reported [26]. The present work focuses upon the synthetic methodology, comparing a conventional heating method to microwave irradiation, which has additional features, including higher yields and an ecofriendly approach. Complementary quantum chemical calculations enhance our understanding of the present experimental data.
2 Experimental details
2.1 Materials and equipments
All chemicals and solvents were reagent grade and were used as purchased without further purification. Analytical data were obtained with a Thermo Finnigan Flash EA 1112 analyzer. Molar conductance was measured on a WPA CMD750 conductivity meter in dimethyl sulfoxide (DMSO) at 25 °C. Thermogravimetric analysis (TGA) data were obtained with a Seiko SII TG–DTA 6300 TG/DT analyzer heated from 20 to 900 °C under nitrogen atmosphere. FT–IR spectra were recorded (mid as KBr pellets and far in polyethylene tablets) on a Jasco FT/IR-600 Plus Spectrometer. Routine 1H (400 MHz) and 13C (100 MHz) nuclear magnetic resonance (NMR) spectra were recorded at ambient temperature in DMSO-d6. Chemical shifts (δ) are expressed in units of parts per million (ppm) relative to TMS. The analytical data and physical properties are summarized for each experiment.
2.2 Synthesis
2.2.1 Pyridine-2,6-dicarboxylic acid dibutyl ester (a)
Thionyl chloride (3.20 mL, 44 mmol) was added dropwise to n-butanol (25 mL) with ice cooling under argon. A suspension of pyridine-2,6-dicarboxylic acid (3.34 g, 20 mmol) in n-butanol (25 mL) was slowly added to it and the mixture was refluxed for 5 h under argon. The mixture was then cooled and poured into ice-cold water (600 mL). The solid mass was filtered, washed with water and dried in vacuo, giving a white crystalline solid with high purity (5.36 g, 96%) and Mp 62 °C. FT–IR (solid, cm−1): 3062, 2957, 2933 ν (C–H), 2870 ν (C–H), 1740 ν (CO), 1576 ν (CC), 1225 ν (C–O), 1174, 1154, 765 δ (C–H), 694.
2.2.2 2,6-bis(hydroxymethyl)pyridine (b)
To a suspension of (a) (4.19 g, 15 mmol) in ethanol (25 mL), NaBH4 (2.72 g, 72 mmol) in ethanol (20 mL) was added slowly for 1 h with ice cooling and under vigorous stirring. The mixture was stirred at room temperature for 3 h and then was relaxed further for 5 h. After evaporation of the solvent, the residue was dissolved in 50 mL of water, adjusted to pH 4 with 2 M HCl, and then adjusted to pH 9 with a saturated aqueous Na2CO3 solution. The solvent was evaporated and the dry residue was extracted with CH2Cl2 (50 mL) by Soxhlet extraction for 48 h. Evaporation of the solvent produced a white solid (1.70 g, 82%) with Mp 112–114 °C. (115–120 °C in [27]) ATR (solid, cm−1): 3346 ν (O–H), 3099 ν (C–H), 1593 ν (CC). 1H NMR (CDCl3), δH ppm: 4.79 (s, 2CH2, 4H), 7.27 (d, 2H), 7.76 (t, H).
2.2.3 2,6-[(tosyloxy)methyl] pyridine (c)
A solution of (b) (1.39 g, 10 mmol) in 35 mL of CH2Cl2 was added to a 40% aqueous solution of KOH (35 mL). The reaction mixture was cooled at 0 °C and stirred for 30 min, after which a solution of p-toluenesulfonyl chloride (3.81 g, 20 mmol) was added in one portion. The reaction mixture was stirred for 1 h at 0 °C and then at room temperature until complete conversion (TLC, 1/4 methanol/toluene). The reaction mixture was added to water (35 mL). The aqueous phase was extracted with CH2Cl2 (3 × 20 mL), the combined organic phase was dried over anhydrous MgSO4, filtered, and the solvent was removed under reduced pressure to afford a white crystalline solid (3.89 g, 87%) with Mp 119–121 °C (121–122 °C in [28]). FT–IR (solid, cm−1): 3070, 3038 ν (C–H), 1596 ν (CC), 743 δ (C–H). 1H NMR (CDCl3), δH ppm: 2.46 (s, 6H, CH3), 5.08 (s, 4H, CH2), 7.34–7.83 (m, 11H).
2.2.4 2,6-bis(benzimidazol-2′-ylthiomethyl)pyridine (L)
2.2.4.1 Microwave-assisted synthesis
Sodium 2-mercaptobenzimidazolate (1.72 g, 10 mmol) was mixed well with an adsorbent silica gel (3.50 g, 60 meshes) in CH2Cl2 (5 mL). The mixture was evaporated under vacuum using a microwave oven for complete removal of the solvent. The powder mixture and (c) (2.24 g, 5 mmol) were mixed thoroughly and irradiated at 250 W with pulses of 45 s for 5 min. After completion of the reaction (monitored by TLC), the mixture was cooled and the product was extracted with dichloromethane (3 × 10 mL). The combined organic phase was dried over anhydrous MgSO4, filtered, evaporated under reduced pressure and dried in vacuum, giving a white solid product with high purity (1.85 g, 92%).
2.2.4.2 Conventional heating method
To a solution of 2-mercaptobenzimidazole (1.50 g, 10 mmol) in absolute EtOH (8 mL) containing Na (0.24 g, 10.5 mmol), (c) (2.44 g, 5 mmol) was added in small portions. The mixture was stirred at 60 °C for 3 h. The solvent was evaporated under vacuum and the residue was poured into 500 mL of ice-cold distilled water. The solid mass was filtered, washed with water. The product was recrystallized from EtOH, giving a white crystalline solid (1.51 g, 73%) with Mp 214–215 °C ([25] 72%). Found (calculated), C21H15N5S2: C, 62.07 (62.51); H, 4.39 (4.25); N, 17.15 (17.36); S, 15.68 (15.89). FT–IR (KBr): see Table 1. 1H NMR (DMSO-d6), δH ppm: 4.70 (s, 4H, CH2), 7.14 (td, J = 6, J = 3.5 Hz, 4H), 7.46 (d, J = 7.5 Hz, 2H), 7.49 (br, s, 4H), 7.72 (t, J = 7.6 Hz, H), 12.64 (br, s, 2H, NH). 13C NMR (DMSO-d6), δC ppm: 37.00 (CH2), 110.57 (unresolved), 117.34 (unresolved), 121.45, 121.74, 135.60 (unresolved), 137.89, 143.50 (unresolved), 149.69, 156.70.
Selected IR spectra characteristics of the ligand (L) and its complexes: experiment vs calculation results in terms of scaled frequencies in cm−1 and IR intensities in parenthesisa.
| Assignment | L Exp | LAb Calc. | LC Calc. | LD Calc. | [PdLCl]Cl Exp | [PdLCl] Calc. | [PdLBr]Br Exp | [PdLBr] Calc. | [PdLI]I Exp | [PdLI] Calc. | ||||
| ν (N–H) | – | – | 3596 (65.8); 3319 (858.0) | 3600 (82.6) | 3599 (125.8) | 3245 | 3583 (169.5) | 3240 | 3583 (169.0) | 3242 | 3583 (192.6) | |||
| ν (CH) | 3054 | m | 3138 (20.1) | 3147 (24.4) | 3139 (19.6) | 3071 | m | 3154 (4.5) | 3068 | m | 3154 (4.8) | 3054 | m | 3154 (4.6) |
| ν (C–H) | 2872 | m | 3030 (8.7) | 3043 (5.2) | 3045 (7.1) | 2905 | m | 3083 (0.8) | 2905 | m | 3080 (0.9) | 2902 | m | 3080 (0.9) |
| ν [(CC) + (CN)] | 1590 | m | 1604 (49.6) | 1600 (18.7) | 1600 (19.8) | 1602 | m | 1612 (35.8) | 1600 | w | 1608 (24.1) | 1595 | m | 1607 (29.0) |
| δ (CH) | 1401 | vs | 1404 (94.9) | 1402 (165.7) | 1402 (95.9) | 1414 | s | 1418 (252.0) | 1412 | s | 1417 (243.0) | 1411 | s | 1416 (228.9) |
| δ (CH) | 738 | vS | 734 (113.1) | 736 (67.2) | 736 (64.9) | 743 | vs | 745 (44.2) | 743 | 745 (55.4) | 743 | 746 (50.6) | ||
| δ (CCS) | 528, 522 | w, sh | 516 (3.2) | 528 (2.7) | 516 (0.7) | 523 | W | 521 (3.7) | 522 | w | 521 (4.0) | 521 | w | 521 (4.2) |
| δ (CH) | 429 | m | 428 (15.9) | 428 (13.3) | 428 (13.7) | 397 | W | 397 (2.8) | 396 | w | 397 (2.6) | 396 | w | 398 (2.7) |
| τ (C–CH2) + δ (N–H) | 398 | m | 388 (21.7) | 392 (17.9) | 392 (17.4) | 372 | m | 358 (15.6) | 370 | m | 358 (7.6) | 367 | w | 358 (7.1) |
| δ (CCS) | 329, 315 | m | 333 (4.8); 317 (5.7) | 316 (8.0) | 330 (10.8) | 352 | w, sh | 359 (4.9) | 351 | w, sh | 359 (3.0) | 352 | vw | 359 (2.1) |
| ν (Pd–Ni) + δ (CCS) | 331 | s | 329 (15.6) | 333 | w | 327 (14.6) | 327 | w | 324 (13.2) | |||||
| ν (Pd–Np) + ν (Pd–X) + τ (S–CH2) | 331 | s | 328 (11.1) | 329 | w | 319 (1.7) | 316 | vw | 316 (0.9) | |||||
| ν (Pd–X) | 300 | w | 309 (2.0) | 226 | w | 264 (10.7) | 215 | m | 223 (2.4) | |||||
| ν (Pd–Np) + ν (Pd–X) + ν (Pd–Ni) + τ (S–CH2) | 253 | w, sh | 262 (1.3) | 256 | m | 256 (1.4) | 254 | vw | 260 (1.0) | |||||
| ν (Pd–Ni) | 248 | w | 260 (10.3) | 256 | m | 260 (8.8) | 247 | w | 261 (7.5) | |||||
| ν (Pd–X) | 279 | w | 292 (13.8) | 215 | w | 234 (3.1) | 205 | w | 201 (2.3) | |||||
| ν (Pd–Ni) + δ (CCS) | 207 | w | 208 (2.6) | 204 | w | 204 (2.1) | 196 | w | 199 (1.9) | |||||
| τ (S–CH2) + {ν (Pd–Ni)} | 291, 279 | w, w | 264 (3.1) | 286 (1.93) | 267 (0.7) | 186 | w | 179 (0.2) | 186 | w | 174 (0.4) | 187 | w | 167 (0.4) |
a For a given type of internal motion: (i) only the modes of the highest intensity are given; (ii) if the modes of comparable intensities are within 3 cm−1, only one of them is given; otherwise, the strongest two are given.
b The spectra of LA and LB are very similar to each other. Thus, only LA is given.
2.2.5 [Pd(L)Cl]Cl
A suspension of PdCl2 (177 mg, 1 mmol) in dry acetonitrile (2 mL) and dry benzene (10 mL) was deoxygenated and stirred at 85 °C under argon for 3 h to form monomeric PdCl2(MeCN)2. The solvent was evaporated to dryness and the residue was charged under argon with (L, 202 mg, 0.5 mmol) in absolute ethanol (10 mL) and stirred at 85 °C for 48 h. The reaction mixture was cooled to room temperature and allowed to stand to form a linden-green solid product, which was filtered, washed with ether, and dried under vacuum (284 mg, 75%). Dec. over 260 °C. Molar conductivity: 72.8 Ω−1·cm2·mol−1, 1:1 electrolyte. Found (calculated), C21H17Cl2N5PdS2; C, 42.98 (43.42); H, 3.10 (2.95); N, 11.89 (12.06); S, 10.92 (11.04). FT–IR: see Table 1. 1H NMR (DMSO-d6), δH ppm: 5.23 (d, J = 14.0, 4H, CH2), 7.33 (td, J1 = 7.8, J2 = 1.2 Hz, 2H), 7.40 (td, J1 = 7.8, J2 = 1.3 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.84 (d, J = 8.0 Hz, 2H), 8.15 (t, J = 7.8 Hz, H), 8.51 (d, J = 7.8 Hz, 2H), 14.19 (s, 2H, NH). 13C NMR (DMSO-d6), δC ppm: 35.35 (CH2), 111.47, 119.47, 122.99, 124.03, 125.49, 133.17, 140.63, 143.40, 149.01, 157.34.
2.2.6 [Pd(L)Br]Br
A brick-red solid complex was synthesized in a manner similar to that used for [Pd(L)Cl]Cl using a mixture of PdBr2 (133 mg, 0.5 mmol) and (L) (101 mg, 0.25 mmol). The yield was (168 mg, 72%). Dec. over 250 °C. Molar conductivity: 68.5 Ω−1 cm2 mol−1, 1:1 electrolyte. Found (calculated), C21H17Br2N5PdS2; C, 37.34 (37.66); H, 2.67 (2.56); N, 10.21 (10.46); S, 9.36 (9.58). FT–IR: see Table 1. 1H NMR (DMSO-d6), δH ppm: 5.20 (d, J = 13.99, 4H, CH2), 7.34 (t, J = 7.5 Hz, 2H), 7.41 (t, J = 7.8 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 7.84 (d, J = 7.8 Hz, 2H), 8.16 (t, J = 7.8 Hz, H), 8.51 (d, J = 8.0 Hz, 2H), 13.99 (s, 2H, NH). 13C NMR (DMSO-d6), δC ppm: 35.24 (CH2), 111.53, 120.11, 122.99, 124.11, 125.62, 133.17, 140.93, 143.42, 148.90, 156.91.
2.2.7 [Pd(L)I]I
The black solid was synthesized in a manner similar to that used for [Pd(L)Cl]Cl using a mixture of PdI2 (180 mg, 0.5 mmol) and (L) (101 mg, 0.25 mmol). The yield was (205 mg, 73%). Dec. 250 °C. Molar conductivity: 65.8 Ω−1·cm2·mol−1, 1:1 electrolyte. Found (calculated), C21H17I2N5PdS2 C, 32.92 (33.02); H, 2.35 (2.24); N, 8.93 (9.17); S, 8.12 (8.40). FT–IR: see Table 1. 1H NMR (DMSO-d6), δH ppm: 5.12 (d, J = 14.02 Hz, 4H, CH2), 7.35 (td, J1 = 7.3, J2 = 1.3 Hz, 2H), 7.41 (td, J1 = 7.3, J2 = 1.3 Hz, 2H), 7.44 (d, J = 7.5 Hz, 2H), 7.80 (d, J = 7.8 Hz, 2H), 8.14 (t, J = 7.8 Hz, H), 8.47 (d, J = 8.0 Hz, 2H), 14.04 (br, s, 2H, NH). 13C NMR (DMSO-d6), δC ppm: 34.85 (CH2), 111.60, 121.38, 122.96, 124.19, 125.73, 133.15, 141.47, 143.35, 148.75, 156.20.
3 Theoretical details
The optimized gas phase structures and the force fields of (L) and its complexes have been obtained at the B3LYP [29,30] hybrid density functional level by employing the triple-zeta quality 6-311++G** basis sets at the O, S, N, C and H atoms [31–33] and double-zeta quality DZVP basis sets of DGauss (abbreviated as DGDZVP) at the heavier Pd, Cl, Br, and I atoms [34,35]. To assess the effect of the solvent (DMSO) on the relative stabilities, the ligand (L) and its complexes were placed in a cavity formed by a set of overlapping spheres in the frameworks of the solvent reaction field and then DMSO was treated implicitly by using the polarizable continuum model (PCM) [36]. All calculations were performed by using Gaussian 03 program package and its fine numerical grid as well as tight SCF convergence criteria [37].
As a result of systematic errors in the calculations due to the fact that solid-state packing effects and anharmonicity have been neglected, and to the basis set and electronic correlation incompleteness, the calculated vibrational frequencies are in general overestimated. To incorporate these effects, the calculated frequencies are generally scaled [38,39]. The scaling factor of the frequencies below 1700 cm−1, i.e., the correlation coefficient between the experimental and calculated frequencies, is obtained as 0.985 for the free ligand (L), in agreement with previous studies [40–43]. The modes with frequencies above 1700 cm−1 demonstrate in general huge shifts from the experimental frequencies, even if their frequencies are scaled [38–43]. Thus, such modes were not considered in deriving a scaling factor of 0.985. The modes with the frequencies below 650 cm−1 couple strongly with Pd–N and Pd–X vibrations, which mostly downshift the frequencies. In this region of the complexes, the scaling factor is obtained as 0.910, whereas the same scaling factor as that of the free (L) is used in the remainder of the spectra of the complexes. The far-IR spectra were simulated in terms of the calculated frequencies (scaled) and IR intensities by using pure Lorentzian band shapes with a bandwidth (FWHH) of 3 cm−1. The present frequency and IR intensity calculations allow assigning the discussed vibrational modes to internal motions.
4 Results and discussion
4.1 Structure and energetics of the free ligand (L)
To determine the structure of the free ligand computationally, we performed extensive potential energy surface scans around each labile bond (ArC–CH2, S–CH2, and ArC–S) in the gas phase. These analyses show that there are several energetically very closely lying stable conformers of (L) that are connected to each other with very small rotational barriers (6 ± 2 kcal/mol). Thus, (L) is found at room temperature as a mixture of its several conformers. Four of the located lowest-energy conformers of (L) (labeled as LA–LD) are shown in Fig. 1 along with their key geometry parameters and their relative stabilities. An isolated single (L) in the gas phase has the most stable structure, with one benzimidazole NH moiety pointing to the pyridine nitrogen (LA). When the electrostatic and polarization effects of DMSO is included in the energies of the gas phase structures implicitly, the LC and LD conformers that belong to the Ci and Cs point groups, respectively, are stabilized by ∼ 1 kcal/mol and become equally stable with LA. The benzimidazole NH moieties do not orient toward pyridine nitrogen in LC and LD, and thus, effectively interact with the solvent. Geometrical relaxations in the solvent environment may further stabilize these conformers. Analogously, in the solid phase, non-bonded intermolecular interactions are expected to inhibit the rotation of benzimidazoles, which may result in most stable conformers as LC and LD (Fig. 1).
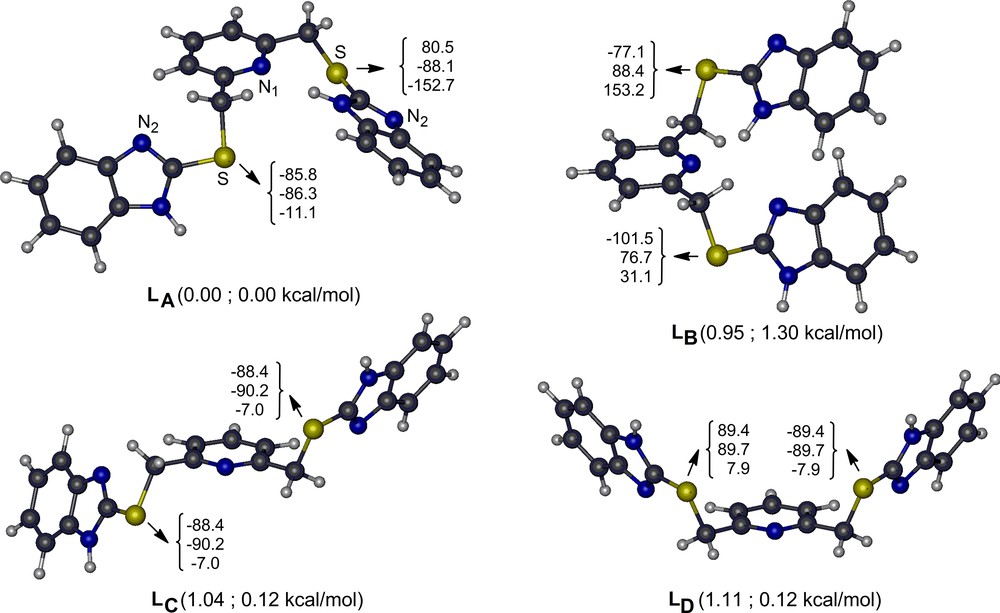
(Color online.) Four of the lowest-energy conformers of L in gas phase along with their key dihedral angles (the triple numbers in degrees are respectively for the bridging N1–C–C–S, C–C–S–C and C–S–C–N2 dihedral angles) and their ground-state energies with respect to LA in the gas phase (the first entry in parenthesis) and in the DMSO environment but at the gas-phase geometry (the second entry in parenthesis).
4.2 Molar conductance analyses
The measured molar conductivities of the Cl, Br, and I complexes are 72.8, 68.5, and 65.8 Ω−1·cm2·mol−1, respectively. These values agree with those for 1:1 electrolytes in DMSO solution (1 × 10−3 M) [44] and thus ensure the formation of the complexes of [Pd(L)X]X composition.
4.3 Nuclear magnetic resonance
1H and 13C NMR results are given as in the Experimental section. The 1H NMR spectra of the complexes are very similar to each other. Thus, in addition to that of the free ligand (L), we only display the 1H spectrum of the Cl complex in Fig. 2. A broad and unresolved NH proton signal in the free ligand's spectrum at 12.64 ppm changes for a well-resolved singlet at ∼ 14 ppm in the complexes. Therefore, coordination occurs via the nitrogen atoms of the imine that inhibits the fluxional behavior of the benzimidazole rings in the free ligand. The significant changes of bridging CH2 protons from a sharp singlet at 4.70 ppm to sharp doublets with considerable deshielding effect at ∼ 5.20 ppm and with high coupling J values of ∼ 14 Hz signify the presence of geminal protons, which strongly suggests a cyclic formation around palladium ion.
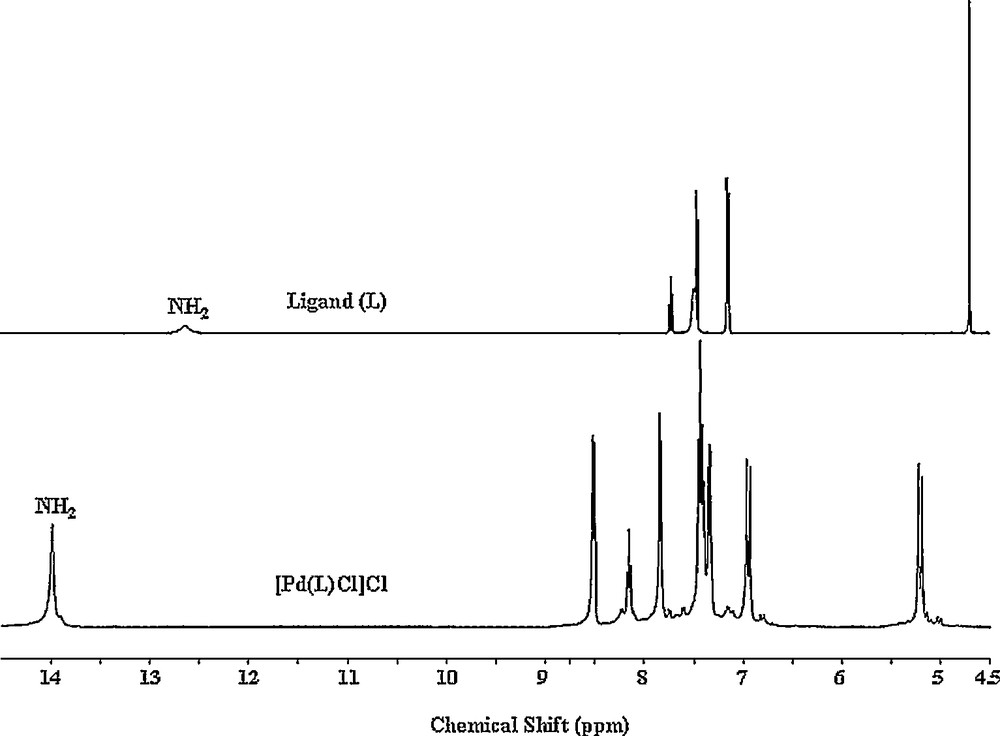
1H NMR (DMSO-d6) spectra of the free L (top) and [Pd(L)Cl]Cl complex (bottom).
Changes are also observed for the aromatic protons in the complexes compared with the free ligand. A downfield resonance of one of these protons (doublets at ∼ 8.50 ppm) is most probably caused by the strong intramolecular effect from an electronegative atom, in these cases halogen ions [45].
The 13C NMR spectrum (Fig. 3) of the free ligand exhibits only five well-resolved signals for the aromatic units due to a fast tautomeric equilibrium in the benzimidazole part of the molecule, indicated by the low rotational barriers calculated. Upon complexation, the fluxional behavior of the NH proton atoms is inhibited and the complexes exhibit 11 well-resolved signals, out of which seven signals belong to the benzimidazole unit. The CH2 resonance shifts from 37.44 ppm to around 35 ppm in the complexes. This also supports coordination via two nitrogen atoms of the benzimidazole moieties as well as the nitrogen atom of the pyridine unit. Thus, the NMR spectra suggest that the complexes are in the [Pd(L)X]X form.
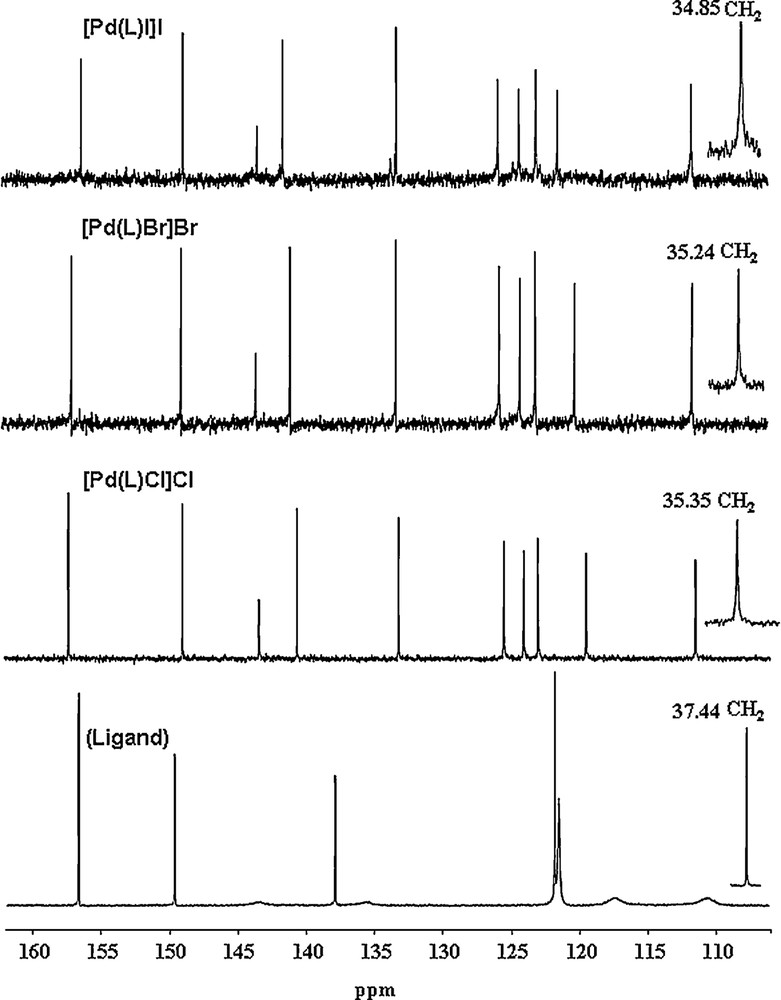
13C NMR (DMSO-d6) spectra of the free L and its complexes.
4.4 Structure and energetics of the complexes
To determine the structure of the complexes computationally, we performed extensive conformational analyses in the gas phase and single-point calculations on the resulting stationary points in the presence of DMSO.
The lowest-energy conformer of the [Pd(L)X]X complexes coordinates to the Pd ion via the nitrogen atom of pyridine (N1), two nitrogen atoms of the benzimidazole moieties (N2 atoms), and one halogen atom (X) both in vacuo and in DMSO environment (Fig. 4) in a slightly distorted square planar arrangement around the metal ion (Table 2). This coordination scheme is also consistent with the NMR results in DMSO. The DMSO environment stabilizes the lowest-energy conformer of the [Pd(L)X]X complexes (Fig. 4) significantly over its other conformers. In addition to N1 and one halogen atom, rather than two N2 atoms:
- • if the complexation occurs via one sulfur atom and one benzimidazole nitrogen atom, the resulting complex becomes less stable in vacuo (in DMSO) by 12 kcal/mol (18 kcal/mol);
- • if two sulfur atoms are involved in the coordination, then it is less stable in vacuo (in DMSO) by 15 kcal/mol (22 kcal/mol).
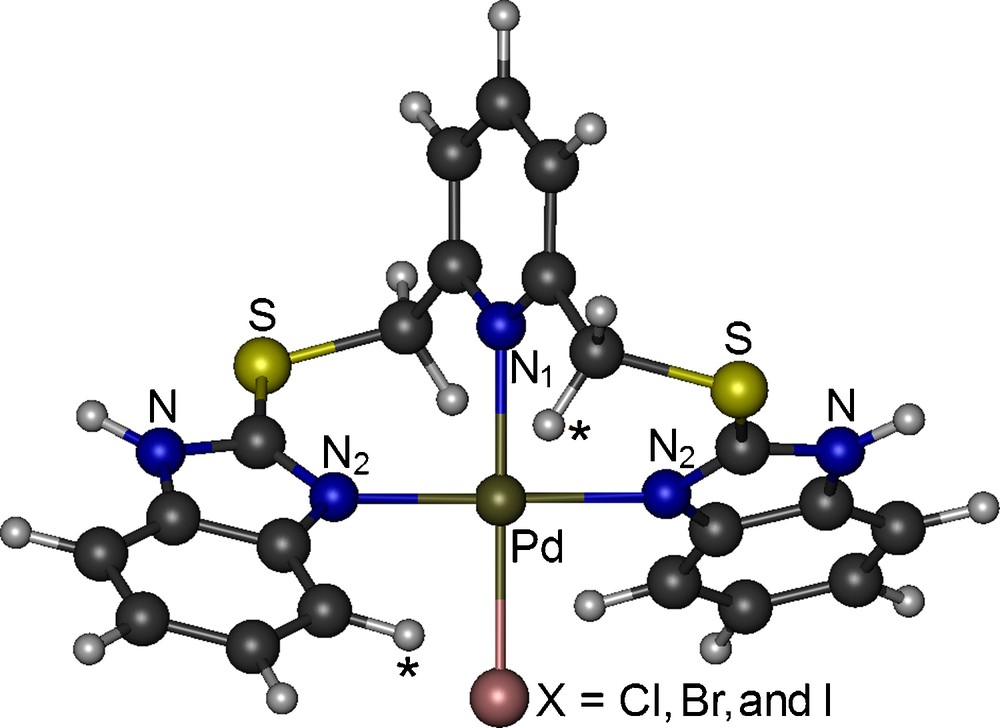
(Color online.) Molecular structure of the Pd complexes.
Calculated geometry parameters (distances in Å and angles in degree) of the [PdLX] complexes around the Pd center in the gas phase.
| Complex | Pd–N1 | Pd–N2 | Pd–X | N2–Pd–N2 | N1–Pd–X |
| [PdLCl] | 2.143 | 2.092 | 2.322 | 178.0 | 179.9 |
| [PdLBr] | 2.158 | 2.094 | 2.454 | 177.0 | 179.9 |
| [PdLI] | 2.181 | 2.095 | 2.648 | 175.7 | 179.8 |
The other possible coordination arrangements of the [Pd(L)X]X complexes lay at least at 23 kcal/mol (27 kcal/mol) above the preferred conformation in vacuo (in DMSO).
The relaxed potential energy surface scans result in a unique position for the non-coordinated X atoms of the [Pd(L)X]X complexes. This position is stabilized through electrostatic interactions with one of the H atoms of CH2 and of the benzimidazole ring shown with (*) in Fig. 4, which is consistent with proton NMR results (see above). In the lowest-energy conformer of the Cl/Br/I complex, it is 2.15/2.37/2.71 Å, 2.43/2.62/2.84 Å and 3.62/3.89/4.60 Å away from the hydrogen atoms of CH2 and benzimidazole moieties and from the Pd atom, respectively. Additionally, it distorts the square planar arrangement of the coordination site in the gas phase. However, in the solution and solid phases, the other couple of CH2 and benzimidazole hydrogen atoms should also interact with the non-bonded X atom of the neighbouring complex, cancelling the distortion caused by the unbalanced electrostatic in the isolated single complex. Thus, the structural and vibrational features of the [PdLX]X complex in the solution and solid phases should be better resembled by the gas phase [Pd(L)X] complex than by the isolated gas phase [Pd(L)X]X complex. Otherwise, there would be several additional signals in the 13C NMR spectra. Therefore, in the following, we discuss the results on the gas phase [Pd(L)X] complex only.
It is worth to note that the coordination site is elongated (Table 2) when the atomic radii of the halogen atom (I > Br > Cl) increases. Thus, the Pd–N and Pd–X vibrations are expected to be shifted toward lower frequencies with increasing the halogen's size.
4.5 Vibrational spectra
The present vibrational spectra can be discussed in terms of three characteristic wave regions: 3250–2870 cm−1 for ν (N–H) and ν (C–H) stretching modes, 1660–650 cm−1 for ν (CC), ν (C–N), δ (C–H) and δ (N–H) vibrations, and 650–180 cm−1 for ν (Pd–N), and ν (Pd–X) stretches as well as several torsional and out-of-plane vibrations (see Table 1). Although N–H stretching is not detected in the free ligand, it appears as a medium band at about 3240 cm−1 in the complexes. The present IR spectra calculations also find that the intensity of the N–H stretching band increases significantly upon complexation. Thus, in addition to the 1H and 13C NMR spectral results, the vibrational spectra suggest coordination through the nitrogen atoms of the benzimidazole moiety.
The characteristic ν(C–H) modes of the ring residues and aliphatic groups are observed in the wave region 3070–2870 cm−1 for both the free ligand and the complexes. Upon complexation, the frequencies of these modes are upshifted by ∼ 20 cm−1 (Table 1). The ν (CC) and ν (CN) stretching vibrations couple to each other and give their strongest band at 1590 cm−1 for the free ligand and ∼ 1600 cm−1 for the complexes. Analogously, the in-plane δ (CH) deformation appears at 1401 cm−1 in the free ligand's case and at ∼ 1415 cm−1 in the complexes’ case. Such small but sizable differences between the experimental frequencies of the free ligand and the complexes are consistent with the calculated shifts due to complexation (Table 1).
Torsional and bending vibrations involving the CH2 carbons are calculated at below 650 cm−1, which is consistent with the literature [40]. The frequencies of these vibrations (Table 1) are sensitive to the conformers of the free ligand (Fig. 5), which differ in the CH2 carbon related dihedral angles (Fig. 1). For example, the calculations estimate a medium intensity C–CH2–S bending at 316 cm−1 for LC and at 330 cm−1 for LD. However, it appears at both frequencies in LA and LB as well as in the experiment. The weak CH2–S torsion (exp.: 291 and 279 cm−1) is estimated at 286 cm−1 for LC and at ∼ 265 cm−1 for LA, LB and LD. Such examples indicate that the experimental IR spectrum of (L) should not belong to a single conformer, but to the superposition of several conformers of (L), in agreement with the calculated ground-state energetics. This conclusion is also apparent from Fig. 5. Rather than the simulated far-IR spectrum of a single conformer, the overlap of the calculated spectra for the lowest-energy conformers of (L) agrees reasonably well with the solid-state experimental spectrum, except the strong and broad band calculated between 360 and 400 cm−1, corresponding to out-of-plane N–H bending. As the NH hydrogen is stabilized through H-bond interactions with its neighbourhood in the solid-state, this band appears very weak in the experimental IR spectra. The corresponding broad band is calculated at ∼ 450 cm−1 in the complexes.
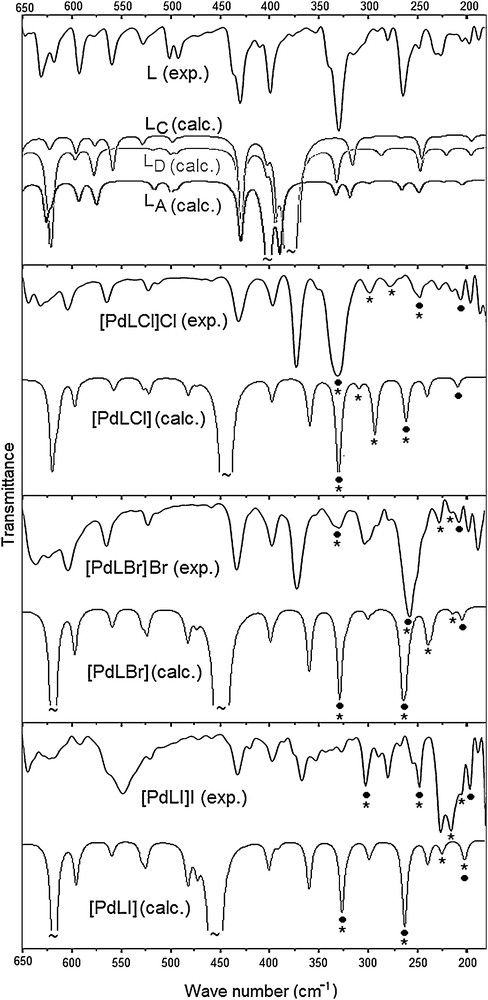
Experimental (solid phase) and calculated (gas phase) far-IR spectra of the free (L) and its complexes (• and * correspond to Pd–N and Pd–X stretching vibrations, respectively). The calculated spectrum of LB is not shown as it is very similar to that of LA.
The stretching frequencies of transition metals with N or X are expected and calculated below the 450 cm−1 frequency [46–52]. Each band at ∼ 320 and ∼ 255 cm−1 in the spectra of the complexes is actually composed of two overlapped bands, one of which is almost pure ν (Pd–N) stretching, and the other is ν (Pd–N) coupled with ν (Pd–X) stretching (Fig. 5 and Table 2). The remaining ν (Pd–N) stretching appears at ∼ 200 cm−1. The pure ν (Pd–X) stretching frequencies are sensitive to the halogen (X) type. When the size of X increases (Cl < Br < I), the ν (Pd–X) frequencies are downshifted (for X = Cl/Br/I, 300/226/215 cm−1 and 279/215/205 cm−1).
4.6 Thermal analyses of the complexes
The TG/DTA behaviors of the complexes are very similar to each other, and thus the temperature dependence of the complexes is shown only for the [Pd(L)Cl]Cl complex in Fig. 6. The percent weight losses with the temperature increase indicate that the softer SCH2 moiety is the first decomposed part of the complexes. The full decomposition of the SCH2 moiety occurs at ∼ 350 °C. The remaining organic content of the complexes decomposes at several overlapped stages in the 350–480 °C temperature range. The DTA curve in this range exothermic and the phase transition starts at ∼350 °C. This temperature is the one at which PdO is prepared by heating the Pd sponge [53]. Thus, Pd is oxidized in the 350–480 °C range. Due to oxidation, the weight of the residue remained at ∼ 480 °C is by ∼ 6% heavier than the Pd content of the complexes. The endothermic phase transition in the DTA curve at ∼ 820 °C is the melting of the PdO lattice (literature: 750 °C in the air [54]).
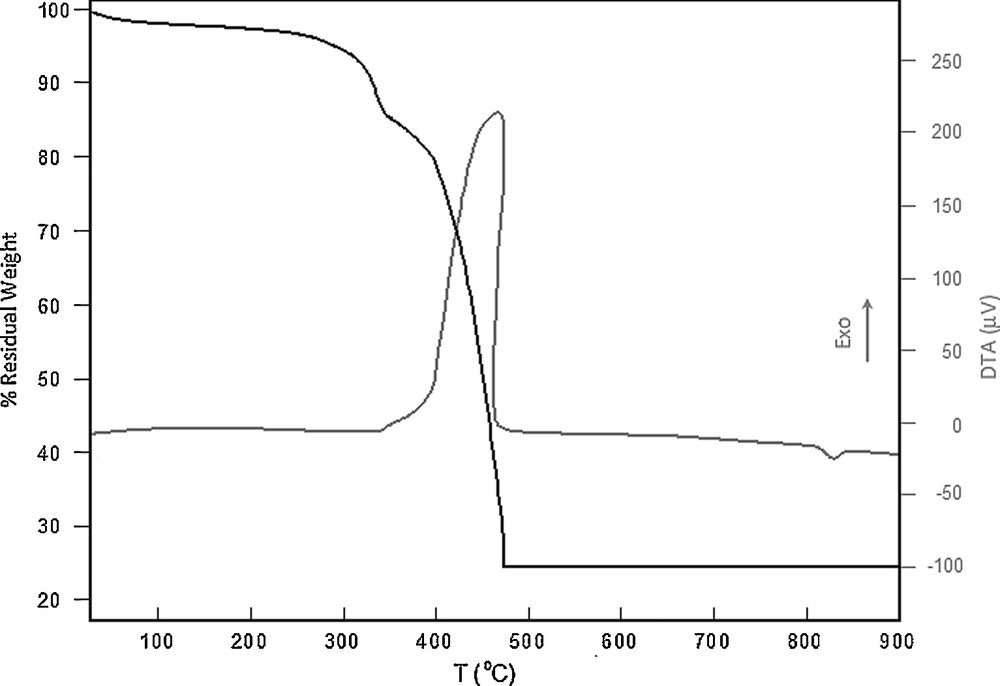
TG and DTA curves for the [PdLCl]Cl complex.
5 Conclusions
A bis-benzimidazole derivative (L) and its three Pd(II)X2 complexes (X = Cl, Br, and I) have been synthesized and characterized by elemental and thermal (TGA and DTA) analyses, molar conductivity measurements, and spectral studies of 1H NMR, 13C NMR mid-IR and far-IR. For the synthesis of the free ligand, microwave irradiation appears more effective than conventional heating. According to the calculated ground-state energetics and IR spectra (both measured and calculated) of the free ligand, the rotations around each labile bond are quite facile at room temperature. The measured molar conductivities of the complexes indicate that the complexes have a Pd(L)X2 composition. The NMR spectra give additional features consistent with the formation of the complexes in the [Pd(L)X]X form. According to NMR spectra, coordination around the Pd ion occurs through three nitrogen atoms and one X atom. Quantum chemical calculations demonstrate that this coordination style has a slightly distorted square planar arrangement around the Pd ion and is significantly more stable than the other coordination arrangements. The agreement between the calculated and experimental FT–IR spectra of the complexes provides further evidence of the complexation. The complexes are stable up to ∼ 250 °C and their first decomposed moiety is the softer SCH2 unit. The complexes oxidize in their second decomposition range in which their remaining organic contents are decomposed.
Acknowledgments
This work was supported by the Scientific Research Fund of Fatih University under project numbers P50021202_Y(2363) and P50011102 _Y(1679).