

1 Introduction
In recent years, considerable interest has been devoted to finding new methodologies for the synthesis of numerous organic compounds under solvent-free conditions. The toxicity and volatile nature of many organic solvents, particularly chlorinated hydrocarbons that are widely used in large amounts for organic reactions have posed a serious threat to the environment [1]. Thus, the design of solvent-free catalytic reactions has received tremendous attention in recent times in the area of green synthesis [2]. Imidazoles are common scaffolds in highly significant biomolecules, including biotin, the essential amino acid histidine, histamine, the pilocarpine alkaloids [3], and other alkaloids, which have been shown to exhibit interesting biological activities, such as antimicrobial, anticryptococcal, cytotoxic activities and inhibition of nitric oxide synthase [4], Many of the substituted imidazoles are known as inhibitors of P38 MAP kinase [5], fungicides and herbicides [6], plant growth regulators [7] and therapeutic agents [8]. Owing to the wide range of pharmacological and biological activities, the development of synthetic methods enabling facile access to this heterocycle is still desirable. Several methods have been reported for the synthesis of imidazoles. 2,4,5-Triaryl-1H-imidazoles are generally synthesized by three-component cyclocondensation of a 1,2-diketone, α-hydroxyketone or α-ketomonoxime with an aldehyde and ammonium acetate, which comprises the use of microwaves [9], ionic liquids [10] europium triflate [11], oxalic acid [12], TBAB [13], CAN [14], refluxing in AcOH [15] and silica sulfuric acid [16]. On the other hand, the synthesis of 1,2,4,5-tetraaryl-1H-imidazoles are carried out by four-component condensation of a 1,2-diketone, α-hydroxyketone or α-ketomonoxime with an aldehyde, a primary amine and ammonium acetate using microwaves [17], heteropolyacid [18], silica gel/NaHSO4 [19] InCl3·3H2O [20] or HClO4–SiO2 [21]. In addition, they can also be accessed by hetero-Cope rearrangements [22] and condensation of a 1,2-diketone with an aryl nitrile and primary amine under microwave irradiation [23]. Despite their potential utility, these methods have a number of drawbacks, including poor yields, utilization of excessive glacial AcOH, prolonged reaction times, high cost of most conventional room temperature ionic liquids, use of hazardous and expensive catalysts and use of toxic organic solvent that limit their utility.
Hence, the development of clean, safe, effective, economical, high-yielding and environmentally benign protocols is still desirable and much in demand.
Silica chloride (SiO2–Cl) is gaining importance as a heterogeneous catalyst and is used in various organic transformations. It is a good solid acid in terms of convenience, cheapness, easy production, and insolubility in all organic solvents. Due to its insolubility in organic solvents, excellent in situ proton generation, and accepting different nucleophiles, it can be used for different purposes in organic chemistry. It has been used as a dehydrating agent [24], and some cyclocondensations yielding dihydropyrimidinone [25], 2-aminothiazoles [26], 6-methyl-3-propynylthio-1,2,4-triazin-5(2H)-one [27], pyranoquinolines, or furanoquinolines [28] are catalyzed by silica chloride. The use of silica chloride to expedite oxidation of alcohol [29], esterification and transesterification [30] transdithioacetalization of acetals [31], deprotection of oximes/hydrazones/semicarbazones [32] synthesis of nitriles and amides [33], synthesis of sulfoxide [34], synthesis of bisindolylmethanes [35], etc. has been well explored.
Considering the significance of the silica chloride and in continuation of our interest in developing new and rapid synthetic methods for the construction of biologically important structural scaffolds [36], we report herein a rapid, efficient, economic, environmentally benign and easy to scale-up method for the synthesis of 2,4,5-triaryl-1H-imidazoles using silica chloride as a heterogeneous and recyclable catalyst under solvent-free conditions (Scheme 1).
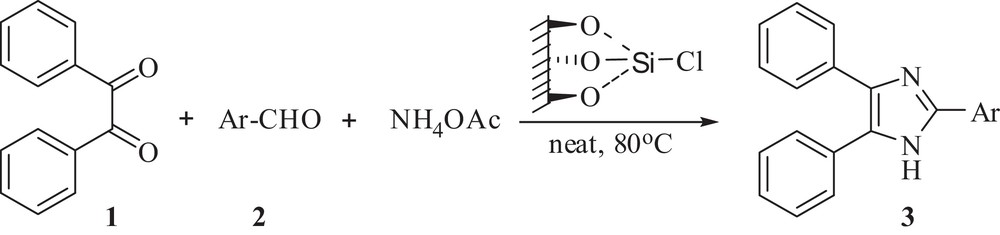
Silica chloride catalyzed synthesis of 2,4,5-triaryl-1H-imidazoles.
2 Results and discussion
Initially, to optimize the amount of catalyst and the reaction temperature, the reaction of benzil (1, 1 mmol), benzaldehyde (2a, 1 mmol), and NH4OAc (10 mmol) was studied under solvent-free conditions in the presence of SiO2–Cl at different temperatures; the results are summarized in Table 1. They revealed that, when 250 mg/mmol SiO2–Cl at 80 °C were used, the reaction proceeded smoothly and gave product 3a in highest yield (80%). Further increase in the amount of silica chloride above 250 mg/mmol did not lead to substantial change in the yield and the reaction time (Table 1). Then, we examined the effect of solvents, like CH3CN, toluene, EtOH, MeOH and THF for the above reaction. The results in Table 2 indicate that solvents affected the efficiency of the reaction and very poor yields were obtained after 1 h. The best results were obtained under solvent-free conditions (Table 2).
Synthesis of 2,4,5-triaryl-1H-imidazole(3a) under various conditions.
| Entry | SiO2–Cl (mg/mmol)a | Temperature (°C) | Time (min) | Yield (%)b |
| 1 | – | 80 | 180 | 00 |
| 2 | 50 | 80 | 120 | 45 |
| 3 | 100 | 80 | 120 | 58 |
| 4 | 150 | 80 | 90 | 65 |
| 5 | 200 | 80 | 60 | 72 |
| 6 | 200 | 100 | 60 | 75 |
| 7 | 250 | 60 | 60 | 55 |
| 8 | 250 | 80 | 30 | 80 |
| 9 | 250 | 100 | 30 | 80 |
| 10 | 300 | 80 | 30 | 79 |
| 11 | 300 | 100 | 30 | 80 |
| 12 | 300 | 120 | 30 | 78 |
a Reaction conditions: benzil (1 mmol); benzaldehyde (1 mmol); ammonium acetate (10 mmol); neat.
b Yields of isolated products.
Effect of solvent for SiO2–Cl catalyzed synthesis of 2,4,5-triaryl imidazole (3a).
| Entry | Solventa | Temperature (°C) | Time (min) | Yield (%)b |
| 1 | CH3CN | Reflux | 60 | 20 |
| 2 | Toluene | Reflux | 60 | 40 |
| 3 | EtOH | Reflux | 60 | 30 |
| 4 | MeOH | Reflux | 60 | 35 |
| 5 | THF | Reflux | 60 | 25 |
| 6 | none | 80 | 30 | 80 |
a Reaction conditions: benzil (1 mmol); benzaldehyde (1 mmol); ammonium acetate (10 mmol), solvent (5 mL).
b Yields of isolated products.
Under optimized reaction conditions, a mixture of benzil (1 mmol), an aromatic aldehyde (1 mmol), NH4OAc (10 mmol) and SiO2–Cl (250 mg) was stirred at 80 °C for the appropriate time (Table 3). After completion of the reaction (TLC), CH2Cl2 (20 mL) was added, and the solid catalyst was recovered by filtration. The solvent was distilled off and the crude product was purified by column chromatography using (SiO2, hexane:AcOEt) as an eluent to afford pure 2,4,5-trisubstituted imidazoles in good to excellent yields. Moreover, the catalyst recovered from the reaction mixture was reused for successive reactions without appreciable loss of activity (Table 3, entry 1). With the optimized procedure in hand, we have extended the same reaction up to 10-fold scale, and similar yields of product were obtained with slight increase in reaction time. A plausible mechanism for the formation of triaryl imidazoles in the presence of silica chloride is postulated in Fig. 1, indicating that silica chloride is an excellent source for the generation of HCl.
Synthesis of 2,4,5-triaryl-1H-imidazole derivatives.
| Entry | Ar | Time (min) | Yield (%)a | MP (°C) | ||
| Found | Reported | |||||
| 1 | C6H5 | 3a | 30 | 80, 78, 77b | 275 | 275–276 [9] |
| 2 | 3-NO2C6H4 | 3b | 35 | 76 | 314 | 313–315 [9] |
| 3 | 4-NO2C6H4 | 3c | 30 | 78 | 236 | 235–238 [9] |
| 4 | 2-ClC6H4 | 3d | 30 | 80 | 198 | 199–201 [10] |
| 5 | 4-ClC6H4 | 3e | 30 | 82 | 261 | 258–260 [9] |
| 6 | 2-BrC6H4 | 3f | 40 | 75 | 202 | 201–202 [10] |
| 7 | 4-BrC6H4 | 3g | 30 | 84 | 250 | 248–249 [10] |
| 8 | 2-OHC6H4 | 3h | 25 | 80 | 205 | 204–205 [10] |
| 9 | 4-OHC6H4 | 3i | 30 | 85 | 266 | 265–267 [10] |
| 10 | 4-MeOC6H4 | 3j | 30 | 90 | 227 | 226–228 [9] |
| 11 | 3,4-(MeO)2C6H3 | 3k | 35 | 87 | 145 | 142–143 [13] |
| 12 | 3-MeO,4-OHC6H3 | 3l | 40 | 84 | 199 | 196–198 [10] |
| 13 | 3,4,5-(MeO)3C6H2 | 3m | 45 | 82 | 260 | 261–262 [13] |
| 14 | 1-Naphthyl | 3n | 35 | 80 | 298 | 297–298 [13] |
| 15 | 2-Furyl | 3o | 30 | 86 | 232 | 233–235 [13] |
a Yields of isolated products.
b SiO2–Cl was recovered and reused for three consecutive runs.
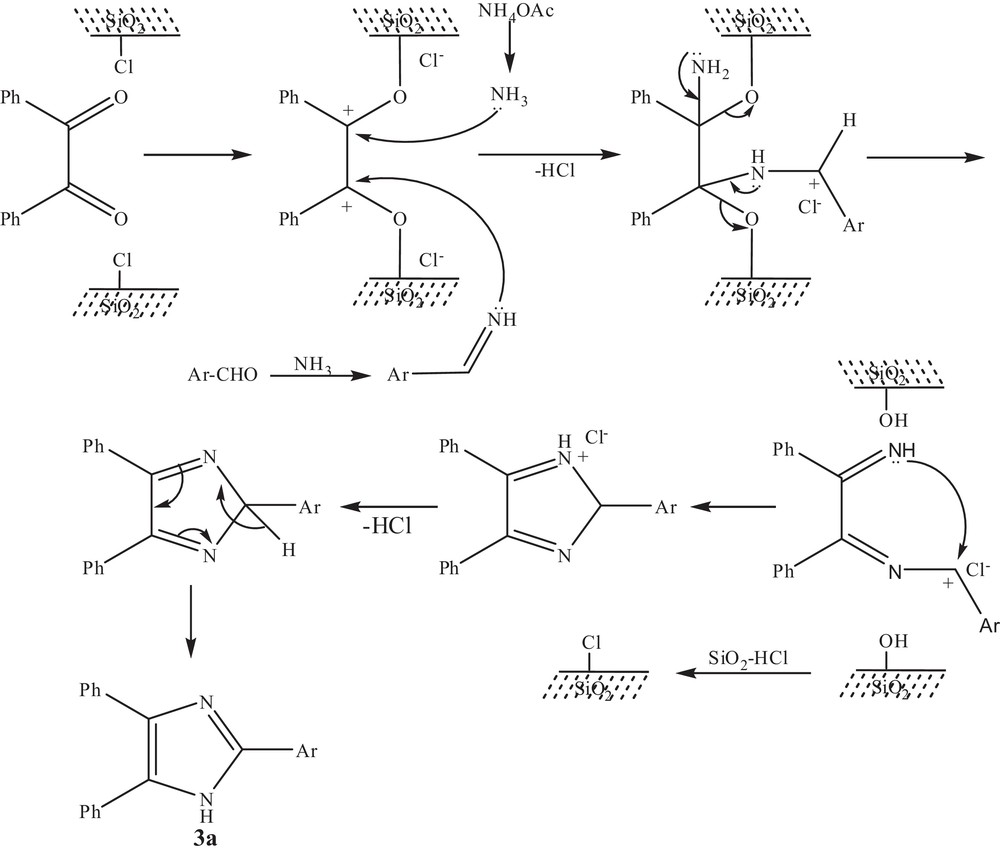
Plausible mechanism for the formation of trisubstituted imidazoles in the presence of SiO2–Cl.
In order to demonstrate the versatility of this protocol, we extended our study to the synthesis of a wide variety of 2,4,5-triaryl imidazoles (3a–o) using different aromatic aldehydes under optimized reaction conditions. All these reactions showed rapid formation of 2,4,5-triaryl imidazole derivatives with high efficiency. The nature (electron-withdrawing or electron-donating) and position of the substituent on the aromatic ring did not show any significant differences in the yield of 2,4,5-triaryl imidazole derivatives. The products were characterized by IR, 1H NMR, and mass spectroscopy data.
In the next attempt, identical reaction conditions were applied to the synthesis of 1,2,4,5-tetraaryl-1H-imidazoles via the one-pot, four-component condensation of benzil 1, aldehydes 2, aniline 4 and NH4OAc (Scheme 2). To our delight, the 1,2,4,5-tetraaryl-1H-imidazoles were also obtained in high yields (Table 4).
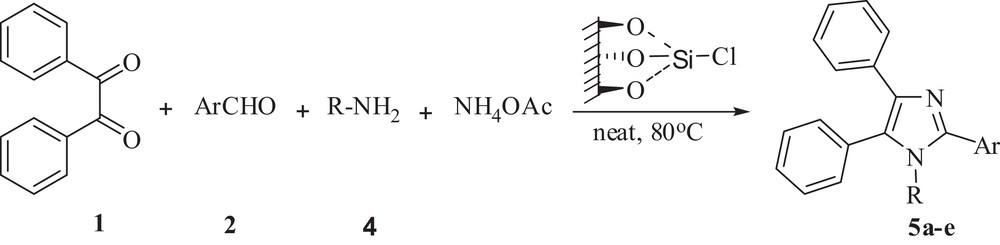
Silica chloride catalyzed synthesis of 1,2,4,5-tetraary-1H-imidazole.
Synthesis of 1,2,4,5-tetraaryl-1H-imidazole derivatives.
| Entry | Ar | R | Time (min) | Yield (%)a | MP (°C) | ||
| Found | Report | ||||||
| 1 | C6H5 | C6H5 | 5a | 40 | 78 | 220 | 221–222 [18] |
| 2 | 3-NO2C6H4 | C6H5 | 5b | 30 | 85 | 245 | 244–246 [18] |
| 3 | 4-MeOC6H4 | C6H5 | 5c | 35 | 87 | 186 | 184–185 [18] |
| 4 | 4-OHC6H4 | C6H5 | 5d | 30 | 82 | 282 | 280–281 [18] |
| 5 | 4-ClC6H4 | C6H5 | 5e | 30 | 85 | 190 | 187–189 [18] |
a Yields of isolated products.
3 Conclusion
In conclusion, we have developed a highly efficient, multicomponent protocol for the synthesis of 2,4,5-triaryl imidazoles and 1,2,4,5-tetraaryl imidazoles under solvent-free conditions using silica chloride as a heterogeneous catalyst in excellent yields. This method offers many attractive features, such as short reaction time, high yield, recovery and reusability as well as economic availability of the catalyst.
4 Experimental
4.1 General procedure for the preparation of SiO2–Cl
To a well-stirred silica gel (20 g) in CH2Cl2 (50 mL) was added dropwise SOCl2 (20 g) at room temperature. Evolution of copious amounts of HCl and SO2 occurred instantaneously. After stirring for another 6 h, the solvent was removed to dryness under reduced pressure (1 Torr). The resulting white-grayish powder was flask-dried and stored in a tightly capped bottle.
4.2 General procedure for the preparation of 2,4,5-triaryl-1H-imidazoles (3a–o)
A mixture of benzil (1 mmol), aldehyde (1 mmol), ammonium acetate (10 mmol) and SiO2–Cl (250 mg) was stirred at 80 °C for the appropriate time (Table 4). After completion of the reaction (TLC), dichloromethane (20 mL) was added, and the solid catalyst was recovered by filtration. The solvent was distilled out and the crude product was purified by silica gel column chromatography using (hexane:ethyl acetate) as an eluent to afford pure 2,4,5-trisubstituted-1H-imidazoles in good to excellent yields.
4.3 General procedure for the synthesis of 1,2,4,5-tetraaryl-1H-imidazoles (5a–5f)
A mixture of benzil (1 mmol), aldehyde (1 mmol), aromatic primary amine (1 mmol), ammonium acetate (5 mmol) and SiO2–Cl (250 mg) was stirred at 80 °C for the appropriate time. After completion of the reaction (TLC), dichloromethane (20 mL) was added, and the solid catalyst was recovered by filtration. The solvent was distilled out and the crude product was purified by silica gel column chromatography using (hexane:ethyl acetate) as an eluent to afford pure 1,2,4,5-tetrasubstituted-1H-imidazoles in good to excellent yields.
4.4 Spectral data of representative compounds
4.4.1 2,4,5-Triphenyl-1H-imidazole (3a)
IR (KBr, cm−1) ν = 1216, 1638, 2470, 2993, 3434; 1H NMR (CDCl3, 200 MHz): δ 7.45–8.25 (m, 15H), 12.61 (bs, 1H); MS (ESI): m/z 296 (M+).
4.4.2 4-(4,5-Diphenyl-1H-imidazol-2-yl)-phenol (3i)
IR (KBr, cm−1) ν = 1216, 1638, 2465, 2998, 3432, 3596; 1H NMR (CDCl3, 200 MHz) δ 7.05 (d, J = 7 Hz, 2H), 7.45–7.60 (m, 10H), 8.04 (d, J = 7 Hz, 2H), 12.58 (bs, 1H); MS (ESI): m/z 312 (M+).
4.4.3 2-(4-Methoxyphenyl)-4,5-diphenyl-1H-imidazole (3j)
IR (KBr, cm−1) ν = 1216, 1636, 2465, 2893, 3428; 1H NMR (CDCl3, 200 MHz) δ 3.89 (s, 3H), 7.24 (d, J = 7 Hz, 2H), 7.48–7.57 (m, 10H), 8.18 (d, J = 7 Hz, 2H), 12.52 (bs, 1H); MS (ESI): m/z 326 (M+).
4.4.4 2-(4,5-Diphenyl-1H-imidazol-2-yl)-2-methoxyphenol (3l)
IR (KBr, cm−1) ν = 1230, 1450, 1605, 2924, 3512, 3614; 1H NMR (CDCl3, 200 MHz) δ 3.88 (s, 3H), 6.98 (d, J = 6.8 Hz, 1H), 7.39–7.56 (m, 10H), 7.65 (d, J = 1.8 Hz, 1H), 7.75 (s, 1H) 12.54 (bs, 1H); MS (ESI): m/z 342 (M+).
4.4.5 2-(3,4,5-Trimethoxyphenyl)-4,5-diphenylimidazole (3m)
IR (KBr, cm−1) ν = 3424, 2934, 2835, 1588, 1495, 1485, 1241, 1128, 1006, 770, 699; 1H NMR (CDCl3, 200 MHz) δ 3.81 (s, 3H), 3.93 (s, 6H), 7.51–7.61 (m, 12H), 12.22 (s, 1H); MS (ESI): m/z 387 [M+H]+.
4.4.6 2-(1-Naphthyl)-4,5-diphenylimidazole (3n)
IR (KBr, cm−1) ν = 3422, 3051, 2960, 1599, 1502, 1443, 768, 695; 1H NMR (CDCl3, 200 MHz) δ 7.18–7.78 (m, 13H), 7.80–7.98 (m, 3H), 9.18 (d, J = 7 Hz, 1H), 12.43 (s, 1H); MS (ESI): m/z 347 [M + H]+.
4.4.7 2-(2-Furyl)-4,5-diphenylimidazole (3o)
IR (KBr, cm−1) ν = 3440, 3021, 2922, 2819, 2722, 1523, 1484, 1445, 1225, 1169, 766, 742, 696. 1H NMR (CDCl3, 200 MHz) δ 6.58 (dd, J = 2.34 Hz, J = 1.56 Hz, 1H), 6.99 (d, J = 3.12 Hz, 1H) 7.20–7.62 (m, 11H), 12.50 (s, 1H); MS (ESI): m/z 287 [M+H]+.
4.4.8 1-(4-Methoxyphenyl)-2,4,5-triphenyl-1H-imidazole (5c)
IR (KBr, cm−1) ν = 3060, 2956, 1606, 1574, 1482. 1H NMR (CDCl3, 200 MHz) δ 7.60 (d, 2H, J = 8.0 Hz), 7.02–7.37 (m, 15H), 6.76 (d, 2H, J = 8.0 Hz), 3.77 (s, 3H); MS (ESI): m/z 402 (M+).