

1 Introduction
Direct alcohol fuel cells operating at low temperature have attracted interest as power sources for numerous applications. Among several alcohols, which can be used in a direct fuel cell, methanol and ethanol are the most promising ones [1–4]. As a liquid they are easier to store and to handle compared to hydrogen. However methanol is toxic and the CC bond of ethanol leads to an incomplete oxidation into CO2. The use of ethers such as dimethylether (DME: CH3OCH3) and dimethoxymethane (DMM: CH3OCH2OCH3) as fuels appears to be a good alternative to the utilization of alcohols because they do not have CC bonds [5–9]. Unlike methanol, DME and DMM have a low toxicity; however DME is a gas at room temperature, which makes its handling and storage more difficult than for a liquid. According to the thermodynamic data (ΔrH° = −1945.9 kJ mol−1, ΔrG° = −1903.2 kJ mol−1, ΔrS° = −143.4 kJ mol−1 K−1) related to DMM, the equilibrium potential is 1.23 V for its complete oxidation into CO2, with n = 16 electrons.
DMM electrooxidation has been previously investigated [8–14]. The acetal reaction indicates that the acid hydrolysis of DMM gives two molecules of methanol and one molecule of formaldehyde. Previous works of the authors, obtained at room temperature, carried out on platinum poly- [8] and single-crystal [9] electrodes, demonstrated a complex mechanism, and excluding the direct hydrolysis of DMM in the operating conditions used.
The use of platinum-based nanoparticle electrodes (Pt, PtRu, PtMo, PtSn, PtRuMo) completes the studies made on poly- and single-crystal platinum electrodes. The main advantage to associate the second and the third metals to metallic Pt is their ability to activate water at lower potentials (formation of OHads) and consequently to improve COads oxidation. Electrode nanoparticles based on platinum with other metals such as ruthenium, molybdenum, and tin have been thoroughly studied for the electrooxidation of small organic molecules [15–18], like methanol which is an intermediate during DMM electrooxidation.
In this work, electrochemical results, fuel cell experiments and in situ infrared (IR) reflectance spectroscopy measurements during DMM adsorption and electrooxidation on PtRuMo/C, PtM/C (M = Ru, Sn, Mo) and Pt/C electrocatalysts are carried out in order to:
- • evaluate the behavior of this fuel at monometallic and plurimetallic electrodes in terms of reactivity;
- • provide results on activity and mechanism observed in previous studies done on poly- and single-crystal electrodes.
2 Experimental
2.1 Electrochemical measurements
The electrochemical material and experimental conditions were described previously [8]. The electrolytic solutions were prepared from 70% HClO4 (Suprapur, Merck) in ultra-pure water (MilliQ, Millipore, 18 MΩ cm). The working electrode was a rotating glassy carbon disc (0.071 cm2 geometric surface area) on which the catalytic powder was deposited, the counter electrode was a glassy carbon plate and the reference electrode was a Reversible Hydrogen Electrode (RHE). All potentials are referred to RHE. Electrochemical characterizations were performed at 50 mV s−1, and in order to avoid the formation of unwanted oxides of Mo, Sn and Ru, the upper potential limit is set at 300 mV vs. RHE for PtMo(90–10)/C and PtSn(90–10)/C catalysts, and 500 mV vs. RHE for PtRu(80–20)/C and PtRuMo(85–15–5)/C catalysts. In this way, the electroactivity of the different catalysts are compared for the DMM electrooxidation.
2.2 Synthesis of the colloidal precursors of PtM/C and Pt/C electrocatalysts and characterizations
Pt and Pt-based particles [Pt/C, PtMo(90–10)/C, PtSn(89.3–10.7)/C, PtRu(82.9–17.1)/C, PtRuMo(80.2–13.2–6.6)/C] with 40% metal loading, were prepared from colloidal precursors and dispersed on carbon powder (Vulcan XC72). The technique used is based on the Bönneman method [19]. The synthesis is carried out under controlled atmosphere (argon) free of oxygen and water, with non-hydrated metal salts in an organic solvent (THF). The first step consists of the preparation under stirring of a tetra-alkyl triethylborohydride reducing agent (Nalk4)+(Bet3H)−, which will also act as a surfactant after metal reduction, preventing any agglomeration of the metallic particles:
The colloidal precursors are then dispersed on a carbon support (carbon black, Vulcan XC72). The mixture is sonicated under atmospheric conditions then calcined at 300 °C for 1 h under the ambient atmosphere to remove the organic surfactant.
The Bönneman method allows several approaches for the synthesis of multimetallic supported catalysts.
The synthesis under stirring of catalysts with controlled atomic ratio occurs by co-reduction of different metal salts before the reduction step and formation of the precursor colloid:
This method was used for the preparation of PtRu/C, PtMo/C and PtRuMo/C.
The synthesis of catalysts with controlled atomic ratios occurs by co-deposition of different metal colloids before the calcination step and formation of the catalytic powder:
This method was used for the preparation of PtSn/C.
Then electrodes were prepared using ink made from the carbon-supported catalyst and a Nafion® solution according to a method described previously [20].
For voltammetry and in situ IR reflectance measurements, this ink is deposited on a vitreous carbon disk and after evaporation of the solvent at 40 °C (30 min), the electrode is ready to be used for the experiment.
Characterizations of the catalyst nanoparticles were carried out by different physical techniques: transmission electron microscopy (TEM) for a morphological observation of their surface with estimation of the particle size by X-ray energy dispersive analysis (EDX) with a Philips CM 120 microscope/EDX analyser equipped with a LaB6 filament with a nanoprobe (5 nm). X-ray diffraction (XRD) is used to determine the catalyst's structure. Powder diffraction patterns are recorded on a Bruker D5005 Bragg–Brentano diffractometer operated with a powered copper tube (40 kV and 40 mA) at ambient temperature with 2θ ranging from 30° to 90° by 0.02° steps. The metallic composition of the catalyst was determined by TG-DTA experiments using a SDT Q 600 TA Instruments. The temperature ranged from 0 to 600 °C at a rate of 5 °C min−1, under ambient atmosphere and in a platinum crucible.
2.3 In situ infrared reflectance spectroscopy
In order to better understand the overall mechanism of the oxidation of dimethoxymethane on the different electrocatalysts, an in situ IR reflectance spectroscopic study was performed. With this technique, it is possible to observe the adsorbed species at the electrode surface. In the case of dimethoxymethane, it was previously seen that adsorbed CO [8,9] is a reaction intermediate on poly- and monocrystals of Pt, i.e. during DMM electrooxidation:
- • on polycrystalline platinum a slow production of adsorbed CO was observed at low potentials [8];
- • on Pt(1 0 0) and Pt(1 1 0) COads formation is favored, whereas on Pt(1 1 1) COads formation was not observed.
IR spectra were obtained with a Bruker IFV 66 spectrometer with acquisition techniques, allowing performing single potential alteration infrared reflectance spectroscopy (SPAIRS) and subtractive normalized interfacial Fourier transform infrared reflectance spectroscopy (SNIFTIRS) experiments, as described in details previously [21].
2.4 Fuel cell tests
The fuel cell tests in a single cell of 5 cm2 geometric surface area were carried out with a Globe Tech test bench. The E versus j and P versus j curves were recorded using a high-power potentiostat (Wenking model HP 88) interfaced with a computer to apply the current and store the data, and a variable resistance in order to monitor the current applied to the cell. The concentrations of DMM used are 1 mol L−1 and 2 mol L−1. The fuel cell was fed with a DMM solution without pre-heating. Backpressure valves allowed us to control DMM and oxygen pressures in the cell.
Anode for fuel cell tests were prepared from an ink consisting of a mixture of 100 μL of Nafion® (5 wt. % from Aldrich) solution, 1 mL of H2O MilliQ, and 25 mg of catalytic powder (2.0 mg cm−2 metal loading, 40% metal/C). Cathode is a commercial E-TEK electrode loaded with 40wt%Pt supported on Vulcan carbon (2.0 mg cm_2 Pt). Anode and cathode were brushed on a carbon gas diffusion electrode. Carbon gas diffusion electrodes were house-made using a carbon cloth from Electrochem. Inc., on which was brushed an ink made of Vulcan XC72 carbon powder and a PTFE/water emulsion in isopropanol. The gas diffusion electrodes were loaded with 4 mg cm−2 of a mixture of carbon powder and 15 wt. % PTFE. Prior to the preparation of the membrane electrode assembly (MEA), the electrodes were heated at 150 °C to recast the Nafion® film. The metal loading of the electrodes was 2 mg cm−2 and the Nafion® loading of the electrode was 0.8 mg cm−2. The MEAs were prepared, by hot pressing at 130 °C for 3 min under a pressure of 35 kg cm−2, a pre-treated Nafion® 117 membrane with the house-made electrodes. The Nafion® membranes were preliminary treated according to a standard method described by Büchi and Srinivasan [22].
3 Results
3.1 Characterizations of PtM/C and Pt/C electrocatalysts
Characterizations are given in Fig. 1 for a Pt/C catalyst with 40% metal loading. Fig. 1(a) represents a TEM picture of the catalyst dispersed on Vulcan XC72. In Fig. 1(b), the distribution of the particle size is shown, with a mean value of 2.54 ± 0.12 nm. In Fig. 1(c), TG–DTA results show that the metal is well loaded, with 40% of Pt. XRD spectra in Fig. 1(d) indicate that the expected crystalline structure of platinum is obtained, i.e. face-centred cubic crystalline platinum. Five peaks of platinum are observed: peak of (111) plane at 2θ = 39.8°, peak of (200) plane at 2θ = 46.3°, peak of (220) plane at 2θ = 67.7°, peak of (311) plane at 2θ = 81.6°. Concerning the structure of PtSn, PtRu, PtMo and PtRuMo catalysts, Pt is decorated by the second and third metals. In Table 1, particle sizes of the different supported catalysts (dTEM), EDX results, lattice parameters and TG experiments are presented. The results indicate that the different catalysts present characteristics of the face-centred cubic crystalline structure; nanometric size and metal compositions are in good agreement with the bulk compositions: PtSn(89.3–10.7)/C for PtSn(90–10)/C, PtRu(82.9–17.1)/C for PtRu(80–20)/C, PtRuMo(80.2–13.2–6.6)/C for PtRuMo(80–15–5)/C. Concerning the composition of PtMo/C, the signal of Mo was too low to make it possible to distinguish precisely the Mo quantity. For a greater convenience the different catalysts will be named without their metal composition.
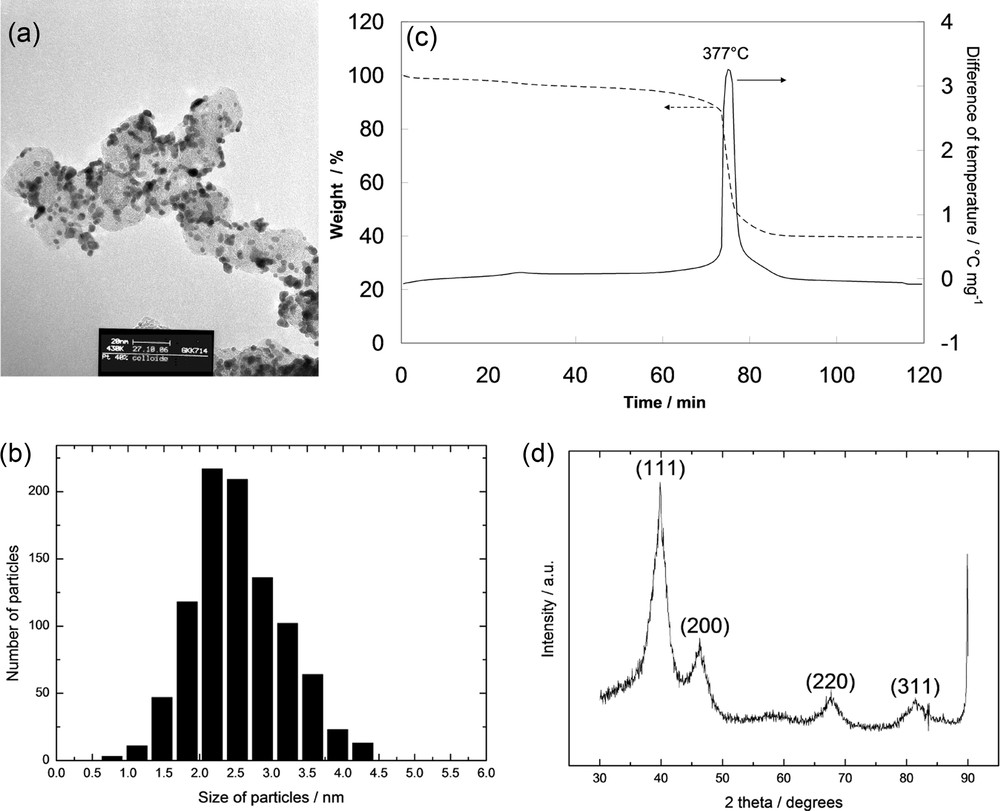
Physical characterization of a 40% Pt/C catalyst. (a) transmission electron microscopy image, (b) particle size distribution, (c) TG-DTA graphs, (d) X-ray diffraction spectrum.
Results of mean particles size (dTEM), EDX results, and lattice parameters of the different catalysts.
| a/Å | dTEM/nm | EDX | TG | |
| Ptcolloïdal | 3.915 | 2.54 ± 0.12 | 40 wt.% Vulcan | |
| PtMo(90–10) | 3.916 | 2.75 ± 0.19 | 40 wt.% Vulcan | |
| PtRu(80–20) | 3.91 | 1.93 ± 0.07 | 82.9/17.1 | 40 wt.% Vulcan |
| PtRuMo(80–15–5) | 3.908 | 1.97 ± 0.12 | 80.2/13.2/6.6 | 40 wt.% Vulcan |
| PtSn(80–20) | 3.922 | 2.60 ± 0.11 | 89.3/10.7 | 40 wt.% Vulcan |
Fig. 2 presents the XRD data of the whole catalysts. Addition of another metal to platinum refines the peak of the (111) plane according to the diffraction peak width and does not allow a shift of the diffraction peak position. This result suggests that Pt particles are decorated by the second metal particles, so that the catalysts cannot be considered as alloys.
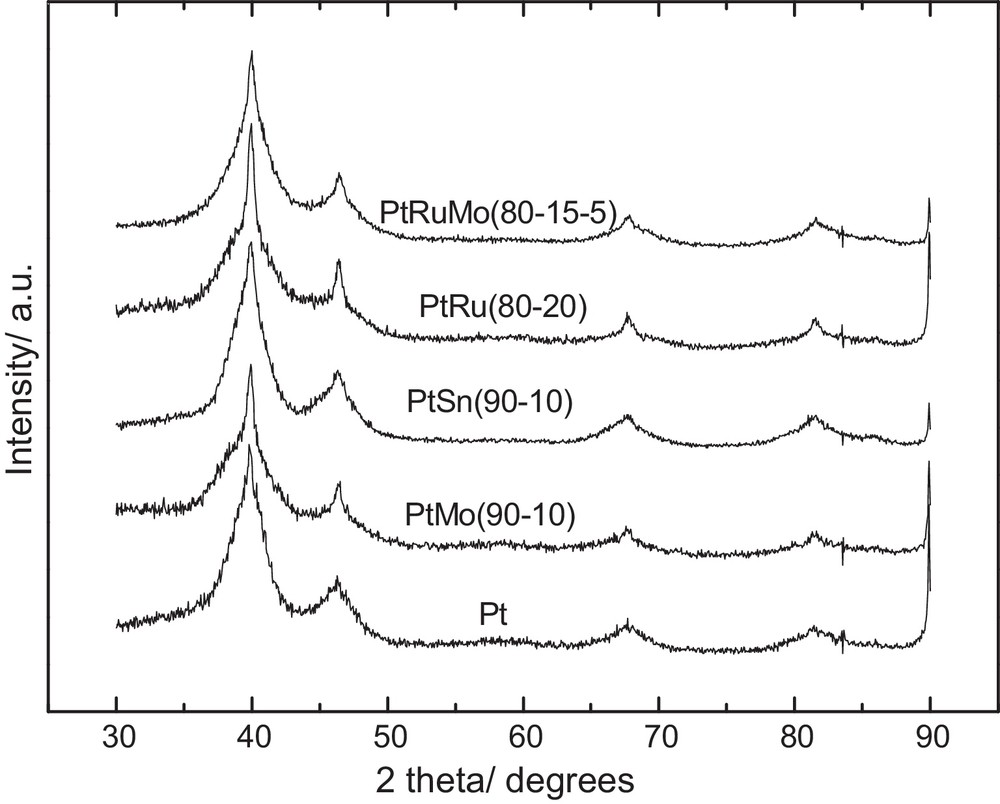
X-ray diffraction spectra of Pt/C, PtMo(90–10)/C, PtSn(90–10)/C, PtRu(80–20)/C, PtRuMo(80–15–5)/C with 40% metal loading.
3.2 Electrochemical measurements
Fig. 3 shows voltammetric results (positive going sweep) of 0.1 M DMM electrooxidation on Pt, PtMo, PtSn, PtRu, PtRuMo electrodes recorded in 0.2 M HClO4. Curves are corrected with background current subtraction measured without DMM.
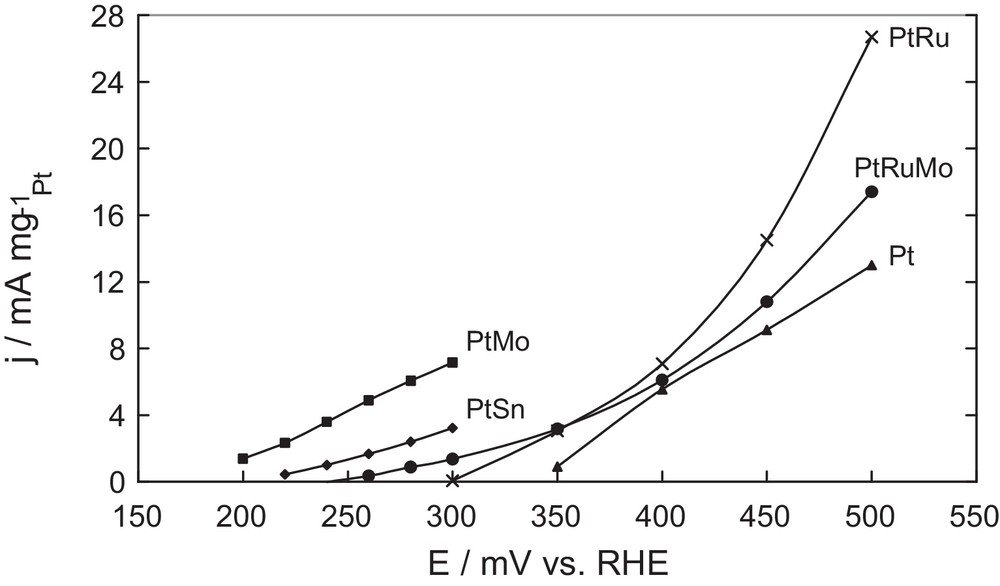
Background-current-subtracted voltammograms for 0.1 M dimethoxymethane in 0.2 M HClO4, at 50 mV s−1 and 20 °C. Pt/C, PtMo(90–10)/C, PtSn(90–10)/C, × PtRu(80–20)/C, PtRuMo(80–15–5)/C.
As shown in Fig. 3, the systems PtRu, PtMo, PtSn and PtRuMo exhibit higher oxidation rates of DMM compared to pure Pt, at all potentials. This should be due to the ability of metallic Ru, Mo and Sn to dissociate water molecules. DMM oxidation begins at low potentials (E < 200 mV) for PtMo catalyst, at 220 mV for PtSn catalyst, around 250 mV for PtRuMo catalyst, at 300 mV for PtRu catalyst, whereas for Pt catalyst it starts only at 350 mV.
At 300 mV, current density reached 8 mA mg−1 with catalyst PtMo, whereas PtRu and Pt catalysts do not present any oxidative current at this potential. Conversely, at 500 mV, PtRu catalyst presents a current density of 27 mA mg−1. Concerning PtMo and PtSn, these catalysts are subject to an irreversible oxidation at high potential. Mo and Sn can form irreversible oxides (according the Pourbaix diagram in our experimental conditions: Sn4+ and Mo6+), which are inactive for the reaction and can be dissolved in acid medium as observed by Lima et al. [16]. However, catalyst PtRuMo seems to be more stable at 500 mV; this phenomenon has also been observed during the electrooxidation of methanol [16].
3.3 In situ FTIR spectroscopic study of DMM adsorption and oxidation
In situ IR reflectance spectroscopy is a convenient method used to identify adsorbed species, intermediates, and reaction products. In latter studies, IR experiments on polycrystalline and single-crystal platinum were performed during adsorption and oxidation of DMM in order to propose a DMM oxidation mechanism [8,9].
In this paper we focused IR experiments more particularly on the study of bands of COads species (2050 cm−1) and CO2 (2345 cm−1), because it is well known that the expected main product of fuel cell tests is CO2, which is due to the oxidation of CO species. CO and CO2 IR bands have been also observed by other researchers during DMM electrooxidation [12,23,24].
Fig. 4 shows examples of SPAIR and SNIFTIR spectra for Pt/C and PtMo/C. In Fig. 4A, the adsorbed CO band (2050 cm−1) is observed as soon as 150 mV, whereas the CO2 band (2350 cm−1) appears later at 550 mV for the Pt catalyst during SPAIR experiments. The presence of the COads species is also detected during SNIFTIR experiments in Fig. 4B for Pt catalyst.

Column A. Single potential alteration infrared reflectance spectroscopy spectra resulting from the adsorption and oxidation of 0.1 M dimethoxymethane (DMM) on Pt/C and PtMo/C electrodes in 0.2 M HClO4 for different potentials. Spectra were calculated with a reference spectrum taken at 50 mV vs. RHE. Column B. Subtractive normalized interfacial Fourier transform infrared reflectance spectroscopy spectra of species formed during the adsorption and oxidation of 0.1 M DMM at Pt/C and PtMo/C electrode potential in 0.2 M HClO4 as a function of potential.
For PtMo catalyst, the COads species (2050 cm−1) appears as soon as 100 mV and the CO2 band (2350 cm−1) is detected sooner than with Pt catalyst, and, a short time afterwards, COads species at 400 mV (Fig. 4A).
Fig. 5 summarizes the in situ IR reflectance spectroscopic results obtained during the adsorption and oxidation of DMM with Pt/C, PtM/C (M = Ru, Mo, Sn) and PtRuMo/C. The curves in Fig. 5 represent the intensities of IR bands for adsorbed CO and CO2 in the vicinity of the electrode versus potential obtained by SNIFTIRS and SPAIRS respectively for 0.1 M DMM in 0.2 M HClO4. In Fig. 5, the authors considered that the absorption band of CO2 depends on the thin layer, each curve corresponding to a same experiment, assuming that the thin layer is the same for data of a same curve. Concerning the band intensities of CO, it is known that the band intensities do not reflect the concentration of molecules adsorbed at the electrode surface because of dipole–dipole coupling [25]. However as each set of curve representing the intensities of the CO band has been obtained during a same single experiment, a relative comparison is possible. In this way the intensities of the CO and CO2 band can be compared only within a single set of curves. The intensity of the CO band corresponds to the difference between the intensities peak to peak of the bipolar band. Concerning the corresponding potential, SNIFTIRS consists in the acquisition of interferograms at two different potentials, E1 and E2. The change of reflectivity, ΔR/R, is measured as a function of the applied potential where is the reflectivity taken at potential E1 and the reflectivity recorded at E2. All the potential range of interest was studied while keeping the difference (E2–E1) constant (E2–E1 = 200 mV). We used the average values of E1 and E2 for the SNIFTIRS potential range. We can note that previous researches used similar graphs to compare the potentials of band formation of CO and CO2 [17,26].
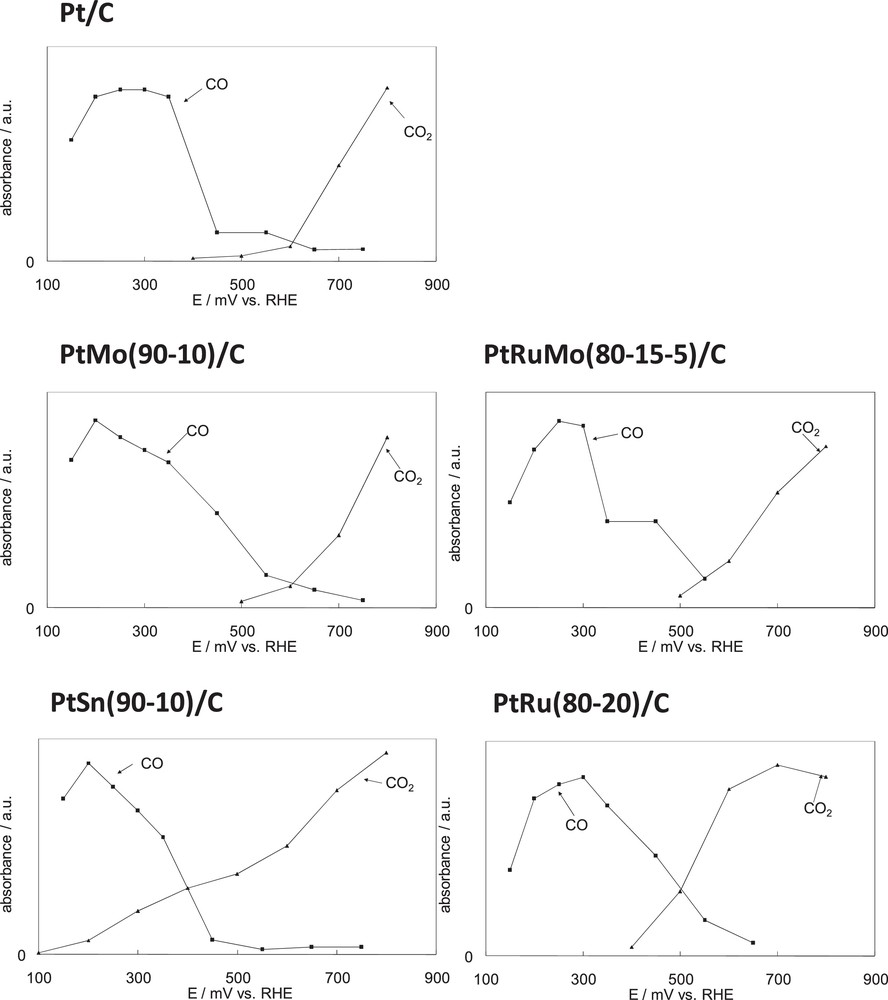
Comparison of the intensity of the COads and CO2 infrared band for different catalysts, obtained from subtractive normalized interfacial Fourier transform infrared reflectance spectroscopy (COads) and single potential alteration infrared reflectance spectroscopy (CO2) results for 0.1 M dimethoxymethane in 0.2 M HClO4.
According to Fig. 5, with Pt, PtMo and PtRuMo catalysts, it is obvious that the formation of carbon dioxide appears at potentials where COads has been removed from the surface of the electrode (400–500 mV). Similar results were observed during the electrooxidation of ethanol and methanol with PtSn and PtRu catalysts respectively [17,18]. The formation of CO2 increases until 800 mV. We can note that the kinetics of CO2 formation is stronger for PtRuMo > PtMo > Pt.
Unlike PtMo and PtRuMo catalysts, the formation of CO2 is visible before the disappearance of the adsorbed CO with PtSn and PtRu catalysts (100 mV for PtSn and for PtRu around 400 mV) with a faster kinetics than for Pt, suggesting that the reaction between adsorbed species CO and OH takes place at 100 mV on PtSn and at 400 mV on PtRu (as on Pt), which is a lower potential than that of PtMo and PtRuMo catalysts (i.e. 500 mV). Similar results have been observed for ethanol electrooxidation with Pt catalyst [18].
3.4 Fuel cell tests
Even if voltammetry and IR spectroscopy are very useful to check the activity of an electrocatalyst, it is necessary to observe the performance of the same catalysts in a complete fuel cell. The working conditions such as catalyst composition, pressure, temperature and flux of the reactants are crucial to estimate the actual performance of a system.
3.4.1 Influence of the catalyst nature
Fig. 6 reports the potential and power versus current density curves of a direct DMM fuel cell (DDMMFC). This figure presents a comparison of the performance of 1 M of DMM obtained with Pt, PtRu, PtMo, PtRuMo catalysts at 50 °C.
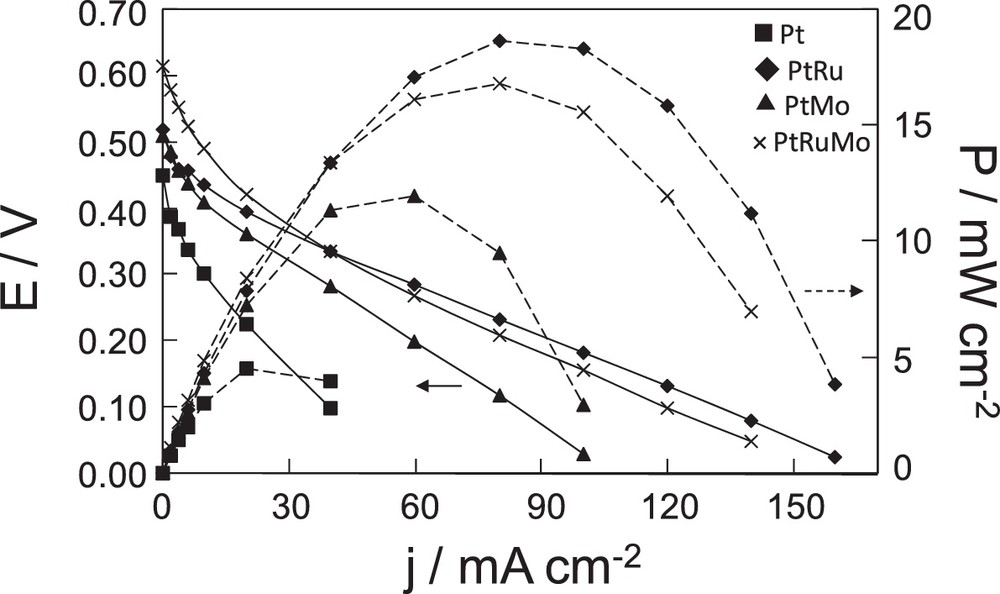
Fuel cell characteristics of a dimethoxymethane fuel cell with Pt, PtRu(80–20), PtMo(90–10), × PtRuMo(80–15–5) anodes [dimethoxymethane (DMM)] = 1 M and PDMM = 2 bar, .
Polarization curves show that the slopes are different according to the catalyst used. The PtRu anode presents a lower loss compared to other catalysts. The difference observed between open-circuit voltages (OCV) indicates a different activity of electrocatalysts for DMM electrooxidation and a possible effect of poisoning.
Fig. 6 allows us to evidence the highest activity of PtRuMo with an OCV of 0.61 V and PtRu with the best power density of 19 mW cm−2. This is in accordance with voltammetry results, in which the trimetallic anode presents the best activity at lower current density, whereas PtRu presents the best activity at 500 mV. The Pt anode shows the lowest performances, as observed from voltammetry experiments. Its OCV is the lowest one and its maximum power density reaches only 5 mW cm−2 at 50 °C. Performances of PtMo catalyst have been evaluated too. The OCV (0.51 V) is close to that of PtRu (0.52 V) and higher than that of Pt (0.45 V). The maximum power density of this catalyst is 12 mW cm−2 at 50 °C. PtSn catalyst was not tested in DDMMFC due to its low current intensity observed by voltammetry. PtMo was preferentially studied in a fuel cell because it presented a higher current in the same potential range.
The activity improvement in DDMMFC obtained with PtRu and PtRuMo anodes is clearly visible compared to the Pt anode as shown in Fig. 6. This tendency confirms the obtained results, presented in Fig. 3, where the highest current densities are observed from 350 mV on PtRu and PtRuMo catalysts. The highest performances of PtRu and PtRuMo electrocatalysts are visible for low values of difference of potential, with a shift of 100 mV with the trimetallic catalyst, and 50 mV with PtRu catalyst. However, the results obtained with PtMo catalyst are not in agreement with voltammetry experiments. During voltammetry, PtMo nanoparticles are shown to be able to electrochemically oxidize adsorbed CO at low potential, which coincides with the H2 oxidation potential. These electrochemical results suggested a better activity of PtMo catalyst at low potential compared to PtRu and PtRuMo catalysts. However, the observed OCV is only 0.51 V in Fig. 6. Previously the long-term stability of PtMo catalyst had been observed and defined as a critical effect for the fuel cell [27,28]. Lebedeva et al. observed that PtMo catalyst suffers from the gradual loss of Mo because of its dissolution into the electrolyte [27]. Papakonstantinou et al. [28] have shown that the oxidation of CO into CO2 at low potential depends on the oxidation state of the Mo surface species and especially on the reactive adsorbed OH species of MoO(OH)x due to the spontaneous dissociation of H2O. However, they postulated that at high potential, the formation of MoO3 is observed, leading to the deactivation in the low-potential range. The possible Mo dissolution could explain that the OCV of PtMo is not higher than the OCV of PtRu and PtRuMo during fuel cell tests.
As a conclusion, higher activities are shown on PtRuMo and PtRu, whereas PtMo and Pt present the lower activities during DDMMFC. In the next paragraph, the effect of temperature and DMM concentration on PtRuMo and PtRu catalysts is presented.
3.4.2 Influence of the reaction temperature
Fig. 7 displays potential and power versus current density curves of DDMMFC on PtRu (Fig. 7A), PtRuMo (Fig. 7B). This study was conducted at different temperatures in order to observe the effect of temperature during fuel cell tests with 1 M DMM. DMM pressure is adjusted so as to use liquid DMM.
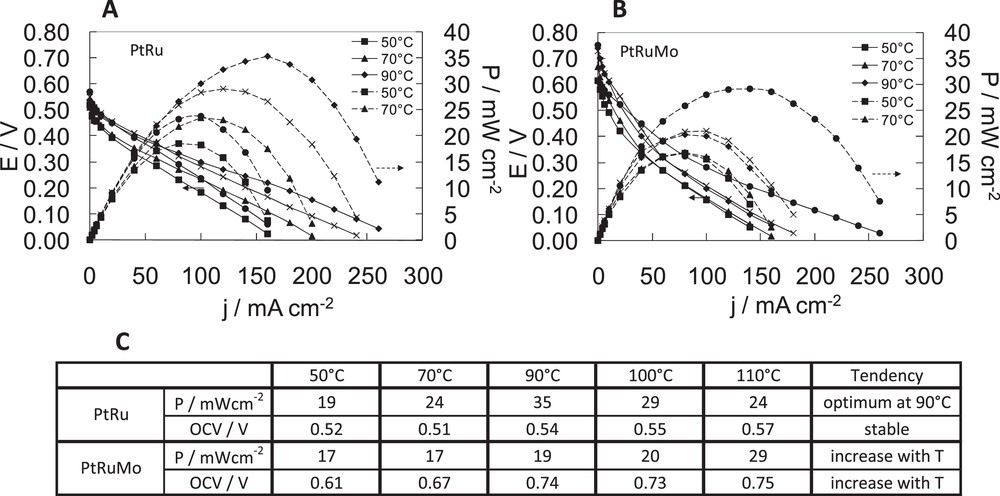
Fuel cell characteristics of dimethoxymethane fuel cell with A PtRu(80–20), B PtRuMo(80–15–5) anodes, C comparison of the effect of temperature for PtRu(80–20), PtRuMo(80–15–5) anodes. With [dimethoxymethane (DMM)] = 1 M and , PDMM = 2 bar at 50 °C and 70 °C, PDMM = 3 bar at 90 °C, PDMM = 4 bar at 100 °C, PDMM = 5 bar at 110 °C.
In Fig. 7B, a temperature increase allows the improvement of DMM oxidation on the PtRuMo anode. The lower performances are obtained at 50 °C and 70 °C with 17 mW cm−2. The higher performances are obtained at 110 °C with 29 mW cm−2 and 150 mA cm−2. Increased temperatures improve the OCV: 0.61 V at 50 °C, 0.67 V at 70 °C, 0.74 V at 90 °C, 0.73 V at 100 °C, 0.75 V at 110 °C.
In Fig. 7A, a temperature increase allows an improvement of DMM oxidation on the PtRu anode. The temperature increase leads to a slight increase of the OCV: 0.52 V at 50 °C, 0.51 V at 70 °C, 0.54 V at 90 °C, 0.55 V at 100 °C and 0.57 V at 110 °C. Then at low temperature (50 °C), the reaction is less activated (19 mW cm−2), the beneficial effect of temperature on the DMM activation process being especially visible at 90–100 °C. However, at 110 °C, a decrease of fuel cell performances, which are comparable to the ones obtained at 70 °C, is observed. The best performance is obtained with PtRu catalyst at 90 °C: 35 mW cm−1 with 150 mA cm−2. This result is quite similar to the one obtained by Chetty et al. [29], where power reached 29.4 mW cm−2 for PtRu/C in the corresponding current density range of 100–130 mA cm−2 at 60 °C. A possible explanation of the decrease of activity at the highest temperature could be a possible hydrolysis of DMM. As observed by Watanabe et al., DMM is a stable solution at room temperature, but at high temperature, it could be easily hydrolyzed into methanol [11]. Then, at 100–110 °C a chemical hydrolysis should occur on the PtRu catalyst and explain this phenomenon. In Fig. 7C, we can note that PtRuMo is not affected by this phenomenon at the highest temperatures because OCV and power performances increase with the temperature. Previous authors observed for the methanol electrooxidation that PtRuMo is superior to PtRu with respect to catalytic activity, durability and CO-tolerance [16,30,31].
3.4.3 Influence of DMM concentration
Fuel cell tests performed previously show the effect of the composition of catalysts and of the temperature on direct DMM fuel cell with 1 M DMM. Fig. 8 reports potential and power versus current density curves of DDMMFC on PtRu and PtRuMo respectively, with 1 M and 2 M DMM to check the effect of DMM concentration on direct DMM fuel cell.
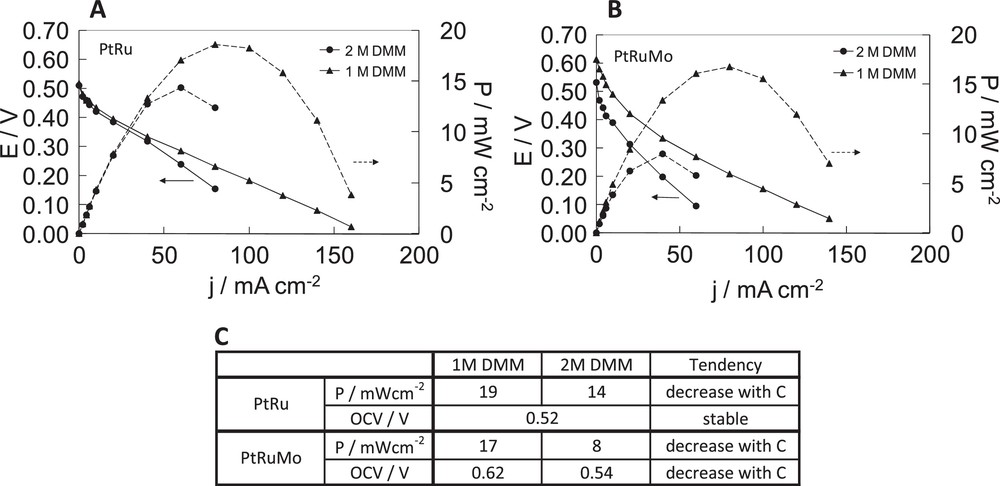
Fuel cell characteristics of dimethoxymethane fuel cell with A PtRu(80–20), B PtRuMo(80–15–5) anodes, C comparison of the effect of temperature for PtRu(80–20), PtRuMo(80–15–5) anodes. With [dimethoxymethane (DMM)] = 1 M and , PDMM = 2 bar at 50 °C and 70 °C, PDMM = 3 bar at 90 °C, PDMM = 4 bar at 100 °C, PDMM = 5 bar at 110 °C.
In Fig. 8A, DMM concentration does not change the OCV, during fuel cell tests carried out with PtRu catalyst. On the other hand from 25 mA cm−2 onward, the activity decreases when using 2 M DMM. The use of 1 M DMM allows higher current densities than that of 2 M DMM.
In Fig. 8B, DMM concentration has an effect on OCV during fuel cell tests carried out with PtRuMo catalyst. It decreases with the increase of DMM concentration. The use of 1 M DMM allows higher current densities than the use of 2 M DMM.
The maximum power density obtained at 50 °C varies from 19 mW cm−2 with 1 M DMM to 14 mW cm−2 with 2 M DMM, which corresponds to a loss of 26% with PtRu catalyst. The maximum power density varies from 17 mW cm−2 with 1 M DMM to 8 mW cm−2 with 2 M DMM, which corresponds to a loss of 53% with PtRuMo catalyst.
For both catalysts, the slopes increase with DMM concentration suggesting an increase of Ohmic resistance in DDMMFC and a smaller mass transfer with 2 M DMM. In a fashion similar to the results observed at different temperatures during DDMMFC on PtRu, the influence of DMM concentration shows a decrease of activity with higher DMM concentrations, which can be due to a possible chemical hydrolysis occurring with higher DMM concentrations. In Fig. 8C, we can see the effect of DMM concentration on the catalytic activity (power performances and OCV). As for the effect of temperature, PtRuMo catalyst is less affected than PtRu catalyst by a chemical hydrolysis of DMM into methanol. As explained previously PtRuMo catalyst is more electro-catalytically active for the methanol electrooxidation and more CO-tolerant than PtRu catalyst [16,30,31].
4 Discussion
IR spectroscopy experiments were previously performed on polycrystalline and single-crystal platinum electrodes [8,9] and in this paper on Pt-based nanoparticles supported on C. In this paper IR spectroscopy focused on the expected main intermediate and the product of reaction of DDMMFC, i.e. COads species and CO2. On Pt, PtMo and PtRuMo, the formation of carbon dioxide appears at potentials where COads had been removed, suggesting that COads acts as a poisoning species which blocks the active sites. Conversely, for PtSn catalyst, carbon dioxide formation starts when large fraction of CO are still adsorbed on the electrode. COads seems to be a reaction intermediate and not a poisoning species due to the fact that the amount of CO2 is related to COads oxidation during IR experiments. Concerning PtRu catalyst, COads acts as a poison up to 400 mV vs. RHE and as a reaction intermediate after 400 mV vs. RHE. A similar phenomenon is observed during the electrooxidation of methanol (PtRu is an active catalyst for direct methanol fuel cell) [17,18], in which COads species is a poisoning species at low potential.
Usually, COads is oxidized with the help of OHads produced by water dissociation on the catalyst close to the COads species. Catalysts that produce OHads at lower potential should raise the activity in voltammetry and DDMMFC. By voltammetry, below 300 mV, PtMo and PtSn give the largest currents, suggesting that they easily produce OHads. Conversely, Pt catalyst, which produces OHads only above 400 mV, gives the smallest current. PtRu and PtRuMo allow an intermediate state: they are more active than Pt and less active than PtMo and PtSn at low potential, but as they can be used at higher potential without damages, they give the overall largest currents. More precisely, at low potential (< 350 mV), PtRuMo is better than PtRu and the opposite is true at higher potential (> 350 mV).
Watanabe et al. have observed during DMM electrooxidation on polycrystalline Pt and PtRu alloy that the oxidation current and the onset potential were comparable to those of methanol electrooxidation for a temperature range from 17 to 120 °C [11]. During our previous study on polycrystalline Pt and single crystals [8,9], authors observed that DMM electrooxidation is a complex mechanism via methanol formation. It is known in the case of methanol oxidation on Pt/C that COads is the poisoning species, whereas in the case of PtRu/C, COads is both a poisoning species and a reactive intermediate. The enhancement of catalyst activity is due to the ability to activate water at lower potential in the presence of Ru. This phenomenon should occur probably in our case and explains why PtRu is an active catalyst in voltammetry (at high potential) and in DDMMFC.
The results of fuel cell tests are slightly different, especially for PtMo catalyst. Voltammetry results let expect that it had the highest OCV but during fuel cell tests, performances are far from that of PtRuMo and equal to the one of PtRu; moreover an increase of the current gave worse results. This observation can be explained by the fact that the potential of the anode is higher than 300 mV vs. RHE, leading to the irreversible formation of higher oxidation states of Mo (Mo6+) as explained in [28], lowering the ability to produce OHads. Moreover, the dissolution of Mo into the electrolyte cannot be excluded [27]. Pt catalyst is the least active one due to its OCV and it does not produce high current and high electrical power in agreement with its voltammetry results, i.e. Pt starts oxidizing DMM at the highest potential and gives the smallest current. PtRu and PtRuMo behave similarly, with the best performances. PtRuMo has the highest OCV, corresponding to an oxidation of DMM starting at lower potential by voltammetry, but at larger current PtRu has a best electrical power, which can be related to the fact that PtRu has the larger current at 500 mV by voltammetry.
However, at high temperature and high DMM concentration conditions with PtRu anode and at high DMM concentration condition with PtRu and PtRuMo anodes, a deactivation is observed at high temperature, which can be explained by the hydrolysis of DMM in solution, out of the electrode, as explained by Watanabe et al. [11]. As demonstrated previously during DMM oxidation on bulk polycrystalline platinum [8], the addition of a small amount of methanol in the solution quickly deactivated the electrode by producing COads. PtRuMo is less affected by this chemical hydrolysis of DMM into methanol because it was observed for methanol electrooxidation that PtRuMo catalyst is superior to PtRu catalyst with respect to its catalytic activity, durability and CO–tolerance [16,30,31].
5 Conclusion
In this paper, DMM electrooxidation was evaluated by voltammetry, FTIR spectroscopy, and DDMMFC on Pt and PtM (M = Ru, Mo, Sn). In voltammetry experiments, PtSn and PtMo catalysts exhibit the highest current density at low potential (300 mV vs. RHE), whereas at high potential PtRu and PtRuMo catalysts exhibit the highest currents (500 mV vs. RHE). Using in situ infrared reflectance spectroscopy, COads and CO2 bands were observed. COads appears to be a poison for Pt, PtMo, and PtRuMo, and a reaction intermediate on PtSn. Then COads is a poison until 400 mV vs. RHE and a reaction intermediate on PtRu. Moreover, during DDMMFC, PtRu and PtRuMo catalysts, (efficient for methanol electrooxidation) exhibit the best performances for DMM during fuel cell tests. Conversely, PtMo, which shows promising performances at low potential in voltammetry experiments, did not exhibit good results at higher potential during fuel cell tests. This phenomenon could be explained by a possible dissolution of Mo and/or the formation of molybdenum oxides that are not active for the dissociation of water at low potential and did not lead to the formation of CO2.
Acknowledgements
The authors thank the Région Poitou-Charentes for the financial support, Stéphane Pronier, and Sandrine Arrii Clacens for their technical help.