

1 Introduction
Phenothiazine, one of the oldest bioactive heterocyclic compounds, is present as major pharmacophore group in many types of drugs in clinical use (tranquillizers [1,2], sedatives, anthelmintic [3,4], anti-inflammatory, antimalarial, antimicrobial, antiparkinsonian [5,6], anticonvulsant [7], antitubercular drugs, etc.), as well as in other biologically active compounds, such as bactericidal products, pesticides [8], compounds with analytical applications (redox indicators and reagents in spectrophotometric determinations) [9,10], high temperature antioxidants for lubricants and dyes [11].
Nitrones are versatile starting materials, with a wide range of applications, including cycloaddition reactions with alkynes, alkenes and α,β-unsaturated aldehydes, to afford carbacephem skeletons [12], isoxazolines [13] and isoxazolidine [14], or generating new carbon–carbon bonds by nucleophilic additions [15]. The interest for the nitrone derivatives is also supported by the antibacterial, antifungal and antiproliferative effects of the above-mentioned classes of organic heterocycles as well as their own antioxidant [16] or antiproliferative properties [17].
The ability of nitrones to trap free radicals and release nitric oxide in vivo and in vitro is important in the treatment of cerebral ischemia, of neurodegenerative diseases, as well as in the prolongation of lifespan [16,18] – i.e. phenyl-tert-butyl-nitrone (PBN or NXY-059) is a free radical scavenger with neuroprotective properties [19].
The most popular method for the preparation of nitrones is the condensation of aldehydes or ketones with N-monosubstituted hydroxylamines [20,21]. The hydroxylamines are generated in situ via the reduction of nitro compounds with zinc powder in the presence of weak acids (NH4Cl or AcOH). Other methods are the direct oxidation of secondary amines to the corresponding nitrones using various metal salts (copper, silver, lead, selenium, mercury, and ruthenium) [22,23] as well as organic oxidants and transition metal complexes (i.e. (salen)Mn(III) [24,25]), the oxidation of imines with m-CPBA, dimethyldioxirane (DMD) or the photochemical oxidation (λ = 350 nm) of aldimines in the presence of O2 over a TiO2 suspension [26].
Microwave irradiation is an excellent alternative for a wide range of heterocyclic syntheses, due to its well-known advantages: reaction rate enhancement, reaction time shortage, better yields, lesser solvent usage [27–30]. According to literature data, the improvements achieved under microwave irradiation came from different effects generated by the interaction of electromagnetic waves during the reaction process as compared to the temperature effect [31].
The present paper aims at describing an efficient microwave synthesis of aromatic and heteroaromatic nitrone derivatives together with their antifungal and antibacterial activities.
2 Results and discussion
The new phenothiazinyl N-aryl or N-alkyl nitrones 3a–3j, 4 were synthetized with a modified procedure [16] by condensation reactions of the formyl phenothiazine 1a–d and hydroxylamines 2a–h in an ethanol/water mixture (5/1, v/v), with low to good yields (16–84%, Scheme 1).
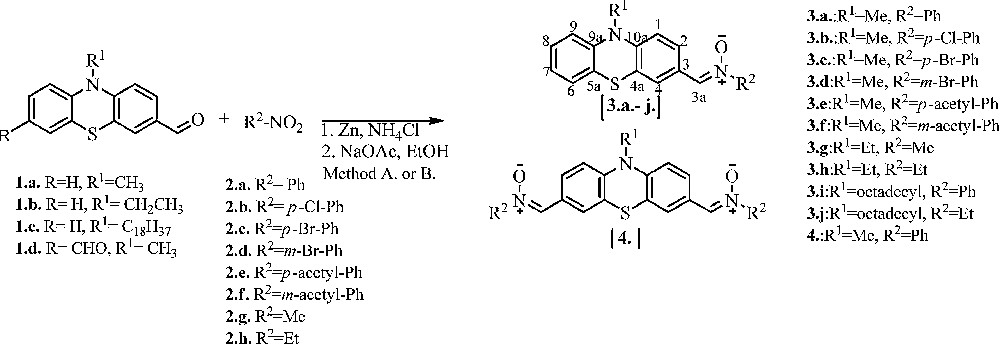
Synthesis of mono-phenothiazinyl 3a–3j, and di-phenothiazinyl 4 nitrones. A. MW, 100 °C, 10 min. B. EtOH, 100 °C, 1–6 h.
Hydroxylamines containing electron-withdrawing and electron-donating groups were obtained by reducing the corresponding nitro compounds with ammonium chloride and zinc (powder). A comparison of the reaction conditions and yields after purification by flash chromatography is presented in Table 1. Irrespective of the reaction conditions (classical or microwave-activated process), nitrones 3a–3d and 4 with electron-withdrawing substituents, such as phenyl, chlorophenyl and bromophenyl, were isolated in low yields: 16 to 35% with classical heating and 28–50% under microwave heating (Table 1). Better yields were obtained for nitrones 3g, 3h, 3j with electron-donating alkyl groups (65–77% with classical heating and 81–84% under microwave heating). The shorter reaction time as well as higher yields obtained under microwave conditions makes this a more advantageous method than the classical synthesis (Table 1).
The synthesis of phenothiazinyl-nitrones by microwave irradiation and by convection heating.
| Entry | Compound | R1 | R2 | Convection heating | Microwave | ||
| Yield (%)a | Reaction time (min) | Yield (%)a | Reaction time (min) | ||||
| 1 | 3a | Me | C6H5 | 20 | 120 | 37 | 10 |
| 2 | 3b | Me | 4-Cl-C6H4 | 16 | 60 | 30 | 10 |
| 3 | 3c | Me | 4-Br-C6H4 | 21 | 60 | 28 | 10 |
| 4 | 3d | Me | 3-Br-C6H4 | 35 | 480 | 50 | 10 |
| 5 | 3e | Me | 4-acetyl-C6H4 | 30 | 90 | 44 | 10 |
| 6 | 3f | Me | 3-acetyl-C6H4 | 41 | 60 | 56 | 10 |
| 7 | 3g | Et | Me | 65 | 75 | 84 | 10 |
| 8 | 3h | Et | Et | 77 | 75 | 81 | 10 |
| 9 | 3i | Octadecyl | C6H5 | 52 | 120 | 74 | 10 |
| 10 | 3j | Octadecyl | Et | 70 | 75 | 84 | 10 |
| 11 | 4 | Me | C6H5 | 34 | 60 | 61 | 10 |
a Isolated yield after column chromatography.
A notable problem associated with many reactive nitrones is dimerization [32]. In the reactions described in this work, no dimerization product was obtained, but the corresponding imines were formed as byproducts during the reaction as well as the purification by flash chromatography. The new class of phenothyazinyl N-substituted nitrones is highly sensitive and decomposes on heating (120–130 °C) without melting.
Our interest in nitrone derivatives is related to their potential antifungal and/or antibacterial properties. According to our goal, a number of previously reported phenyl-nitrones 5a–d [33,34], 6a, b, f [35] 6d [36], 6e, g, h [37,38], 6m, o [39], 6k [40], 6j, n, q [41,42] was also synthesized (see Fig. 1).
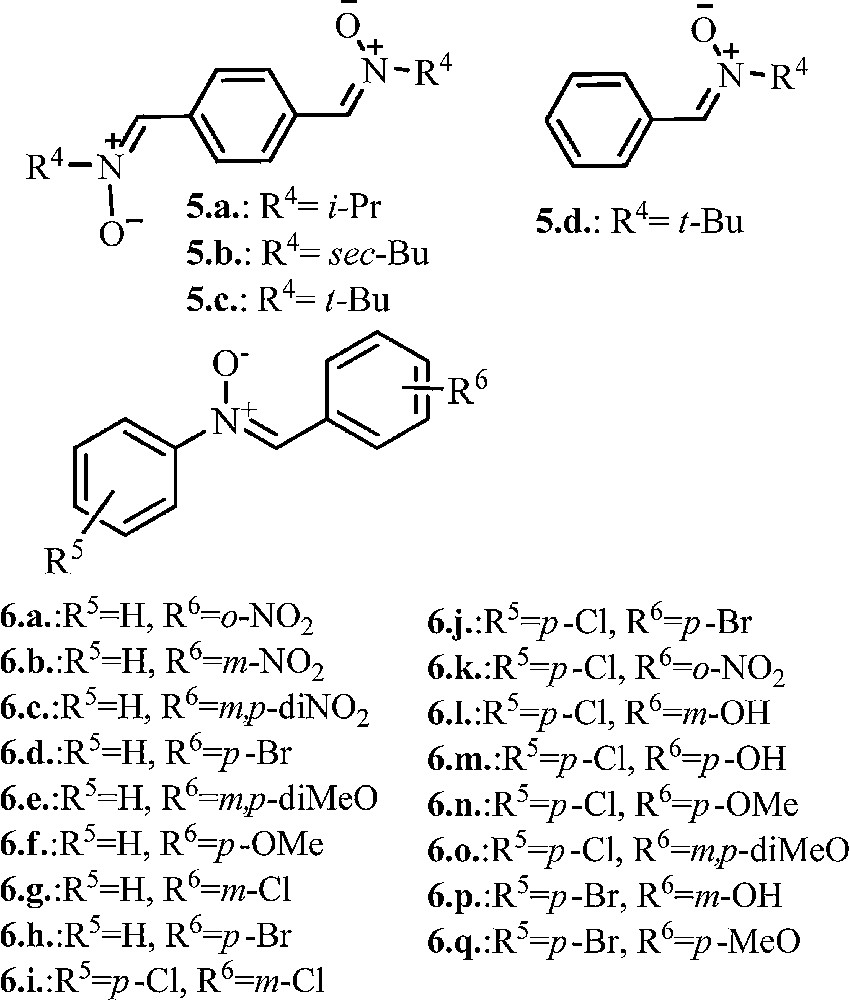
Structure of phenyl mono-, and di-nitrones.
The new phenyl-nitrones 6c, 6i, 6l, 6p were prepared by the one-pot procedure from the corresponding aromatic amines, nitrobenzene derivatives and zinc powder in acetic acid.
The structures of the newly-synthesized nitrones were confirmed by their 1H NMR, MS and FT–IR spectra. In the 1H NMR spectra, the characteristic signal for the phenothiazinyl-nitrones 3a–j and 4 is the singlet generated by the imine proton at ∼ 7.15 ppm, (value for nitrone 3g) and 7.79 ppm, for nitrone 4 (see experimental part).
In the UV spectra, the new phenothiazinyl-nitrones 3a–f, 3i and 4 present three absorption bands, while the nitrones 3g, 3h and 3j present only two. The intense absorption band at 302–318 nm is due to the π–π* electronic transition, while the moderate one, in the range 370–400 nm, with the extinction coefficient (ɛ) between 8 × 103 and 2 × 104, is due to the n–π* electronic transitions within the phenothiazinyl-imino chromophore. The band around 270 nm is correlated with the presence of the phenyl group. The bathochromic shift of the absorption maxima for compound 4 compared to compound 3a is due to the presence of the second imine unit. The presence of the molecular ion, the cleavage of the N-alkyl bond in the phenothiazine unit and the N–O bond in the nitrone unit are common features of the mass spectra of these new nitrones.
All phenothiazinyl-nitrones display light-blue fluorescence with large Stokes shifts (Δν = 7076–7888 cm−1) and the quantum yields (ΦF) between 1.90 for 3i and 4.7% for 3h.
The quantum yields were calculated [43] using perylene in cyclohexane as standard, Table 2.
Optical properties of phenothiazinyl-nitrones.
| Compound | λabs [nm] (ɛ mol−1cm−1)a | λem [nm] | Stokes shift Δν [cm−1] | ΦFb |
| 3a | 315, 272, 393(9517) | 551 | 7255 | 7.83 |
| 3b | 316, 274, 385(8277) | 521 | 7142 | 5.59 |
| 3c | 316, 275, 380(10048) | 518 | 7103 | 5.01 |
| 3d | 317, 275, 397(9561) | 555 | 7535 | 5.43 |
| 3e | 312, 241, 398(8480) | 550 | 7211 | 7.77 |
| 3f | 314, 271, 402(9398) | 542 | 7197 | 8.33 |
| 3g | 302, 377(14026) | 519 | 7275 | 3.50 |
| 3h | 302, 377(21778) | 515 | 7076 | 4.70 |
| 3i | 314, 275, 382(14010) | 569 | 7888 | 1.90 |
| 3j | 302, 376(22408) | 518 | 7235 | 4.20 |
| 4 | 318, 419(16280) | 554 | 7595 | 5.43 |
a UV–Vis measured in CH2Cl2.
b Quantum yields against perylene standard in cyclohexane.
3 Biological assay
The antifungal and antibacterial activities of the new and already reported nitrone derivatives mentioned above were evaluated against several Candida species Gram negative bacteria such as E. coli, Citrobacter spp, Morganella spp, Pseudomonas aeruginosa, Klebsiella pneumoniae (± ESBL), Proteus spp, Acinetobacter spp, and the Gram positive bacterium Staphylococcus aureus.
Antifungal and antibacterial activities were investigated in triplicate, by the disk diffusion method at 8 μM nitrone concentration. Low inhibition potentials (diameter of inhibition area 3–5 mm) were recorded with few exceptions, two Candida strains (Candida albicans and Candida krusei), were sensitive to compounds 5a, 6a and 6p, with the diameter of inhibition area at 10–11 mm.
For the phenothiazinyl-nitrones 3a, 3h and 4, the tests were repeated at higher concentration (25 μg/disk), with the same standard fluconazole concentration on the disk. The fluconazole-resistant Candida strains involved in these tests were Candida parapsilosis, famata and glabrata (isolated from blood), and albicans and krusei, isolated from the urine of patients. For the C. glabrata and C. krusei, the investigated nitrones did not present inhibition activities. In the case of C. famata, compounds 3a and 4 present minimum inhibition activity (d: 3 mm and 4 mm, respectively). A limited inhibition activity (d: 3 mm) was observed for the phenothiazinyl nitrone 3h on C. albicans. Only the C. parpsilosis was sensitive to phenothiazinyl-nitrones, with the inhibition diameter of 6 mm for 3a, 7 mm for nitrone 3h and 6 mm for nitrone 4.
4 Conclusions
In conclusion, a novel class of functionalized phenothiazinyl-nitrones with aromatic and aliphatic chains was synthesized and structurally characterised. Higher yields in a shorter reaction time were obtained under microwave irradiation compared to classical heating. The new nitrones and some known phenyl-nitrones were screened for their antibacterial and antifungal activity against several Candida species and Gram negative or positive bacteria, with moderate results.
5 Experimental
5.1 General information
All reagents and solvents were obtained from commercial sources and were used without purification. Phenyl-nitrones 5a–5d, 6a–6q were prepared by the classical synthesis method according to the literature data [29–38].
The infrared spectra were recorded on a Bruker Vector 22 FT–IR spectrometer from 4000 to 600 cm−1 using KBr pellets. The NMR spectra were recorded at room temperature on a Bruker Avance instrument (1H/13C: 300 MHz/75 MHz) using deuterated solvents; chemical shifts are reported in ppm relative to TMS, J values are given in Hz. For elemental analyses (C, N, H, S), a Thermo Flash 1112 Series elemental analyser was used. Mass spectra were measured on a Shimadzu QP-2010 PLUS GC-MS mass spectrometer (DI, EI-70 eV). The reaction mixtures were irradiated in a CEM Discover LabMate microwave reactor. UV–Vis spectra were recorded in dichloromethane with a PerkinElmer Lambda 35 UV–Vis spectrophotometer; emission spectra were measured in dichloromethane using a PerkinElmer FL 55 fluorescence spectrophotometer. Flash chromatography was performed using silica gel (60 Å, particle size 40–63 μm). The reactions were monitored by thin-layer chromatography (TLC) on aluminium plates precoated with silica gel (60, F254) and visualized by UV light. For the microbial identification, the Vitek 2 bioMerieux system was used.
5.2 Antimicrobial assay
In vitro antifungal and antibacterial activities for the nitrone derivatives were tested according to the disk diffusion method using 5-mm-diameter disk papers [44]. The fungi and bacteria isolated from various body fluids (urine, sputum, wound, blood) were grown [45] onto a Mueller–Hinton culture medium by incubation at 37 °C for 48 h. After identification, according the CLSI standards [46], each colony was suspended in 5 mL of a sterile saline solution. The turbidity of the inoculum was adjusted to McFarland Turbidity Standard No. 0.5, and the inoculums were added to different Petri plates filled with a Mueller–Hinton culture medium, or Mueller–Hinton and methylene blue culture medium for Candida spp. strains. The five sterile filter paper disks were aseptically placed on the inoculated plate, at equal distance from each other. On each plate, the paper disks were impregnated aseptically by pipetting with DMSO (10 μL, the solvent was used as a reference) or with each one of the investigated compounds 3a–j, 6a–q (10 μL, 8 mM solution in DMSO). Agar plates were incubated at 37 °C for 24 h, 48 h and 72 h, respectively, and the diameter of the inhibition area was measured. Tests were performed in triplicate.
5.3 General experimental procedures for the synthesis of phenothiazinyl-nitrones
5.3.1 Preparation of hydroxylamines
The nitro derivative (1 mmol) disolved in ethanol (5 mL), ammonium chloride (0.053 g, 1 mmol) and water (1 mL) were mixed under stirring and cooled on an ice bath to 5 °C, after which zinc dust (0.13 g, 2 mmol) was added slowly. The suspension thus obtained was used without further purification.
5.3.1.1 Method A: microwave-assisted synthesis
In a 10-mL microwave reaction vessel equipped with a stirrer and a cap, the corresponding hydroxylamine solution (1 mmol) and sodium acetate (0.123 g, 1.5 mmol) were added to the corresponding phenothiazine derivative (1 mmol). The reaction mixture was subjected to microwave irradiation using a power level of 100 W at 100 °C for 10 min. After cooling, the mixture was diluted with water and dichloromethane, filtered and the residue washed with the organic solvent. The organic layer was separated, dried on anhydrous sodium sulphate and filtered. The solvent was removed under vacuum and the residue was purified by flash column chromatography on silica with various eluent systems.
5.3.1.2 Method B: classical synthesis
Hydroxylamine was synthesized as described above. To the hydroxylamine suspension (1 mmol), the corresponding phenothiazine derivative (1 mmol) and sodium acetate (0.123 g, 1.5 mmol) were added; the reaction mixture was stirred at 100 °C for 1–6 h. After cooling, the mixture was worked up as described before for microwave-assisted synthesis.
Synthetic procedures for each phenothiazinyl-nitrones and copies of 1H NMR, 13C NMR are included as Supplementary material (S) and are available on the website of this journal.
5.3.2 (Z)-N-((10-methyl-10H-phenothiazin-3-yl)methylene)aniline oxide 3a
Yellowish–green solid; yield 37% (0.12 g) was obtained by microwave-assisted heating, and 20% (0.06 g) by convective heating. Decomposition: 121 °C without melting. IR (KBr) νmax/cm−1 3015, 1550, 679; MS (70 eV), m/z (%): 332 (100, [M]+), 317 (18); 1H NMR (CDCl3) δ 3.41 (s, 3H, N–CH3), 6.86–6.81 (m, 2H, H1, H9), 6.95 (t, 1H, H7, ), 7.13 (d, 1H, H6, ), 7.19 (t, 1H, H8, , ), 7.49–7.44 (m, 3H, Hc, Hd), 7.75 (d, 2H, Hb, ), 7.78 (s, 1H, H3a), 8.18 (d, 1H, H4, ), 8.26 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 147.9, 144.7, 133.6, 133.6, 129.81, 129.3, 129.2, 127.7, 127.6, 127.3, 125.4, 123.4, 123.2, 121.7, 114.5, 113.9, 35.7; anal. calcd. for C20H16N2OS: C, 72.26; H, 4.85; N, 8.43; S, 9.65; O, 4.81; found: C, 72.36; H, 4.89; N, 8.40; S, 9.58.
5.3.3 (Z)-4-chloro-N-((10-methyl-10H-phenothiazin-3-yl)methylene)aniline oxide 3b
Yellowish–green solid; yield 30% (0.11 g) was obtained by microwave-assisted heating, and 16% (0.05 g) by convective heating. Decomposition: 115 °C without melting. IR (KBr) νmax/cm−1 2894, 1611, 812, 734; MS (70 eV, EI), m/z (%): 366 (100, [M]+), 351 (32), 350 (21); 1H NMR (CDCl3) δ 3.42 (s, 3H, N–CH3), 6.87–6.82 (m, 2H, H1, H9), 6.96 (t, 1H, H7, ), 7.18–7.12 (m, 2H, H6, H8), 7.43 (d, 2H, Hb, ), 7.72 (d, 2H, Hc, ), 7.76 (s, 1H, H3a), 8.16 (d, 1H, H4, ), 8.25 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 153.2, 153.1, 148.3, 146.7, 134.2, 133.2, 131.7, 129.3, 127.6, 125.6, 124.9, 122.6, 120.0, 120.1, 119.9, 118.5, 117.3; anal. calcd. for C20H15ClN2OS: C, 65.48; H, 4.12; N, 7.64; S, 8.74; O, 4.36; found: C, 65.47; H, 4.14; N, 7.63; S, 8.73.
5.3.4 (Z)-4-bromo-N-((10-methyl-10H-phenothiazin-3-yl)methylene)aniline oxide 3c
Yellowish–green solid; yield 28% (0.11 g) was obtained by microwave-assisted heating, and 21%, (0.08 g) by convective heating. Decomposition: 125–127 °C without melting. IR (KBr) νmax/cm−1 3124, 1542, 757, 593; MS (70 eV), m/z (%): 410 (100, [M]+), 412 (96), 394 (27), 396 (23); 1H NMR (CDCl3) δ 3.41 (s, 3H, N–CH3), 6.86–6.82 (m, 2H, H1, H9), 6.96 (t, 1H, H7, , ), 7.21–7.11 (m, 4H, H6, H8, Hb), 7.60 (d, 2H, Hc, ), 7.76 (s, 1H, H3a), 8.16 (d, 1H, H4, ), 8.23 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 148.1, 144.6, 133.7, 132.3, 129.4, 129.2, 128.3, 127.7, 127.4, 125.4, 123.2, 114.6, 113.9; anal. calcd. for C20H15BrN2OS: C, 58.40; H, 3.68; N, 6.81; S, 7.80; O, 3.89; found: C, 58.42; H, 3.70; N, 6.82; S, 7.84.
5.3.5 (Z)-3-bromo-N-((10-methyl-10H-phenothiazin-3-yl)methylene)aniline oxide 3d
Yellowish–green solid; yield 50% (0.21 g) was obtained by microwave-assisted heating, and 35% (0.14 g) by convective heating. Decomposition: 127 °C without melting. IR (cm−1, KBr) νmax 3155, 1608, 762, 593; MS (70 eV), m/z (%): 410 (100, [M]+), 412 (98), 394 (31), 396 (26); 1H NMR (CDCl3) δ 3.41 (s, 3H, N–CH3), 6.85–6.82 (m, 2H, H1, H9), 6.97 (t, 1H, H7, ), 7.15 (d, 1H, H6, ), 7.19 (t, 1H, H8, , ), 7.33 (t, 1H, He, , ), 7.76 (d, 1H, Hf, ), 7.71 (d, 1H, Hd, ), 7.77 (s, 1H, H3a), 7.96 (d, 1H, Hb, ), 8.19 (d, 1H, H4, ), 8.24 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 149.3, 148, 144.5, 133.8, 132.7, 130.5, 129.4, 127.7, 127.6, 127.3, 125, 124.9, 123.3, 123.2, 122.8, 122.7, 120.2, 114.5, 113.8, 35.6; anal. calcd. for C20H15BrN2OS: C, 58.40; H, 3.68; N, 6.81; S, 7.80; O, 3.89; found: C, 58.45; H, 3.73; N, 6.78; S, 7.89.
5.3.6 (Z)-4-acetyl-N-((10-methyl-10H-phenothiazin-3-yl)methylene)aniline oxide 3e
Orange-red solid; yield 44% (0.16 g) was obtained by microwave-assisted heating, and 30% (0.10 g) by convective heating. Decomposition: 123 °C without melting. IR (KBr) νmax/cm−1 2986, 1721, 1573, 589MS (70 eV), m/z (%): 374 (100, [M]+), 359 (42), 358 (37); 1H NMR (CDCl3) δ 2.62 (s, 3H, CH3), 3.38 (s, 3H, N–CH3), 6.82–6.79 (m, 2H, H1, H9), 6.94 (t, 1H, H7, , ), 7.11 (d, 1H, H6, ), 7.11 (t, 1H, H8, ), 7.85 (d, 2H, Hb, ), 7.84 (s, 1H, H3a), 8.01 (d, 2H, ), 8.19 (d, 1H, H4, ), 8.23 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 196.8, 151.5, 148.2, 144.4, 137.7, 134.3, 129.6, 129.4, 127.7, 127.7, 127.3, 124.8, 123.3, 122.7, 121.7, 114.6 113.8, 35.6, 26.8; anal. calcd. for C22H18N2O2S: C, 70.57; H, 4.85; N, 7.48; S, 8.56; O, 8.55; found: C, 70.59; H, 4.87; N, 7.47; S, 7.57.
5.3.7 (Z)-3-acetyl-N-((10-methyl-10H-phenothiazin-3-yl)methylene)aniline oxide 3f
Orange-red solid; yield 56% (0.21 g) was obtained by microwave-assisted heating, and 41% (0.14 g) by convective heating. Decomposition: 123–126 °C without melting. IR (KBr) νmax/cm−1 3086, 1811, 1636, 593; MS (70 eV), m/z (%): 347 (100, [M]+), 359 (32), 358 (22); 1H NMR (CDCl3) δ 2.65 (s, 3H, CH3), 3.40 (s, 3H, N–CH3), 6.85–6.80 (m, 2H, H1, H9), 6.95 (t, 1H, H7, , ), 7.12 (d, 1H, H6, ), 7.17 (t, 1H, H8, , ), 7.57 (t, 1H, He, , ), 7.86 (s, 1H, H3a), 8.02–7.98 (m, 2H, Hd, Hf), 8.25–8.22 (m, 2H, Hb, H2), 8.32 (d, 1H, H4, ); 13C NMR (75 MHz, CDCl3) δ 195.9, 170.4, 149, 148.1, 144.5, 137.9, 133.9, 129.7, 129.5, 129.4, 127.7, 127.3, 126, 124.9, 123.4, 123.3, 122.8, 121.1, 114.5, 113.8; anal. calcd. for C22H18N2O2S: C, 70.57; H, 4.85; N, 7.48; S, 8.56; O, 8.55; found: C, 70.56; H, 4.80; N, 7.50; S, 8.59.
5.3.8 (Z)-N-((10-ethyl-10H-phenothiazin-3-yl)methylene)methanamine oxide 3g
Green oil; yield 84% (0.22 g) was obtained by microwave irradiation, and 65% (0.17 g) by convective heating. Decomposition: 126 °C without melting. IR (KBr) νmax/cm−1 2874, 1645, 1468, 670; MS (70 eV), m/z (%): 284 (100, [M]+), 269 (62), 268 (45); 1H NMR (CDCl3) δ 1.33 (t, 3H, CH3, ), 3.75 (s, 3H, CH3), 3.84 (q, 2H, CH2, ), 6.80–6.74 (m, 2H, H1, H9), 6.85 (t, 1H, H7, , ), 7.02 (d, 1H, H6, ), 7.08 (t, 1H, H8, , ), 7.15 (s, 1H, H3a), 7.91 (d, 1H, H4, ), 7.98 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 146.3, 143.6, 134.4, 128.2, 127.4, 127.3, 127.1, 124.7, 123.5, 123.4, 122.8, 115.1, 114.3, 53.9, 42, 12.8; anal. calcd. for C16H16N2OS: C, 67.58; H, 5.67; N, 9.85; S, 11.28; O, 5.63; found: C, 67.23; H, 5.62; N, 9.8050; S, 11.09.
5.3.9 (Z)-N-((10-ethyl-10H-phenothiazin-3-yl)ethylene)methanamine oxide 3h
Green solid; yield 81% (0.23 g) was obtained by microwave irradiation, and 77% (0.18 g) by convective heating. Decomposition: 127 °C without melting. IR (KBr) νmax/cm−1 3019, 1501, 1468, 751; MS (70 eV), m/z (%): 298 (100, [M]+), 283 (58), 282 (39); 269 (13); 1H NMR (CDCl3) δ 1.39 (t, 3H, CH3, ), 1.52 (t, 3H, CH3, ), 3.95–3.86 (quin, 4H, CH2), 6.81 (d, 1H, H9, ), 6.82 (d, 1H, H1, ), 6.90 (t, 1H, H7, , ), 7.07 (d, 1H, H6, ), 7.11 (t, 1H, H8, , ), 7.24 (s, 1H, H3a), 7.99 (d, 1H, H4, ), 8.05 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 145.1, 143.5, 132.7, 128.2, 127.2, 127.1, 127, 124.7, 123.4, 123.2, 122.7, 115, 114.2, 61.3, 41.8, 13.4, 12.6; anal. calcd. for C17H18N2OS: C, 68.42; H, 6.08; N, 9.39; S, 10.5; O, 5.36; found: C, 68.42; H, 6.03; N, 9.35; S, 10.09.
5.3.10 (Z)-N-((10-octadecyl-10H-phenothiazin-3-yl)methylene)aniline oxide 3i
Green solid; yield 74% (0.42 g) was obtained by microwave irradiation, and 52% (0.29 g) by convective heating. Decomposition: 121–124 °C without melting. IR (KBr) νmax/cm−1 2919, 1457, 756; MS (70 eV), m/z (%): 570 (74, [M]+), 554 (23), 499 (39), 485 (82), 387 (27), 359 (18), 345 (32); 1H NMR (CDCl3) δ 0.88 (t, 3H, δCH3, ), 1.26 (s, 30H, CH2), 1.79 (q, 2H, βCH2, ), 3.95–3.86 (quin, 4H, CH2, ), 3.83 (t, 2H, αCH2, ), 6.85–6.83 (m, 2H, H1, H9), 6.90 (t, 1H, H7, , ), 7.15–7.06 (m, 2H, H6, H8), 7.43–7.41 (m, 3H, Hc, Hd), 7.76–7.72 (m, 3H, H3a, Hb), 7.13(d, 1H, H4, ), 8.25 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 148.8 8, 147,1, 143.9, 133.5, 129.6, 129.1, 128.8, 127.7, 127.4, 127.3, 125, 124.2, 123.9, 122.9, 121.5, 115.5, 114.7, 47.7, 31.9, 29.7, 29.5, 29.4, 29.2, 26.8, 26.7, 22.7, 14.1; anal. calcd. for C37H50N2OS: C, 77.85; H, 8.83; N, 4.91; S, 5.62; O, 2.80; found: C, 77.84; H, 8.89; N, 4.97; S, 5.67.
5.3.11 (N)-N-((10-octedecyl-10H-phenothiazin-3-yl)ethylene)methanamine oxide 3j
Yellowish–green solid, yield 84% (0.43 g) was obtained by microwave irradiation, and 70% (0.36 g) by convective heating. Decomposition: 125 °C without melting. IR (KBr) νmax/cm−1 3102, 1576, 834; MS (70 eV), m/z (%): 522 (100, [M]+), 507 (26), 506 (41), 451 (21), 395 (56), 311 (100); 1H NMR (CDCl3) δ 0.87 (t, 3H, δCH3, ), 1.25 (s, 30H, CH2), 1.77 (q, 2H, βCH2, ), 3.90 (q, 2H, CH2, , 6.84–6.80 (m, 2H, H1, H9), 6.89 (t, 1H, H7, , ), 7.07 (d, 1H, H6, ), 7.12 (t, 1H, H8, ), 7.23 (s, 1H, H3a), 7.95 (d, 1H, H4, ), 8.10 (d, 1H, H2, ); 13C NMR (75 MHz, CDCl3) δ 146.6, 144.2, 132.7, 128.1, 127.4), 127.3, 127.2, 124.9, 124.3, 124.1, 122.8, 115.5, 114.7, 61.5, 47.6, 29.7, 29.75, 29.6 29.4, 29.3, 26.9, 26.7, 22.7, 14.2, 13.6; anal. cald. for C33H50N2OS: C, 75.81; H, 9.64; N, 5.36; S, 6.13; O, 3.06; found: C, 75.83; H, 9.65; N, 5.33; S, 6.19; anal. calcd. for C33H50N2OS: C, 75.81; H, 9.64; N, 5.36; S, 6.13; O, 3.06; found: C, 75.83; H, 9.65; N, 5.33; S, 6.19.
5.3.12 (N,N′Z,N,N′Z)-N,N′-(10-methyl-10H-phenothiazine-3,7-diyl)bis(methan-1-yl-1-ylidene) dianiline oxide 4
Green solid; yield 61% (0.27 g) was obtained by microwave irradiation, and 34% (0.15 g) by convective heating. Decomposition: 128 °C without melting. IR (KBr) νmax/cm−1 2916, 1442, 624; MS (70 eV), m/z (%): 451 (100, [M]+), 436 (24), 435 (39); 1H NMR (CDCl3) δ 3.42 (s, 3H, N–CH3), 6.85–6.82 (m, 2H, H1, H9), 7.49–7.42 (m, 6H, Hc, Hd), 7.76–7.73 (m, 4H, Hb), 7.79 (s, 1H, H3a), 7.16(d, 2H, H4, H6, ), 8.27 (d, 2H, H2, H6, ); 13C NMR (75 MHz, CDCl3) δ 148.9, 145.5, 133.4, 129.9, 129.2, 129.2, 127.6, 125.9, 122.9, 121.7, 114.2, 35. 9; anal. calcd. for C33H50N2OS: C, 71.82; H, 4.69; N, 9.31; S, 7.10; O, 7.09; found: C, 71.82; H, 4.71; N, 9.31; S, 7.12.
5.3.13 (Z)-N-benzylidene-3,4-dinitroaniline oxide 6c
Light brown solid, yield 85% (0.24 g), mp 120–121 °C. MS (70 eV), m/z (%): 287 (100, [M]+), 271 (65); 1H NMR (CDCl3) δ 7.55–7.53 (m, 3H, H3, H4, H5), 7.80–7.77 (m, 2H, H2, H6), 8.55 (dd, 1H, Hf, , ), 8.73 (s, 1H, CH), 8.90 (d, 1H, Hb), 9.71(d, 1H, He, ), 13C NMR (75 MHz, CDCl3) δ 120.5, 121.8, 126.7, 127.8, 129.5, 129.7, 130.2, 131,4; anal. calcd. for C13H9N3O5: C, 54.36; H, 3.16; N, 14.63; O, 27.85; found: C, 454.27; H, 3.20; N, 14.58.
5.3.14 (Z)-3-chloro-N-(4-chlorobenzylidene)aniline oxide 6i
Beige solid, yield 75% (0.19 g); mp 105–106 °C. MS (70 eV), m/z (%): 265/267/269 (30/7/3, [M]+), 249 (100); 1H NMR (CDCl3) δ 7.51–7.40 (mt, 3H, H1, H2, Hd), 7.75 (d, 2H, H3, H6, ), 7.90 (s, 1H, CH), 8.17 (t, 1H, Hc, , ), 8.26 (d, 1H, Hb, ), 8.55 (s, 1H, Hf); 13C NMR (75 MHz, CDCl3) δ 123.0, 127.1, 127.2, 128.5, 128.9, 129, 129.5, 129.9, 131.1, 131.9,133.2, 134.8, 136.1; anal. calcd. for C13H9Cl2NO: C, 58.67; H, 3.41; Cl, 26.64; N, 5.26; O 6.01; found: C, 58.72; H, 3.48; N, 5.18.
5.3.15 (Z)-N-(4-chlorobenzylidene)-3-hydroxyaniline oxide 6l
Off-white solid, yield 83% (0.2 g); mp 195–196 °C. MS (70 eV), m/z (%): 247/249 (70/21, [M]+), 230/232 (100); 1H NMR (DMSO-d6) δ 6.90 (dd, 1H, Hd, , ), 7.29 (t, 1H, He, , ), 7.60 (d, 2H, H2, H6, ), 7.71 (d, 1H, Hf,, ), 7.95 (d, 2H, H3, H5, ), 8.16 (s, 1H, CH), 8.43 (s, 1H, Hb), 9.68 (broad s, 1H, OH); 13C NMR (75 MHz, DMSO) δ 114.8, 118.2, 120.7, 123.4, 129.0, 129.5, 131.9, 134.1, 134.2, 147.2, 157.2; anal. calcd. for C13H10ClNO2: C, 63.04; H, 4.07; Cl, 14.31; N, 5.66; O, 12.92; found: C, 63.12; H, 4.10; N, 5.62.
5.3.16 (Z)-N-(4-bromobenzylidene)-3-hydroxyaniline oxide 6p
Beige solid, yield 80% (0.23 g); mp 188–190 °C. MS (70 eV), m/z (%): 291/293 (100, [M]+), 275/277 (85); 1H NMR (DMSO-d6) δ 6.91 (dd, 1H, Hd, , ), 7.29 (t, 1H, He, , ), 7.75–7.69 (m, 3H, H2, H6, Hf), 7.89 (d, 2H, H3, H5, ), 8.15 (s, 1H, CH), 8.44 (s, 1H, Hb), 9.68 (broad s, 1H, OH); 13C NMR (75 MHz, DMSO) δ 115, 118.2, 120, 120.7, 122.8, 123.7, 129.5, 131.2, 131.9, 134.1, 147.6, 157.3; anal. calcd. for C13H10NO2: C, 53.45; H, 3.45; Br, 27.35; N, 4.79; O, 10.95; found: C, 53.50; H, 3.49; N, 4.82.
Acknowledgments
Financial support from the Romanian Ministry of Education, Research Youth and Sports (Grant ID PCCE 140/2008), as well as from the Sectorial Operational Programs for Human Resources Development 2007–2013, co-financed by the European Social Fund, under the project No. POSDRU/107/1.5/S/76841 with the title “Modern Doctoral Studies: Internationalization and Interdisciplinarity” is gratefully acknowledged.