

1 Introduction
Nitrones are powerful tools in organic synthesis [1] and are used as useful reagents or sometimes involved as intermediates in the synthesis of a variety of nitrogen-containing compounds which find application as agrochemicals and/or as pharmaceuticals. Generally, nitrones easily undergo 1,3-dipolar cycloaddition reactions with a large variety of substituted alkenes including both electron-rich and electron-poor dipolarophiles [2]. The resulting isoxazolidines are key intermediates for the synthesis of many compounds since they are immediate precursors of 1,3-amino alcohols [3].
In addition to the well-known cycloaddition chemistry of nitrones, there are several reports on nucleophilic additions to nitrones [4]. Many of these reactions are promoted and/or catalyzed by Lewis acids. However, relatively little is known about the influence of the metal atom on the reactivity and even the selectivity of the reaction. Nitrones are isoelectronic with allyl anions and enolates, but the presence of the C=N moiety gives the functionality an iminium character, which is responsible for its reactivity as electrophile with organometallic reagents. Complexes with coordinated nitrones are thought to be intermediates in the addition of organometallic reagents. If nitrones can act as ligands by reaction with metals they may, in turn, be activated by precomplexation with Lewis acids (Fig. 1).

The interest of these reactions resides in the stereocontrolled nucleophilic additions that can be carried out with α-alkoxy and α-amino nitrones 1 and 2, derived from d-glyceraldehyde and l-serine, respectively (Fig. 2).
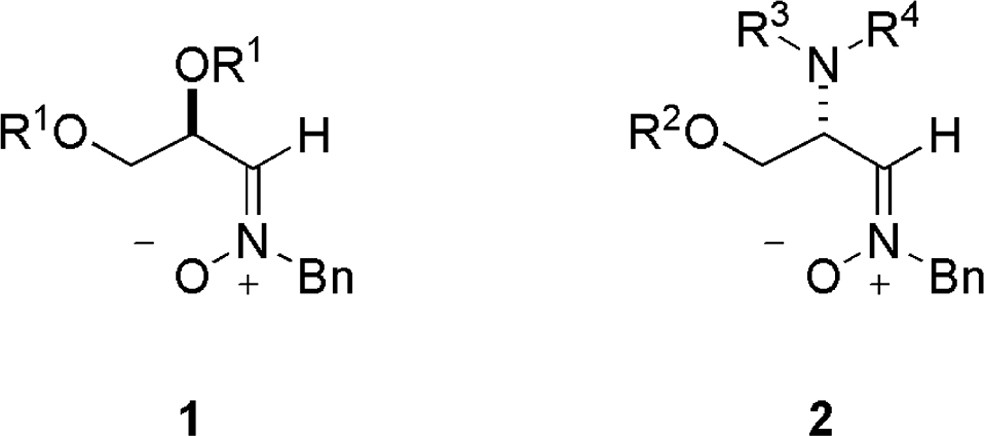
In this review, the synthetic utility of nitrones 1 and 2 will be discussed as a roundtrip from aminoacids to nucleosides, illustrating the enormous potential of those compounds as building blocks for the synthesis of the mentioned biomolecules.
2 Synthesis of nitrones
Nitrones can be prepared by various procedures, the choice of which would depend on the target molecule [1].
In the case of nitrones 1 and 2 the condensation method between an aldehyde and a hydroxylamine is the best choice due to the availability of the parent aldehydes, d-glyceraldehyde [5] and l-serinal [6], in several protected forms. Following the reported procedure for the synthesis of the aldehydes and condensing them with N-benzyl hydroxylamine [7] according to our optimized conditions, nitrones 1 [8] and 2 [9] could be prepared in multigram scale (Scheme 1), the method being applicable to other carbohydrate- and aminoacid-derived nitrones. In all cases, only the Z-isomer was obtained in agreement with previous observations that showed such an isomer as the most stable for aldonitrones.

3 Stereocontrolled nucleophilic additions
It has been known since the 1960s that nitrones smoothly undergo nucleophilic addition of organometallic reagents [10]. When the chiral nitrones are concerned two diastereoisomers (syn and anti) [11] can, in principle, be obtained. In the case of acyclic nitrones 1 and 2 the diastereofacial induction can be controlled in a rather different way depending on the type of α-substitution.
d-Glyceraldehyde derived nitrones 1a and 1b bearing a rigid group at α-position led to syn adducts when they react with a variety of organometallic reagents including lithiated heterocycles, lithium acetylides, Grignard reagents, allenyllithiums and stabilized α-alkoxyanions (Scheme 2) [4]. The same trend is observed when zinc (II) bromide, magnesium (II) bromide, trimethylsilyl triflate or trimethylchlorosilane were used as promoters of the reaction.

On the other hand, a complete reversal of the sterochemical course of the reaction was observed when diethylaluminum chloride, boron trifluoride etherate or titanium (IV) chloride were used as precomplexing agents. In all these cases anti compounds were obtained preferentially. Interestingly, this behavior is of general applicability to other nitrones having an endocyclic oxygen atom at α-position, as those reported in Scheme 2, bottom. In fact, the two only exceptions observed in our laboratories for that Lewis acid modulated stereocontrol correspond to α-alkoxynitrones in which the α-oxygen is not included into a rigid system.
The stereocontrol of nucleophilic additions to α-aminonitrones did not depend on any Lewis acid but on the type of protection of the α-amino group. Thus, diprotected nitrone 2a gave rise to syn adducts both in the absence and in the presence of any Lewis acid (Scheme 3). On the other hand, α-amino monoprotected nitrone 2b led preferentially to anti adducts. Quite probably, this behavior is related not only with the type of protection at the α-nitrogen but also with the presence of a carbamate group at such nitrogen (Figs. 1 and 2).

Recently described models like that showed in Fig. 3 [12] as well as the fact that N,N-dibenzyl-α-amino nitrones showed a poor selectivity and low chemical yields [13], strongly support the hypothesis of the necessity of a coordinating group at the α-position.

4 Synthesis of amino acids
d-Glyceraldehyde derived nitrones are synthetic equivalents of the glycine cation if we consider the equivalence between a dioxolane ring and a carboxyl function. The first is easily transformed into the second by acidic oxidative methods [14]. Application of this methodology to the stereocontrolled addition of Grignard reagents to 1a provided an enantiodivergent synthesis of α-amino acids (Scheme 4) [15].

Alternatively, the vicinal diol functionality can only be oxidized at the primary alcohol, after the appropriate protective groups chemistry. In such a case, access to α-hydroxy-β-aminoacids is given. The synthesis of (2S,3R)-N-Boc phenylisoserine and its (3S) epimer outlined in Scheme 5 constitutes an example of that approach [16].

Similarly, oxidation of the primary alcohol at the resulting hydroxylamines from the addition of C-nucleophiles to α-amino nitrones derived from l-serine allowed the synthesis of epimeric 2,3-diaminoacids [17]. The employed α-amino nitrones can be considered as synthetic equivalents of highly functionalized synthons (Scheme 6).
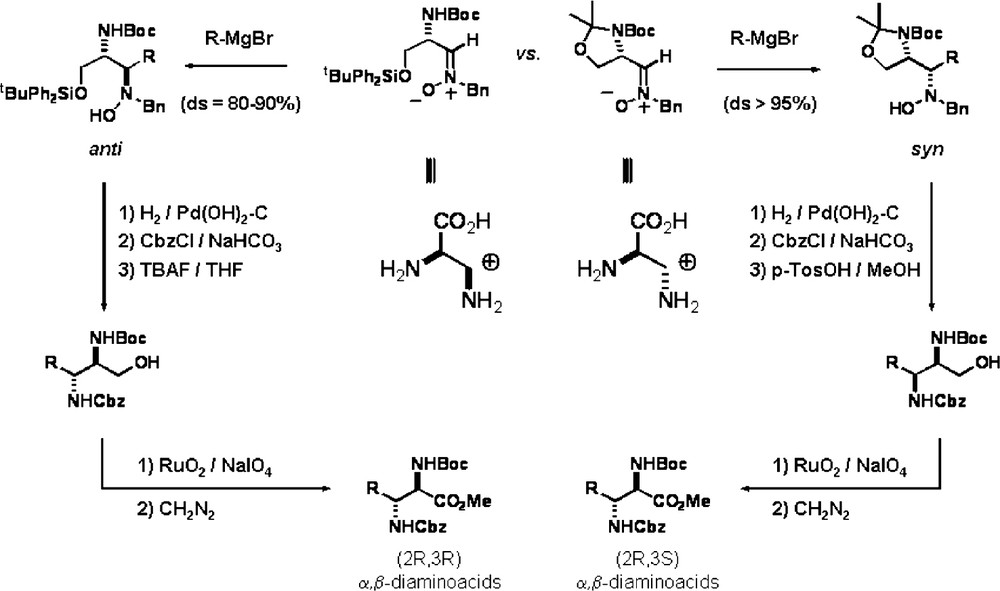
The synthetic approaches illustrated in the previous schemes were based on oxidative processes carried out on the dioxolane/oxazolidine ring. The nucleophilic addition reaction served to introduce the side chain, in most cases without additional functional groups. In order to prepare small molecules with diverse functional groups [18] it is also possible to maintain the functionalities present in the molecule by adding a nucleophile which could be further transformed into a carboxyl group (masked carboxyl). Among these carboxyl surrogates are stabilized α-alkoxyanions [19], vinyl metals [20], lithium acetylides [21] and furan [22]. Both α-alkoxy and α-amino nitrones underwent nucleophilic addition to give diastereomeric hydroxylamines in a stereocontrolled way (Scheme 7). Further oxidation of the introduced group following literature procedures led to the corresponding hydroxylated amino acids.

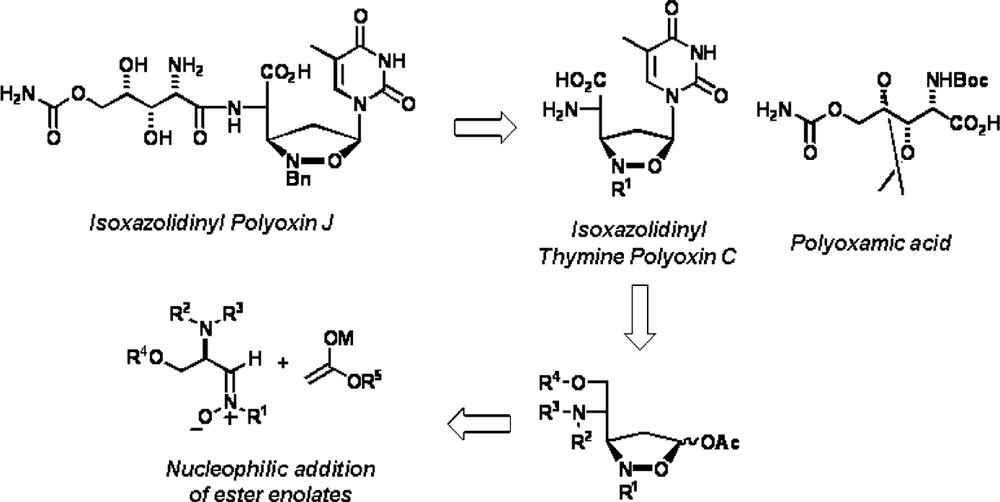
The methodology outlined in Scheme 7 was applied to the synthesis of the important nucleoside antibiotic and antifungal agent Polyoxin J [23]. The retrosynthetic analysis depicted in Scheme 8 shows that Polyoxin J can be prepared from the two aminoacid components, polyoxamic acid and thymine polyoxin C. In spite of the large number of synthesis for these two components that can be found in recent literature [24], only three total syntheses of Polyoxin J have been reported to date [25].
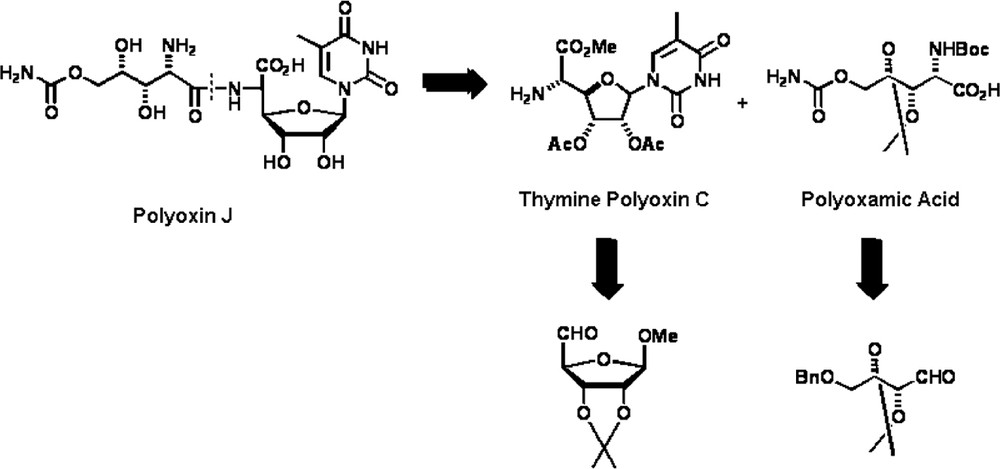
Interestingly, the aminoacids polyoxamic acid and thymine polyoxin C have an opposite relative configuration between the α-amino group and the adjacent stereogenic center (syn vs. anti). Thus, they could be prepared by using the above-mentioned methodology, i.e. incorporation of a carboxyl unit to the parent nitrone, provided that stereocontrol could be exerted at the nucleophilic addition stage.
The polyoxamic acid unit was prepared in 14 steps from natural l-tartaric acid in 11.1% overall yield (Scheme 9) [26]. The key step of the synthesis was the syn addition (in the absence of any Lewis acid) of furyllithium to the starting l-threose derived nitrone.

The synthesis of the thymine polyoxin C fragment started from d-ribose and is completed in 11 steps and 6.9% overall yield (Scheme 10) [27]. In this case the crucial addition of furyllithium was carried out in the presence of one equivalent of diethylaluminum chloride in order to obtain preferentially the desired anti hydroxylamine.

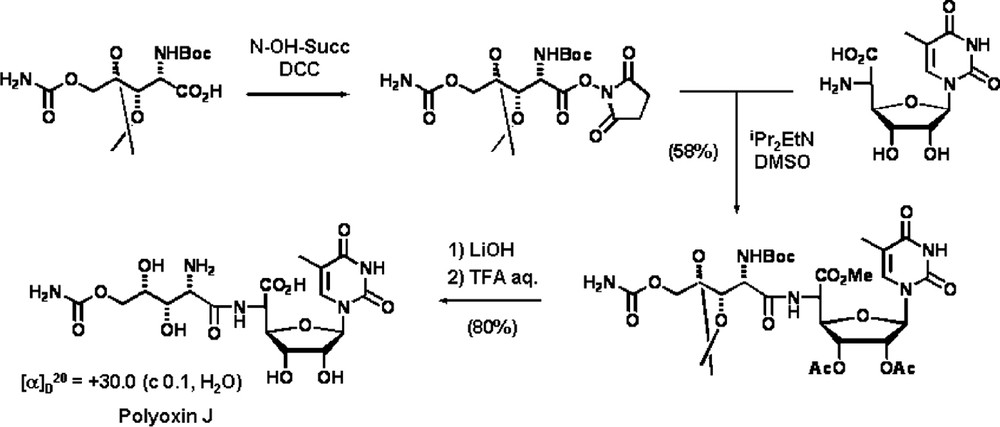
Finally, the coupling between the two aminoacids and deprotection afforded synthetic Polyoxin J (Scheme 11) [28] whose physical and spectroscopic properties were identical to those of the natural product (Fig. 4) [29].

5 Synthesis of nucleoside analogues
In recent years, there has been a growing interest for a particular type of nucleoside analogues in which the furanose ring, that acts as a spacer between the base moiety and the hydroxymethyl group, is replaced by a different heterocyclic ring. This kind of analogues are called heterocyclic nucleosides and several synthetic approaches have been designed for their preparation in an enantiomerically pure form [30]. In particular, isoxazolidinyl nucleosides have emerged as a new family of compounds with potential antiviral activity [31]. These compounds possess in their structure an isoxazolidine ring that can be constructed through the addition of an ester enolate to a nitrone followed by an intramolecular cyclization as illustrated in the retrosynthetic analysis depicted in Scheme 12.

Alternatively, these compounds can also be prepared through a 1,3-dipolar cycloaddition approach but that sort of reactivity is not the object of the present review [32]. The addition of the sodium enolate derived from methyl acetate gives directly the cyclic adduct, no evidence of the open-chain hydroxylamine being observed (Scheme 13) [33].

This is probably due to the basic medium of the reaction which favors the intramolecular cyclization of the intermediate hydroxyamino anion. Since the reaction was conducted in the absence of any additive only the syn adduct was obtained in good agreement with previous experiences with other nucleophiles, as mentioned in this account.
When the reaction was carried out in the presence of one equivalent of diethylaluminum chloride the major isomer was, as expected, the anti one, but in a rather low chemical yield. The reason of this behavior may be due to the in situ formation of a less reactive aluminum enolate by reaction of the sodium enolate and the nitrone–aluminum complex (Scheme 14)1. As an alternative we chose the reaction of the nitrone 1a with the corresponding silyl ketene acetal. This reaction had been described by Kita et al. [34] more than 15 years ago. Although some stereocontrol of the reaction could be exerted, there was no evidence of stereodivergent conditions by changing the group at the nitrone nitrogen atom. In our hands [35], the addition of silyl ketene acetals was more complex than the corresponding reaction with sodium enolates.
We observed up to four different products including open-chain adducts and cyclic adducts. In all cases the nitrone needs activation with one equivalent of Lewis acid in contrast to the catalytic amount of zinc (II) iodide used by Kita et al. [34].
The ratio of the adducts depends on the Lewis acid employed as a promoter. When the activation was carried out with a silyl triflate only silylated adducts were obtained and in all cases the syn isomer was the major product. More interestingly, in the case of activation with boron trifluoride etherate a complete reversal of the stereochemical course of the reaction was observed and the anti isomers were only obtained. Thus, a stereodivergent synthetically useful approach for the addition of silyl ketene acetals to nitrones has also been developed. For synthetic purposes the complexity of the reaction mixture can be circumvented by transforming the crude mixture into the corresponding isoxazolidin-5-ones, in a one-flask procedure consisting of sequential desilylation and induced cyclization (Scheme 15) [35].

The diastereofacial divergency of the reaction is best explained by considering alternative mechanisms on the basis of the different Lewis acids employed. By activating with a silyl triflate, ionic species were generated favoring a stepwise mechanism. On the contrary, activation with diethylaluminum chloride led to a neutral complex which underwent a cycloaddition-type addition (although rather asynchronous). Both mechanisms converged to a common intermediate thus explaining the obtained compounds (Scheme 16).

The mechanism illustrated in Scheme 16 is supported by theoretical calculations [36] and previous empirical results. The calculated transition states for the two pathways are shown in Fig. 5.

In addition it is worthy of note that all reported cycloaddition reactions (1,3-DC) with d-glyceraldehyde derived nitrones and closely related compounds led to anti compounds [37]. By contrast, all organometallic addition reactions (NA) to the same nitrones in the absence of any Lewis acid led to syn adducts [4]. Such a different stereochemical induction for concerted (1,3-DC) and stepwise (NA) reactions clearly supports the hypothesis of Lewis acid-induced mechanisms.
The obtained isoxazolidin-5-ones were the starting point for the enantiodivergent synthesis of the targeted isoxazolidinyl nucleosides. The introduction of the base moiety, according to classical Vörbruggen et al. [38] protocol, and further transformation of the dioxolane ring into a hydroxymethyl group (deprotection–oxidation–reduction) led to enantiomeric nucleoside analogues in good overall yields [35]. The methodology was applied to thymine, uracil, 5-fluorouracil and 6-chloropurine analogues (Scheme 17). Unfortunately, all biological tests assayed showed no activity for these analogues.

The enolate methodology, used for the preparation of nucleoside analogues can also be applied for the preparation of more complex compounds. The above-mentioned Polyoxin J, although active in vitro, presents a considerable diminished activity in vivo as a consequence of its intracellular accumulation failure. This is due to a poor recognition by peptide permeases. Extensive research is being done towards structural modifications of Polyoxins with the aim of developing more effective and safe antifungal agents. Those structural modifications could include the formation of prodrugs [39], modifications of the base moiety (not still explored) and addition/substitution/elimination of substituents at the furanose ring [40]. Taking advantage of our previous experience acquired during the synthesis of Polyoxin J and our methodology for preparing isoxazolidinyl nucleosides, we embarked into the synthesis of isoxazolidinyl Polyoxin J [41]. For the synthesis of this analogue it is needed the previously prepared polyoxamic acid and the corresponding isoxazolidinyl thymine polyoxin C. The retrosynthetic analysis for this compound is showed in Scheme 18.
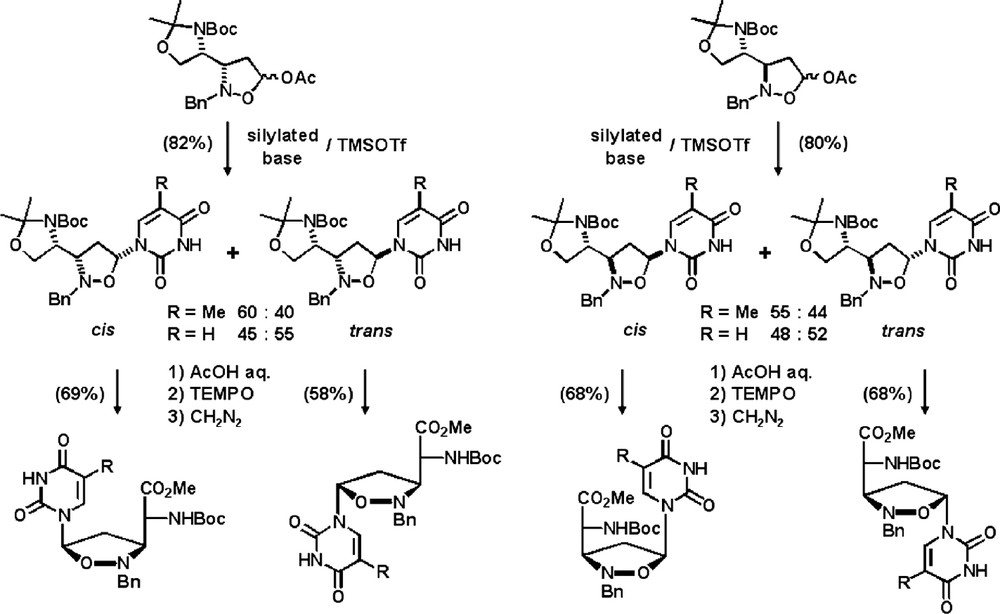
In order to prepare the intermediate isoxazolidinyl amino acids, silyl ketene acetals were made to react with l-serine derived nitrones 2 in the presence of tert-butyldimethylsilyl triflate. As expected, the reaction proceeds with stereodivergency and after reduction, acetylation and deprotection, syn and anti aminoalcohols were obtained. Unfortunately, all attempts of transforming these compounds into the target intermediates failed (Scheme 19) [41].

In order to circumvent this drawback the base moiety was introduced prior the unmasking of the glycine unit. Finally, deprotection and oxidation led separately to the four possible isomers of isoxazolidinyl thymine polyoxin C (Scheme 20).
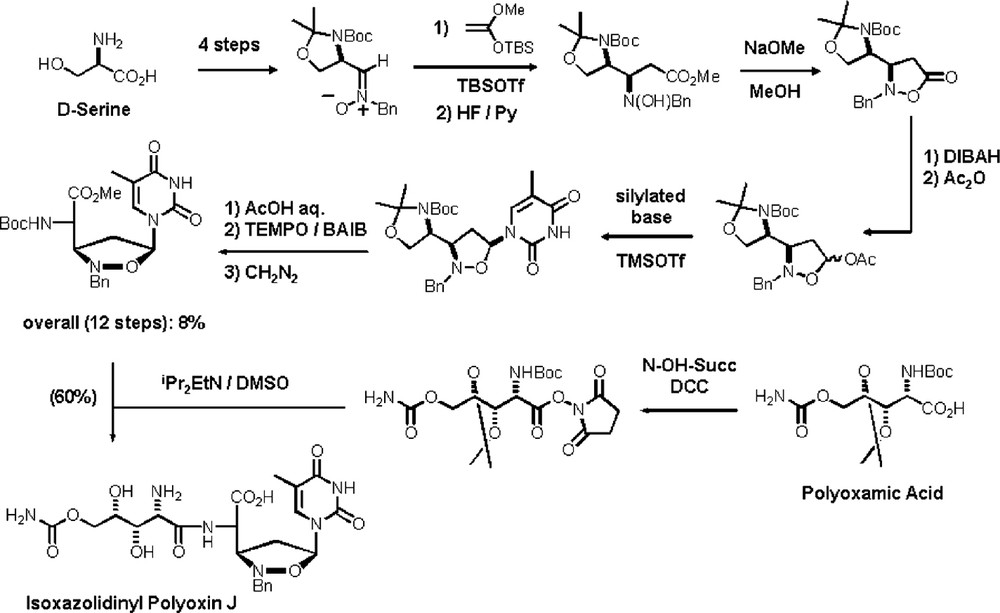
For the synthesis of the analogues of Polyoxin J is necessary to start from d-serine. Because of it, the optimized reaction sequence was repeated with the nitrone derived from that amino acid [42] and, after condensation with synthetic polyoxamic acid, the isoxazolidinyl Polyoxin J was obtained (Scheme 21).
6 Concluding remarks
The chemistry summarized in this revision shows that α-alkoxy and α-amino nitrones, prepared in enantiomerically pure form from d-glyceraldehyde and l-serine, are extremely useful chiral building blocks in a wide variety of diastereoselective preparations of amino acids and nucleosides. These include organometallic nucleophilic additions of Grignard reagents, organolithium compounds, metal enolates and silyl ketene acetals. The success of these reactions is based on both Lewis acid and protective group tuning. Future developments of the discussed approaches are expected to contribute to the synthesis of novel biologically interesting compounds.
Acknowledgements
The author expresses his gratitude to all individuals whose names appear in the publications. He also expresses special gratitude for financial support to the Spanish Government (Projects PB97-1014 and BQU2001-2428) and the Aragon Government (Project P116-2001).
1 In order to verify the formation of the aluminium enolate, it was prepared by a known method and made to react with the nitrone. An identical result was observed, thus demonstrating the starting hypothesis.