

1 Introduction
The enaminones are versatile and useful building blocks for the synthesis of many bioactive compounds, such as taxol [1], 1,5-benzodiazepine [2], 1,4-dihydropyridine [3], pyridinone [4], pyrimidine [5], quinoline [6], furoisoquinoline [7], indole [8], isoxazole [9], and pyrrolo-1,2,4-triazine [10] derivatives. They are valuable precursors in organic synthesis [11,12], in pharmaceutical applications, such as anticonvulsivant [13,14] or anti-inflammatory [14,15] drugs, and as duocarmycine classes of antitumor agents [14,16] as well as antibacterial and antimalarial agents [14,17].
The most well-known route for the synthesis of β-enaminones involves the direct condensation of β-dicarbonyl compounds with amines in refluxing aromatic hydrocabones with azeotropic removal of water [18]. Several catalysts have been applied to achieve this transformation, including P2O5/SiO2 [19], I2 [20], LiHSO4/SiO2 [21], NaAuCl4 [22], Bi(TFA)3 [23], Sc(OTf)3 [24], Yb(OTf)3 [25], ZrOCl2·8H2O [26], HClO4/SiO2 [27], and silica chloride [28]. Recently, we used silica-supported ferric hydrogen sulfate [29] as an efficient, heterogeneous and recyclable catalyst for the synthesis of β-enaminones and β-enamino esters. However, bis-(β-enaminones) are missing in most of the above procedures. Only a few narrow examples are reported in the literature [19–21,30,31].
In this paper, we wish to report an independent synthesis of bis-(β-enaminones) and bis-(β-enamino esters) in the presence of Fe(HSO4)3·SiO2 (Scheme 1). Bis-(β-enaminones) and bis-(β-enamino esters) are used for developing a ligand and chelate chemistry that opens wide possibilities for their use in different areas of industry and engineering. Some of them are metal complex catalysis of polymerization, hydrogenation, and carbonylation, in molecular electronics, in certain magnetoactive materials [32], in extraction, separation, quantitive gas chromatography determination of metals and of their isotopes, and also for chemical vapor deposition applications (CVD) [30–33].
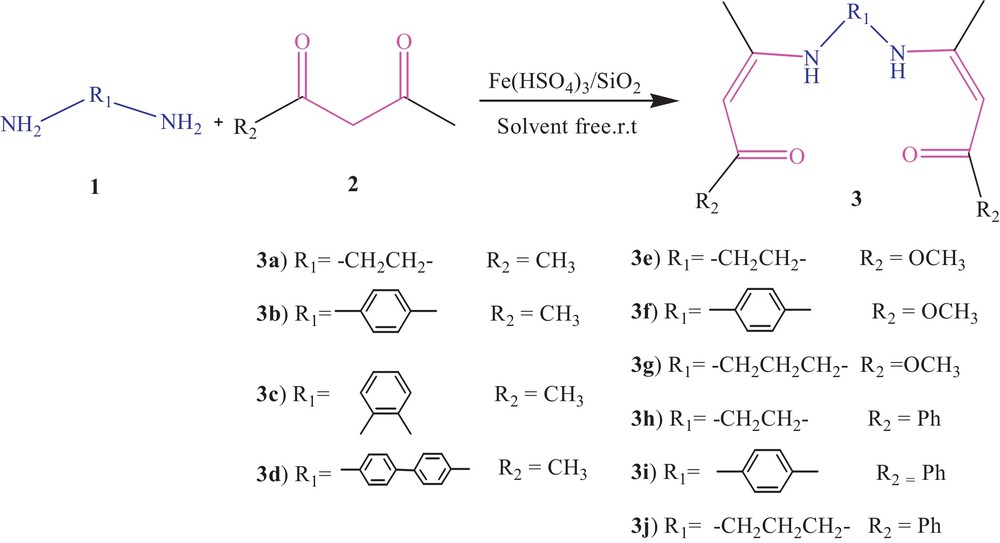
Synthesis of bis-(β-enaminones) and bis-(β-enamino esters).
2 Experimental
2.1 General
Chemicals were either prepared in our laboratories or purchased from Merck, Fluka and Aldrich Chemical Companies. All yields refer to the isolated products. Melting points were recorded on an Electrothermal IA9000 Series Melting Point Apparatus. FT–IR spectra were recorded on a Thermo Nicolet AVATAR-370-FT–IR spectrophotometer. The 1H NMR spectra were recorded on a Bruker AC 100 spectrometer at 100 MHz. Chemical shifts are reported in ppm downfield from TMS as an internal standard; coupling constants J are given in Hertz. Elemental analyses were obtained on a Thermo Finnigan Flash EA micro-analyzer. Silica-supported ferric hydrogensulfate prepared from Fe(HSO4)3 [34] and silica gel, as we have previously reported [35].
2.2 General procedure for the synthesis of compounds 3a–3j
A mixture of the β-dicarbonyl compound (4 mmol), of diamine (2 mmol) and Fe(HSO4)3·SiO2 [0.22 g containing 0.25 mmol of Fe(HSO4)3] was stirred in solvent-free conditions at room temperature for the appropriate time. After completion of the reaction as indicated by TLC, the mixture was diluted by ethyl acetate (15 mL). The insoluble catalyst was separated by filtration and rinsed with ethyl acetate, dried and reused. The solvent was evaporated from the filtrate and the crude product was purified by recrystallization from EtOH/H2O (1:1).
3 Spectral data of the prepared compounds
3.1 (Z)-4-{2-[(Z)-1-Methyl-3-oxo-1-butenyl amino] ethyl amino}-3-penten-2-one (3a)
Yield 90%; mp = 110–111 °C, lit. [19] mp 109–110 °C; 1H NMR (CDCl3, 100 MHz) δ: 10.8 (b, 2H), 5.0 (s, 2H), 3.3 (m, 4H), 1.9 (bs, 12H). IR (KBr, cm−1) υ: 3440, 3084, 2945, 1612, 1577, 1523, 1371, 1288.
3.2 (Z)-4-{4-[(Z)-1-Methyl-3-oxo-1-butenyl amino] anilino}-3-penten-2-one (3b)
Yield 70%; mp = 99–100 °C; 1H NMR (CDCl3, 100 MHz) δ: 12.2 (b, 2H), 6.5-7.3 (m, 4H), 5.1 (s, 2H), 2.07 (s, 6H), 1.9 (s, 6H). IR (KBr, cm−1) υ: 3342, 3227, 1607, 1551, 1521, 1308, 1279.
3.3 (Z)-4-{2-[(Z)-1-Methyl-3-oxo-1-butenyl amino] anilino}-3-penten-2-one (3c)
Yield 60%; mp = 95–97 °C; 1H NMR (CDCl3, 100 MHz) δ: 12.5 (b, 2H), 6.6–7.5 (m, 4H), 5.2 (s, 2H), 2.1 (s, 6H), 1.95 (s, 6H).IR (KBr, cm−1) υ: 3340, 1608, 1563, 1506, 1279.
3.4 (Z)-Methyl-3-((4′-(((Z)-4-oxopent-2-en-2-yl)amino)-[1,1′-biphenyl]-4-yl)amino)but-2-enoate (3d)
Yield 65%; mp = 115–117 °C; 1H NMR (CDCl3, 100 MHz) δ: 12.5 (b, 2H), 7-7.55 (m, 8H), 5.2 (s, 2H), 2.1 (s, 6H), 2.0 (s, 6H). IR (KBr, cm−1) υ: 3333, 3219, 1608, 1551, 1498, 1281.
3.5 (2Z,2′Z)-Dimethyl 3,3′-(ethane-1,2-diylbis(azanediyl))bis(but-2-enoate)methyl (3e)
Yield 98%; mp = 137–139 °C; 1H NMR (CDCl3, 100 MHz) δ: 8.6 (b, 2H), 4.5 (s, 2H), 3.6 (s, 6H), 3.35 (m, 4H), 1.9 (s, 6H). IR (KBr, cm−1) υ: 3308, 2957, 1656, 1582, 1279, 1171.
3.6 (2Z,2′Z)-Dimethyl 3,3′-(1,4-phenylenebis(azanediyl))bis(but-2-enoate) (3f)
Yield 50%; mp = 149–150 °C; 1H NMR (CDCl3, 100 MHz) δ: 10.2 (b, 2H), 6.5-7.3 (m, 4H), 5.0 (s, 2H), 3.65 (s, 6H), 1.9 (s, 6H). IR (KBr, cm−1) υ: 3268, 2937, 1662, 1600, 1257, 1161, 789.
3.7 (2Z,2′Z)-Dimethyl 3,3′-(propane-1,3-diylbis(azanediyl))bis(but-2-enoate) (3g)
Yield 100%; mp = 54–56 °C; 1H NMR (CDCl3, 100 MHz) δ: 8.5 (b, 2H), 4.45 (s, 2H), 3.6 (s, 6H), 3.2–3.8 (m, 6H), 1.9 (s, 6H). IR (KBr, cm−1) υ: 3284, 2949, 1656, 1603, 1310, 1263, 1169, 1132.
3.8 (Z)-3-{2-[(Z)-1-Methyl-3-oxo-3-phenyl-1-propenyl amino] ethyl amino}-1-phenyl-2-buten-1-one (3h)
Yield 72%; mp = 179–181 °C, lit. [19] mp 178–180 °C; 1H NMR (CDCl3, 100 MHz) δ: 11.6 (b, 2H), 7.3–7.9 (m, 10H), 5.7 (s, 2H), 3.6–3.7 (m, 4H), 2.1 (s, 6H). IR (KBr, cm−1) υ: 3342, 3227, 1607, 1551, 1521, 1308, 1279.
3.9 (Z)-3-{2-[(Z)-1-Methyl-3-oxo-3-phenyl-1-propenyl amino] anilino}-1-phenyl-2-buten-1-one (3i)
Yield 80%; mp = 148–150 °C; 1H NMR (CDCl3, 100 MHz) δ: 12.2 (b, 2H), 6.7–7.9 (m, 14H), 5.5 (s, 2H), 1.95 (s, 6H). IR (KBr, cm−1) υ: 3325, 3223, 1596, 1574, 1543, 1324, 1278.
3.10 (Z)-3-{3-[(Z)-1-Methyl-3-oxo-3-phenyl-1-propenyl amino] propyl amino}-1-phenyl-2-buten-1-one (3j)
Yield 100%; mp = 86–88 °C; 1H NMR (CDCl3, 100 MHz) δ: 11.5 (b, 2H), 7.3-7.9 (m, 10H), 5.65 (s, 2H), 3.35–3.6 (m, 4H), 2.1 (s, 6H). IR (KBr, cm−1) υ: 3342, 3227, 1607, 1551, 1521, 1308, 1279.
4 Results and discussion
This paper reports for the first time a regio-and chemo-selective bis-enamination of β-dicarbonyl compounds under solvent-free conditions using Fe(HSO4)3·SiO2 at room temperature in excellent yields and purity. Silica-supported ferric hydrogensulfate was prepared from Fe(HSO4)3 [34] and silica gel as we previously reported [35]. IR spectra of Fe(HSO4)3 and Fe2(SO4)3·x H2O were compared as shown in Fig. 1. As can be seen, the IR spectrum of Fe(HSO4)3 is clearly different from that of Fe2(SO4)3·x H2O. The atomic absorption analysis spectrum shows that the expected percentage of Fe has been recovered, whereas the titration of the aqueous solution of Fe(HSO4)3 with NaOH did not show significant contamination of the catalyst with H2SO4.
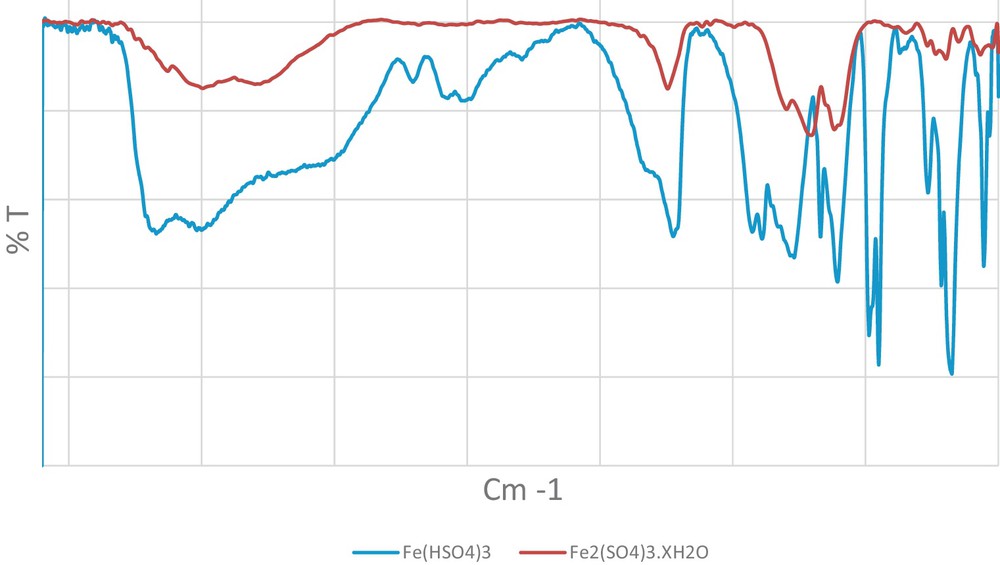
Comparison of IR spectra of Fe(HSO4)3 and Fe2(SO4)3·x H2O.
Initially, the condensation of ethylene diamine with 2 equiv of acetylacetone was selected as a model reaction, and the influence of various reaction parameters, like catalyst, temperature and time, on the isolated yield of 3a was examined (Scheme 1, Table 1). Fe(HSO4)3·SiO2 proved to be the most efficient catalyst because the reaction could be carried out in excellent yield and short reaction time under solvent-free conditions. The best reaction conditions require the presence of a catalytic amount of Fe(HSO4)3·SiO2 (12.5 mol%) under solvent-free conditions at room temperature.
Optimization conditions of the reaction between ethylene diamine and acetylacetone.
| Entry | Solvent | Catalyst | Mol% | Condition | Yield (%) |
| 1 | CH3OH | Fe(HSO4)3 | 10 | Reflux, 1 h | 45 |
| 2 | CH3OH | Fe(HSO4)3 | 12.5 | RT, 1 h | 48 |
| 3 | Solvent-free | Fe(HSO4)3/SiO2 | 5 | RT, 30 min | 70 |
| 4 | Solvent-free | Fe(HSO4)3/SiO2 | 0 | RT, 60 min | 5 |
| 5a | Solvent-free | Fe(HSO4)3/SiO2 | 12.5 | RT, 15 min | 90 |
| 6 | Solvent-free | Fe(HSO4)3/SiO2 | 20 | RT, 15 min | 91 |
| 7b | Solvent-free | Fe(HSO4)3/SiO2 | 12.5 | RT, 15 min | 90 |
| 8b | Solvent-free | Fe(HSO4)3/SiO2 | 12.5 | RT, 15 min | 88 |
| 9b | Solvent-free | Fe(HSO4)3/SiO2 | 12.5 | RT, 15 min | 85 |
| 10b | Solvent-free | Fe(HSO4)3/SiO2 | 12.5 | RT, 15 min | 85 |
| 11b | Solvent-free | Fe(HSO4)3/SiO2 | 12.5 | RT, 15vmin | 80 |
| 12b | Solvent-free | Fe(HSO4)3/SiO2 | 12.5 | RT, 45 min | 80 |
a Optimum conditions.
b Reusability of the catalyst in the new runs.
A variety of diamines, including aliphatic and aromatic diamines were condensed with various 1,3-dicarbonyl compounds, such as acetylacetone, benzoylacetone and methyl acetoacetate. (Scheme 1). The results are summarized in Table 2.
Bis-enamination of β-dicarbonyl compounds in the presence of Fe(HSO4)3·SiO2.
| Entry | R1 | R2 | Product | Time (min) | Yield (%) | Mp (°C) |
| 1 | –CH2CH2– | CH3 | 15 | 90 | 110–111 | |
| 2 | CH3 | 25 | 70 | 99–100 | ||
| 3 | CH3 | 25 | 60 | 95–97 | ||
| 4 | CH3 | 30 | 65 | 115–117 | ||
| 5a | –CH2CH2– | OCH3 | 7 | 98 | 137–139 | |
| 6 | OCH3 | 16 | 50 | 149–150 | ||
| 7 | –CH2CH2CH2– | OCH3 | 12 | 100 | 54–56 | |
| 8 | –CH2CH2– | Ph | 16 | 72 | 179–181 | |
| 9 | Ph | 16 | 80 | 148–150 | ||
10 | –CH2CH2CH2– | Ph | 16 | 100 | 86–88 |
a Isolated yields.
It is clear from our results that silica ferric hydrogensulfate catalyzed the condensation reaction of 1,3-dicarbonyls with diamines, providing a remarkably rapid and viable alternative route for the synthesis of potentially bis-(amino enone) ligands. In all reactions, β-ketoesters are more reactive than β-diketones. In the case of β-ketoesters, the ketone group is more reactive than the ester group. Only dibenzoylmethane is unreactive in this reaction. The isolated products were characterized by their FT–IR and 1H NMR spectral analyses. Ligand behaviors and antibiological activities of these novel compounds are in progress in our laboratories.
In the 1H NMR (100 MHz, CDCl3) spectra of enamino esters 3e–3g, which exist in their enamino esters forms, a broad singlet can be evidenced in the 8.5–8.6 region for the NH group. However, this signal was shifted to 10.8–12.5 ppm for enamino ketones 3a–3d and 3h–3j, which includes an intramolecular hydrogen bonding of the NH group with the ketone group [29]. Compounds 3a–3d show a broad singlet at 12.2–12.5 ppm for the NH group, which is hydrogen bonded with the ketone carbonyl group. In compounds 3h–3j, this signal was shifted to 10.8–11.6 ppm; the conjugation of the carbonyl with the phenyl group is responsible for this upfield shift. The NH group was observed at 13 ppm (enaminones as major tautomers) and the OH group at 14 ppm (enols as minor tautomers). The hydroxyl group is more acidic than the amine group, and so the H·····N hydrogen bonding of the OH group with imine (14 ppm) is stronger than the H·····O hydrogen bonding of the NH group with the carbonyl group (13 ppm), respectively. However, experimental data showed that enamino ketones and enamino esters exist usually under the enamine form. The parent keto forms were not observed. These compounds showed the enol form in the downfield region if monitored with high-resolution NMR spectrometers [29].
The catalyst's reusability is the most significant advantage of our method. For the reaction of ethylene diamine with acetylacetone, no significant loss of the product yield was observed when Fe(HSO4)3·SiO2 was reused after it has been recycled five times (Table 1, entries 7–11). A further new run needs longer reaction times (Table 1, entry 12).
The suggested mechanism for this reaction was shown in Scheme 2. According to it, electron-releasing substituted amines that have nucleophilic identity carry on the first step of the reaction, and then, by stabilizing the cationic intermediate III, intermediate II was transformed into the products; in the meantime, the catalyst was regenerated.
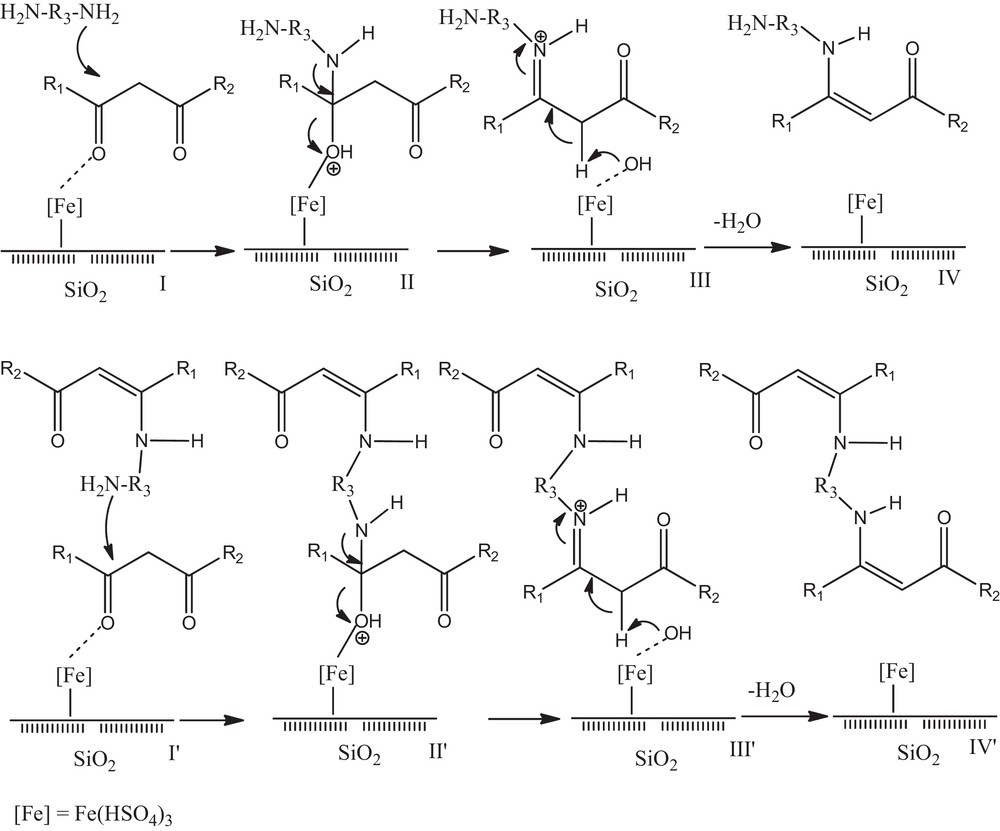
Suggested mechanism for the synthesis of bis-(β-enaminones) and bis-(β-enamino esters).
5 Conclusion
In conclusion, we have developed an efficient protocol for the synthesis of bis-(β-enaminones) and bis-(β-enamino esters) using silica ferric hydrogensulfate as a heterogeneous, recyclable and stable catalyst. The protocol could be employed for the condensation of different dicarbonyl compounds and diamines for the synthesis of potentially bioactive drugs and ligands. The advantages offered by this protocol include high yields of the desired products, under ambient conditions with diverse substrate compatibility, making it an important supplement to the existing methods.
Acknowledgements
We are grateful to the Ferdowsi University of Mashhad for their financial support of this work (GN: 3/18349).