

1 Introduction
Plants are important natural sources of organic molecules used in different sectors, such as the perfume industry as fragrances, the food industry as additives, or the cosmetics and the pharmaceutical industries [1]. Essential oils from plants are complex mixtures of several organic molecules, containing terpenes as notable components. These molecules bear several biological properties (biocides, pesticides), making them interesting biosourced compounds for many applications [2]. However, chemical transformation is crucial to generate derivatives with novel properties and uses [3]. In order to valorize these natural compounds, some of us have long been involved in the study of the sesquiterpenes called “himachalenes” (see Scheme 1), extracted from the essential oil of Cedrus atlantica, an endemic tree in Morocco [4]. Himachalenes have been a topic of investigation for several years. While research has been devoted to their total synthesis [5], other studies have revealed the versatility of natural himachalenes to different types of transformations (especially exploiting the selective reactivity of the double bonds) through reactions in organic solvents, such as Ru-catalysed aerobic epoxidation [6], diol formation using KMnO4 [7] or epoxidation through m-CPBA [8] as well as formal dihalocarbene addition or halogen addition [7], these transformations leading to mono-olefinic compounds. From the formed epoxides, Lewis acid catalysed rearrangements have recently been described [9].
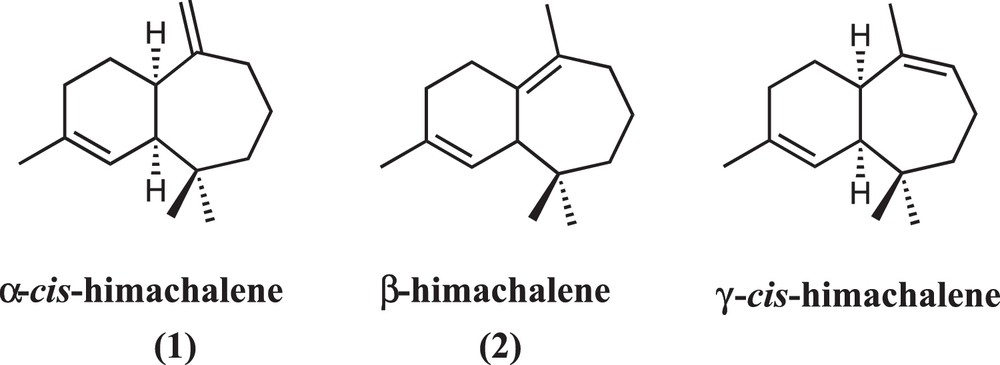
Different himachalene isomers.
Himachalenes and their halogenated derivatives were epoxidized using the classical organic peroxide m-CPBA as an epoxidating agent in organic solvents, often chlorinated ones. With the increasing demand for cleaner and safer processes, the epoxidation of himachalenes and their derivatives should be revisited and hopefully lead to efficient and “greener” transformations. We are currently interested in solvent-free epoxidations and have reported efficient protocols for the epoxidation of cyclooctene using molybdenum- [10,11] and vanadium- [12] based catalysts, as well as polyoxometalates [13]. We present herein the results of the epoxidation reaction of the himachalenes using the solvent-free catalysed method using aqueous TBHP as an oxidant and [MoO2(SAP)]2 (SAP = salicylideneaminophenolato) (Scheme 2) as a catalyst.
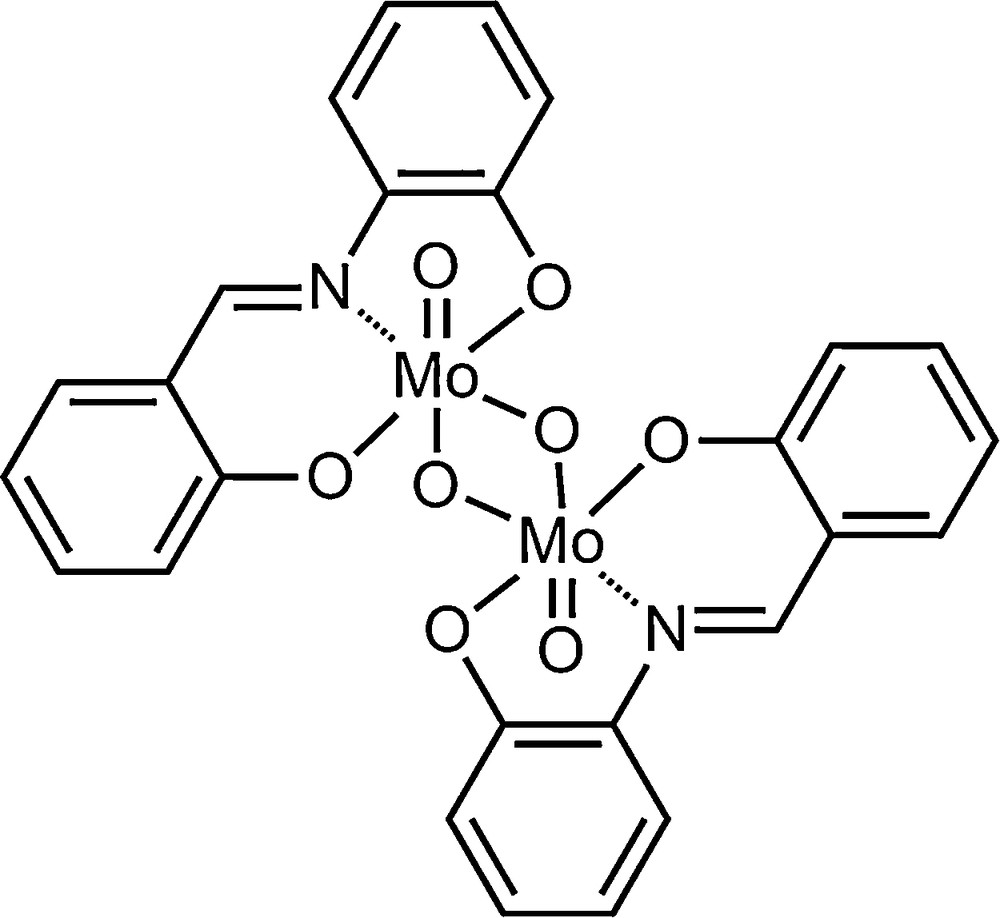
[MoO2(SAP)]2 catalyst.
2 Results and discussion
2.1 Himachalenes and chlorinated derivatives
The “raw” essential oil studied herein was extracted from the grinded wood of Atlantic Cedar (see Experimental part for details). This essential oil contains a high quantity (around 70% wt of the total oil) of the three himachalene isomers, called α-cis (1)-, β- (2) and γ-cis-himachalene, the isomerism depending on the position of the olefinic function of the 7-membered cycle (Scheme 1). The β-himachalene is the most abundant isomer in the mixture followed by the α-cis and a minor quantity of the γ-cis. The present work is focused on the study of the two main compounds 1 and 2.
Two methods were employed to obtain halogenated mono-olefinic compounds from himachalenes. The first halogenated compound, 3-chlorohimachal-7,13-ene, (3, Scheme 3) is the product of formal hydrochlorination of α-cis-himachalene. Action of gaseous HCl on a himachalene mixture followed by recrystallization in hot methanol leads to 3 as a unique species containing an exocyclic double bond [14]. The advantage of this compound in comparison to the himachalenes 1 and 2 is the presence of a unique double bond available for functionalization. The structure of this compound was elucidated by X-ray crystallography and 13C NMR [15].

Synthetic pathway for the formation of 3-chlorohimachal-7,13-ene (3) from the himachalene mixture.
The second investigated halogenated himachalene derivative, (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene (4, Scheme 4), corresponds to the formal addition of a dichlorocarbene to the tetrasubstituted double bond of the β-himachalene (2), using in situ dichlorocarbene generation from chloroform and NaOH in the presence of an ammonium salt [16]. This compound was characterized by different methods, including single-crystal X-ray diffraction [17] and 13C NMR [16].
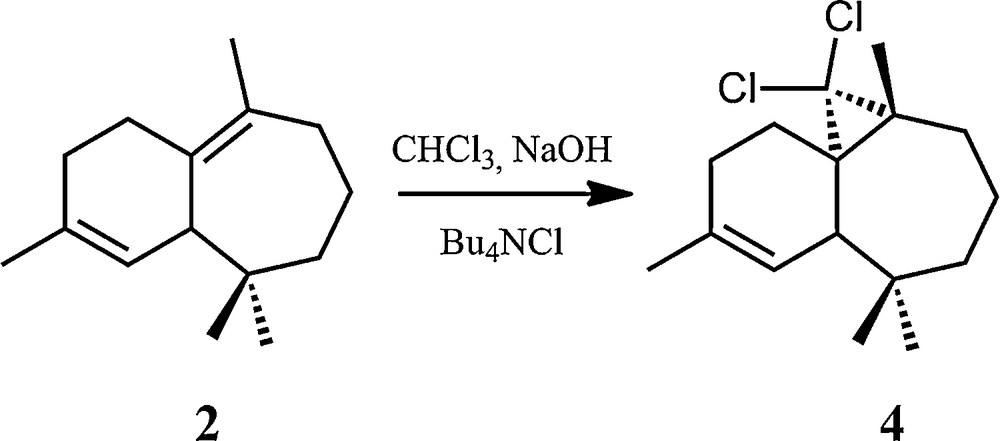
Synthetic pathway for the formation of the (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene (4) from β-himachalene (2).
2.2 Epoxidations
It has to be noticed that a few himachalene epoxides have already been identified as minor components of the essential oils [18]. However, the low natural quantity has stimulated investigations to find efficient synthetic epoxidation methods from the more abundant himachalenes using simple organic reactions (m-CPBA in chloroform or dichloromethane). According to the accepted mechanism of the epoxidation reaction [19,20], the reactivity is related to the electron density of the olefin double bond, leading to the reactivity trend shown in Scheme 5. The reactivity is also related to the electrophilic character of the oxygen atom transferred from the oxidant, hence meta-chloroperbenzoic acid (m-CPBA) is typically chosen as oxidizing agent [21].
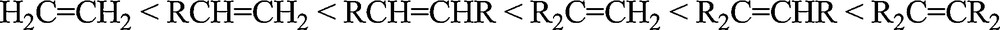
Reactivity trend of olefins towards epoxidation.
All reactions using m-CPBA need the use of organic solvents. In the quest of greener reaction conditions, the recently introduced solvent-free method using [MoO2(SAP)]2 as a catalyst was tested herein [12]. The advantages of this catalytic method are the use of a small quantity of catalyst (1% Mo vs substrate), a smoother oxidant (TBHP in water), and no added organic solvent.
2.3 Epoxidation of himachalenes by the m-CPBA method
Epoxides were obtained using an improved method relative to that already reported in the literature [13], from the α-, β-himachalene mixture. The reaction occurs in dichloromethane at room temperature with one equivalent of m-CPBA, giving the corresponding (2S,3R)-2,3-epoxy-cis-himachal-7,13-ene 5 and (1R,6S,7R)-6,7-epoxy-3,7,11,11-tetramethylbicyclo[5,4,0]undec-2-ene 6 in a 28:72 ratio (Scheme 6). Both products could be separated by column chromatography (see Experimental part for details) and additional IR and NMR characterisation of both epoxides is presented herein. The C–O–C vibration corresponding to the oxirane ring was observed around 890 cm−1 and 1034 cm−1 for 5 and 6, respectively.
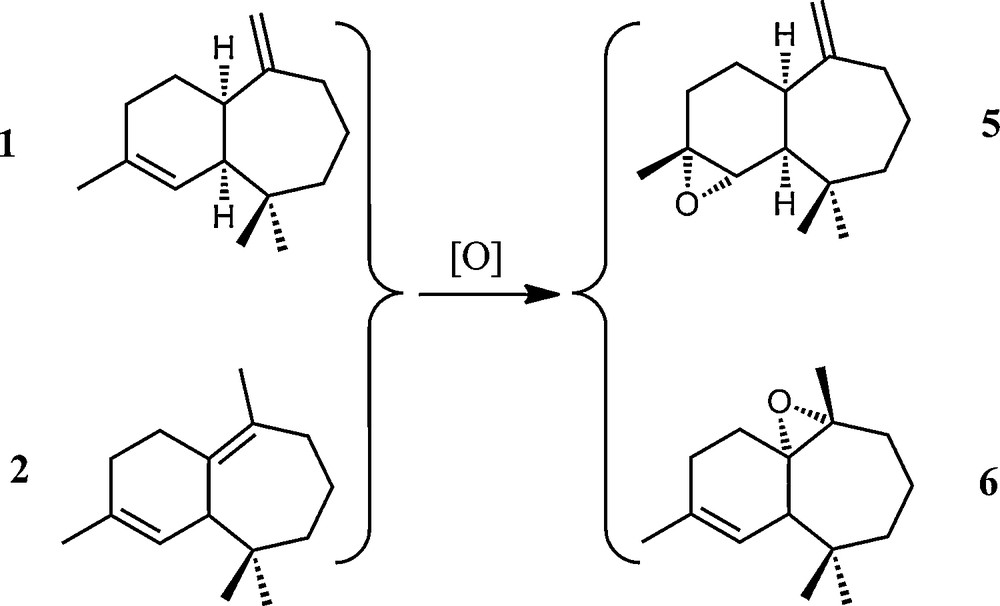
General scheme of epoxidation of (1) and (2) into the corresponding epoxides (5) and (6).
The 1H NMR spectrum of the epoxide 5 shows a doublet at δ 4.78, corresponding to the two exocyclic methylenic protons C13–H and a doublet at δ 2.95, attributable to the proton linked to the oxirane ring C2–H, proving the selectivity of the reaction. The 13C NMR spectrum presents relevant signals situated at δ 155 and 112, attributable to the exocyclic double bond C7C13 and at δ 62 and 58 for the two carbon atoms of the oxirane ring.
The 1H and 13C NMR signals of the epoxide 6 were also completely assigned. Characteristic 1H NMR signals are the doublet at δ 5.38 corresponding to the olefinic C2–H proton coupled with the signal at δ 2.00, which corresponds to C1–H. The relevant 13C NMR signals appear at δ 134 and 112 for the carbon of the endocyclic double bond C3C2–H and two signals around δ 65.0 and 65.1 ppm for the carbon atoms of the oxirane function.
2.4 Epoxidation of himachalenes by the solvent-free method
The catalysed transformation of the himachalenes mixture (Scheme 6) was carried out at 80 °C using an [Mo]/himachalenes ratio of 1:100. The reaction was followed by GC during 4 h with samples that were withdrawn periodically from the mixture and analysed. No extra solvent was added to the reaction mixture. The conversion vs time of the himachalenes and the formation of the corresponding epoxides are shown in Fig. 1 with a TBHP/himachalenes ratio of 4.
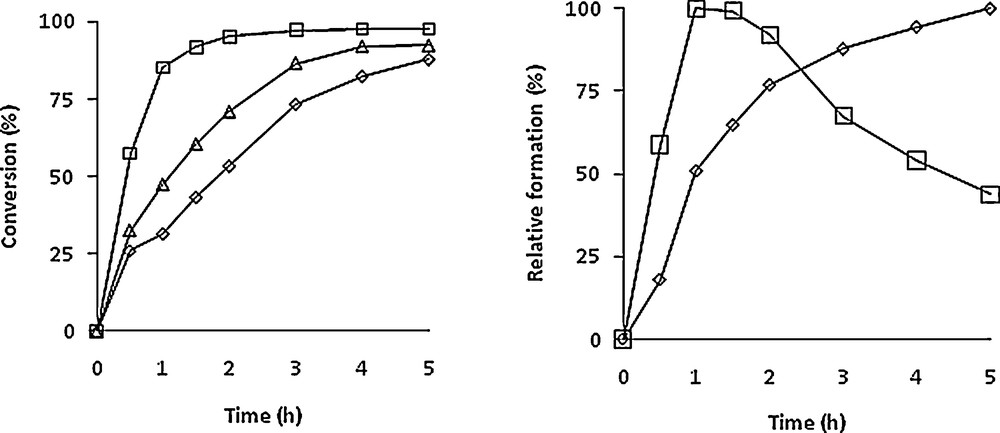
Conversion of himachalenes (left) and relative formation of epoxides (right) of α-cis (◊),β-(□) and γ-cis (Δ) himachalenes vs time. The left-hand-side graph considers the formation of each epoxide separately, the 100 value being the maximal formation for each epoxide. Experimental conditions: Mo/TBHP/himachalenes = 1/400/100. T = 80 °C.
As already shown with m-CPBA [7] and in agreement with the reactivity trend mentioned above, β-himachalene reacts faster than α-himachalene in the presence of TBHP. Indeed, more than 90% of β-himachalene and only 50% of α-himachalene were converted after 120 min. However, the optimal yield for product 6 occurred after 1 h and the subsequent decrease is certainly due to overepoxidation (four equivalents of TBHP were used) as well as to the possible subsequent formation of diols (presence of water from aqueous TBHP), which however were not identified.
Several experiments with different TBHP/himachalenes ratios (0.6 to 4) were carried out using the same [Mo]/himachalenes ratio (1/100). Although the reactions were carried out on himachalene mixtures, the conversion vs time plots of the two most abundant compounds are shown separately in Fig. 2, i.e. α-cis (1) (left) and β-himachalenes (2) (right) as well as the influence of Mo/TBHP ratio on the conversion of both himachalenes in Fig. 3. The best conversion for 1 was observed after 5 h of reaction at 80 °C with four equivalents of TBHP. As observed in Fig. 2, quantitative conversion of the β-himachalene 2 was achieved already after 2 h of reaction when using 4 equivalents of TBHP in the mixture. Both products 5 and 6 could be isolated in 80% and 90% yields (with regard to each compound separately) and in pure form after reaction chromatography carried out under optimized conditions, i.e. after 90 min in a 5/6 ratio of 33/67. The major difference between the m-CPBA and the solvent-free catalysed method consists of the greener conditions of the latter.
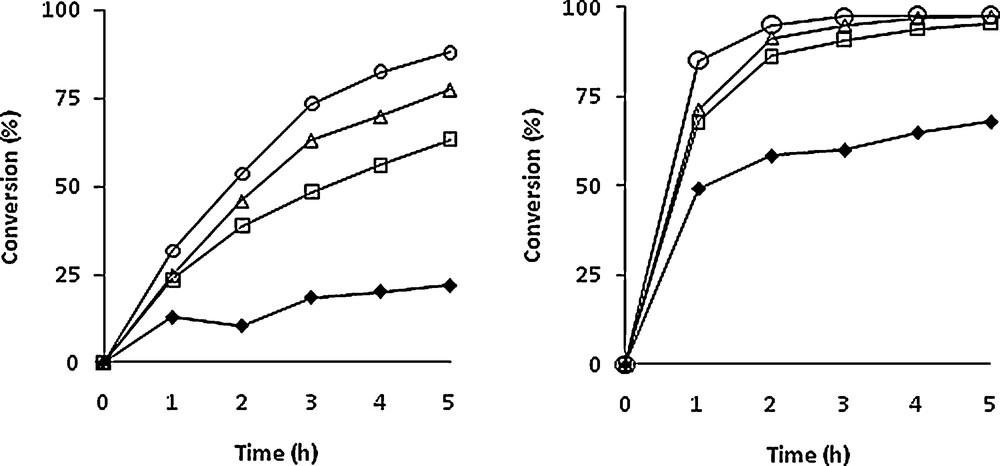
Conversion of α-cis-himachalene (1, left) and β-himachalene (2, right) vs time (in hours) with different THBP amount. Experimental conditions: [Mo]/TBHP/himachalenes = 1/x/100. T = 80 °C. x = 60 (), 150 (□), 250 (Δ), 400 ().
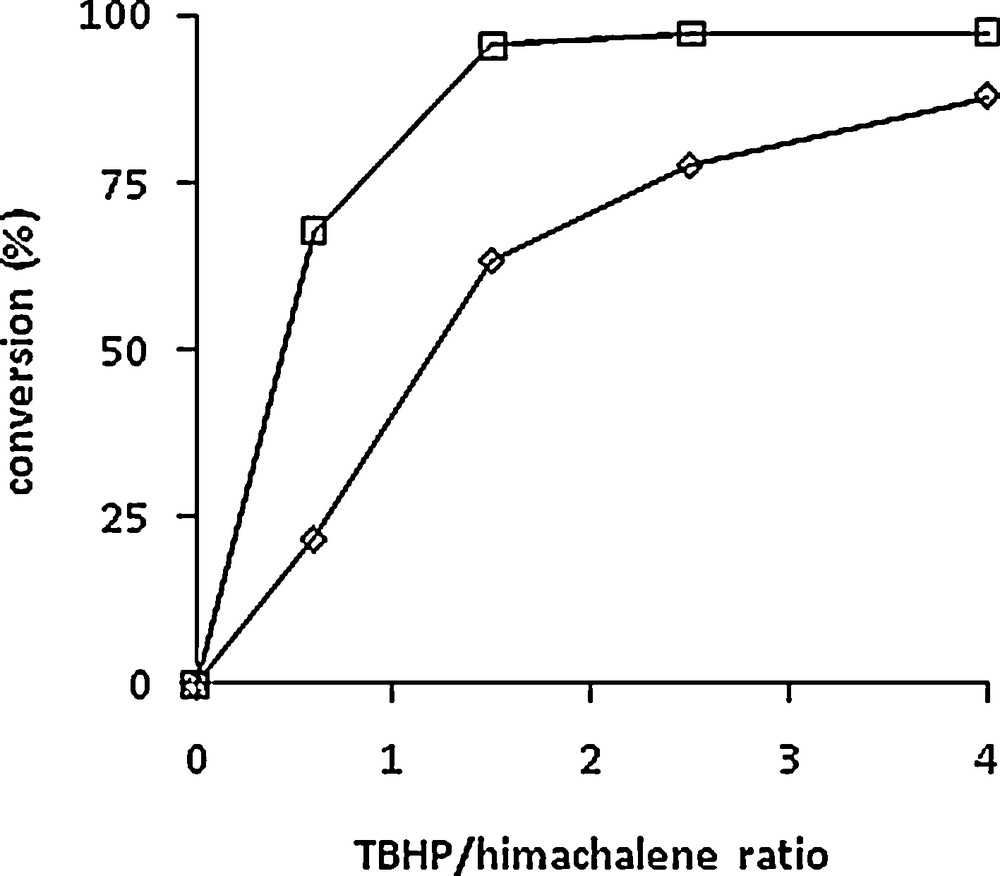
Relative conversion of α-cis-himachalene 1 (◊) and β-himachalene 2 (□) at 5 h vs TBHP/himachalenes ratio. Experimental conditions: [Mo]/himachalenes = 1/100. T = 80 °C.
2.5 Epoxidation of (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene (4)
As already described in the literature [8], treatment of the (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene 4 with a stoichiometric amount of m-CPBA led to two products 7 and 7′ in a 70:30 relative ratio and with a 80% total yield (Scheme 7).
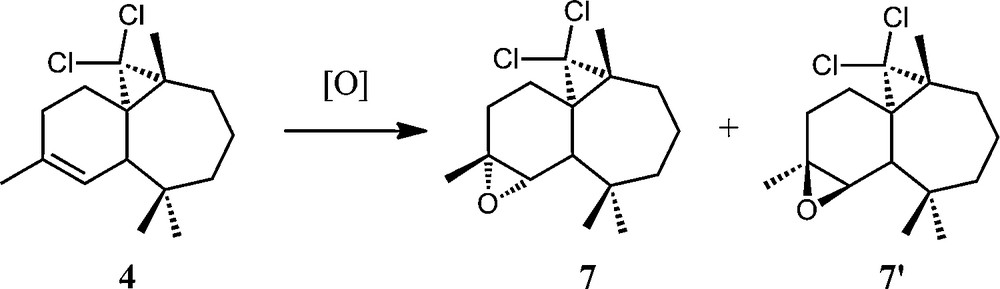
General scheme of epoxidation of (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene (4).
Epoxides 7 and 7′ gave different 1H and 13C NMR spectra. In the 1H NMR spectrum, the doublet corresponding to the ethylenic proton of 4 disappears and a signal appears at δ 3.00 for 7 and 3.10 for 7′, corresponding to oxirane formation. The 13C NMR spectrum reveals in particular two signals at δ 55.0 and 61.9 for 7 (55.8 and 61.8 for 7′) attributable to the carbon atoms of the oxirane ring. The absolute configurations of both compounds have been determined through X-ray crystallography [22].
The epoxidation of 4 with one equivalent of TBHP and 0.5% equivalent of [MoO2(SAP)]2 at 80 °C has been monitored for 7 h (see Fig. 4). An 80% conversion could be obtained after 5 h. The epoxides were isolated as a mixture and analysed by NMR, giving a 50:50 ratio for the two stereoisomers.
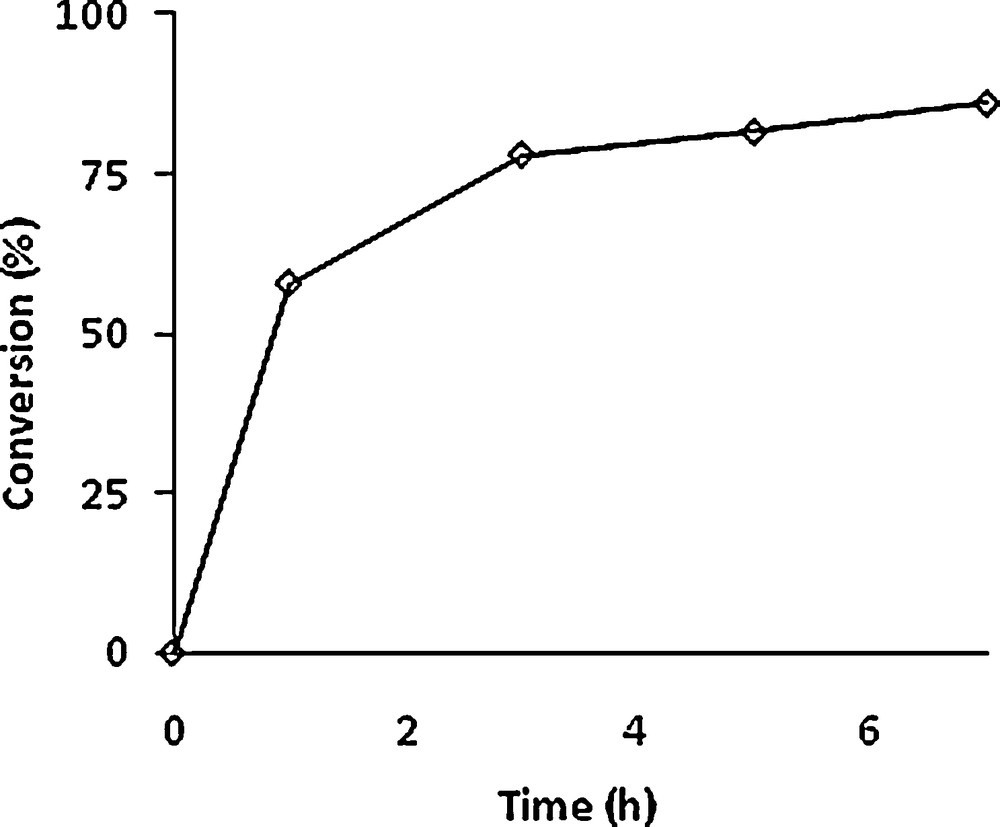
Conversion of (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene (4) vs time (in hours). Experimental conditions: Mo/TBHP/substrate = 1/100/100. T = 80 °C.
2.6 Epoxidation of 3-chlorohimachal-7,13-ene (3)
As previously described [23], 3 reacts in chloroform with one equivalent of m-CPBA at the exocyclic double bond and leads to two stereoisomers 8 and 8′ (Scheme 8) in a 85:15 ratio with a total yield of 85% after 4 h. Both compounds were separated through column chromatography.
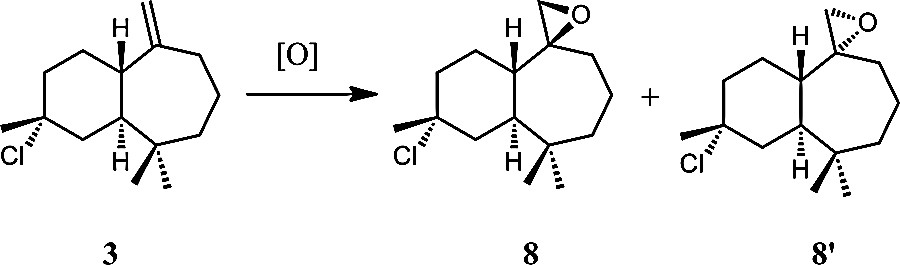
General scheme of epoxidation of 3-chlorohimachal-7,13-ene (3).
Both isomers 8 and 8′ were characterized separately through 1H and 13C NMR spectrometry and gave similar spectra [24]. The two ethylenic protons of 3 disappeared and a signal corresponding to two protons appeared at δ 2.50 for 8 and 8′. In the 13C NMR spectrum, as observed in the previous reaction, the signals corresponding to the ethylenic carbon atoms of 3 disappeared and two signals in agreement with the formation of the oxirane ring appeared at δ 45.3 and 57.0.
The solvent-free Mo-catalysed reaction with THBP was followed under identical conditions as for the other previously described epoxidations. The substrate conversion is indicated in Fig. 5. The conversion was very high even after 1 h and a 70% selectivity was observed at the end of the reaction, yielding the two diastereoisomers 8 and 8′ in a 67:33 ratio.
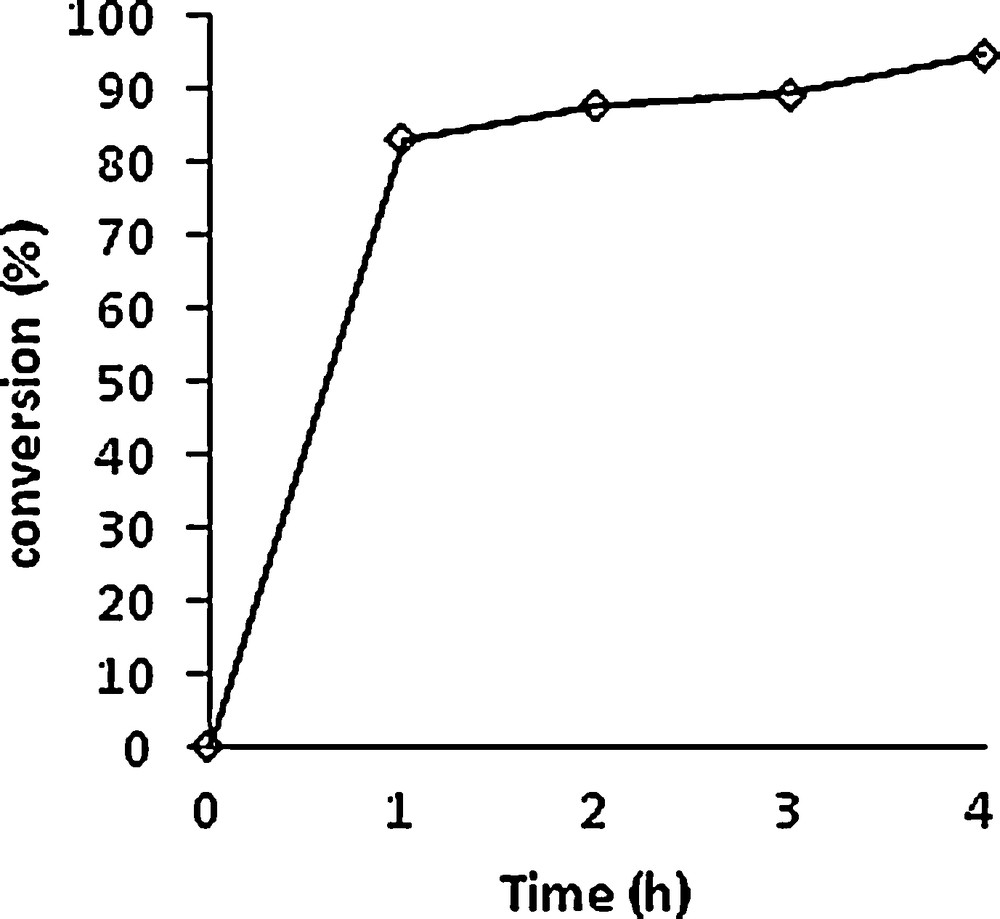
Conversion of 3-chlorohimachal-7,13-ene (3) vs time. Experimental conditions: [Mo]/TBHP/substrate = 1/100/100. T = 80 °C.
2.7 Mechanistic discussion
The epoxidations of the himachalenes with peracids were studied theoretically with α-himachalenes [25] and β-himachalenes [26], explaining the selectivity trends on the basis of the well-known Bartlett mechanism [27]. Concerning the metal-catalyzed olefin epoxidation by TBHP, mostly realized with molybdenum- or vanadium-based catalysts, Mimoun and Sharpless have proposed relevant mechanisms in organic solvents that involve the addition of TBHP to the catalyst in the activation phase [28]. We have recently shown that the olefin epoxidation with the [MoO2(SAP)] fragment as a catalyst and TBHP as an oxidant follows a mechanism relatively similar to that operating in the presence of peracids [11]. On this basis, the slight difference in selectivity that is observed between the m-CPBA epoxidation and the [MoO2(SAP)]-catalyzed epoxidation by TBHP can be attributed to the slightly different steric interaction between the oxygen atom delivering species and the substrate in the transition state (B vs A in Scheme 9).

Transitions states of epoxidation of olefin with m-CPBA (A) and with [MoO2(SAP)]/TBHP (B).
3 Conclusion
We have shown herein for the first time that the solvent-free catalysed epoxidation is relatively efficient for the transformation of natural substrates. The himachalenes 1 and 2 and their derivatives 3 and 4 could be efficiently transformed into epoxide products in close analogy to the classical m-CPBA method, although giving slightly different steroisomeric ratios for the epoxidation products of 3 and 4. The rational use of this method for the epoxidation of a wider range of natural compounds is an interesting perspective of this work.
4 Experimental part
All preparations were carried out in air. Organic solvents (ethanol, diethylether, hexane, ethylacetate), organic (acetophenone, ACROS), and inorganic (MnO2, Aldrich) chemicals were employed as received without any purification. Compounds 3 and 4 were obtained following the known procedures [8]. [MoO2(SAP)]2 was freshly synthesized as described previously [11] prior to use. TBHP (70% in water, ACROS) was used as received. The infrared spectra were recorded in KBr matrices at room temperature with a Nicolet Impact 410 spectrometer in the 4000–400 cm−1 range. Melting points were measured in capillary tubes with a Buchi 510 apparatus. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance DPX-300 spectrometer using TMS as a reference and CDCl3 as a solvent. Catalytic reactions were followed by gas chromatography on an Agilent 6890 A chromatograph equipped with an FID detector, an HP5-MS capillary column (0.30 m × 0.25 mm × 0.25 m) and automatic sampling. The conversions were calculated relatively to an internal standard (acetophenone). The column chromatographic separations were carried out using Merck 60 silica gel.
5 Extraction and separation of sesquiterpenes [29]
Grinded Cedar wood (200 g) was introduced in a glass flask equipped with a Clevenger apparatus containing water. The system was maintained under reflux for 12 h. The obtained oil was purified through column chromatography and a mixture of the three isomers (α-cis(1), β-(2) and γ-himachalene) in (23:61:16) ratio was isolated (4.2 g).
6 General epoxidation procedure of himachalenes and derivatives with m-CPBA
6.1 For himachalenes
In a 100-mL flask containing the himachalene mixture (2 g, 9.79 mmol) solubilized in 50 mL of CH2Cl2 was added the stoichiometric amount of m-CPBA (1.69 g, 9.79 mmol). The mixture was stirred for 2 h at room temperature and then treated with 40 mL of a 10% aqueous solution of sodium hydrogen carbonate. The aqueous phase was extracted three times with 20 mL of diethyl ether. The combined organic phase was dried with anhydrous sodium sulphate and the volatile solvents were removed under reduced pressure. The residue was separated through column chromatography (silica gel) using hexane/ethyl acetate (97/3) as the eluent. The epoxides 5 (0.44 g, 90% yield relatively to the corresponding himachalene 1) and 6 (1.24 g, 95% yield relatively to the corresponding himachalene 2) were recovered as yellow oils after removal of the volatile solvents under reduced pressure. Their identity was confirmed by comparison of the NMR properties with those reported in the literature [8].
6.2 For (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene
The procedure was identical to that used for the himachalenes, using (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene (1.5 g, 5.4 mmol) as a substrate. Eluent: hexane/ethyl acetate (97/3). The final products are white solids. Final yield: 80% (7/7′ = 70:30).
6.3 For 3-chlorohimachal-7,13-ene
The procedure was identical to that used for the himachalenes, using 3-chlorohimachal-7,13-ene (1 g, 4.15 mmol) as a substrate. Solvent: chloroform. Eluent: hexane/ethyl acetate (98/2). The final products are white solids. Final yield: 85% (8/8′ ratio = 85/15).
7 General catalytic epoxidation procedure with TBHP
7.1 Standard catalytic runs
In a typical experiment, one equivalent of the substrate and [MoO2(SAP)]2 (0.005 equiv.) were mixed together in a round-bottom flask and stirred in air. Acetophenone was added as an internal standard. The reaction temperature was regulated at 80 °C and then x equiv. of aqueous THBP was added to the mixture under stirring, setting the zero reaction time. Samples of the organic phase were periodically withdrawn after stopping the stirring and allowing phase separation. For each aliquot, the reaction was quenched by addition of MnO2, followed by the addition of diethylether and removal of the manganese oxide and residual water by filtration through silica before GC analysis.
7.2 Optimised procedure for the solvent-free himachalenes epoxidation
In a flask equipped with a magnetic stirrer and cooler, 1 g (4.9 mmol) of the himachalene mixture and 16.6 mg of [MoO2(SAP)]2 (0.024 mmol, 0.005 equiv) were added. The mixture was warmed to 80 °C and then aqueous TBHP (70%, 2.68 mL, 19.6 mmol, 4 equiv) was added. The mixture was stirred at 80 °C for 90 min. After cooling, the organic residue was extracted with diethylther, concentrated under reduced pressure and the epoxides were then separated by column chromatography through silica gel and recovered as yellow oils (5, 0.19 g, 80% relatively to the corresponding himachalene 1; 6, 0.62 g, 90% relatively to the corresponding himachalene 2) as described above for the m-CPBA epoxidation procedure.
7.3 Optimised procedure for the solvent-free epoxidation of (1S,3R,8R)-2,2-dichloro-3,7,7,10–tetramethyltricyclo[6,4,0,01,3]-dodec-9-ene (4)
In a flask equipped with a magnetic stirrer and a cooler, 1.72 g (5.98 mmol) of dichlorocarbenehimachalene 4 and 20.3 mg of [MoO2(SAP)]2 (0.029 mmol, 0.005 equiv.) were added. The mixture was warmed to 80 °C and aqueous TBHP (70%, 0.82 mL, 5.98 mmol, 1 equiv.) was added. The mixture was stirred at 80 °C for 6 h. After cooling, the mixture was worked up as for the above-described epoxidation of the himachalalene mixture, using hexane/ethylacetate (96/4) as the eluent for the chromatographic separation. The epoxides 7 and 7′ were isolated together as white crystalline powders (1.32 g, 73% yield).
7.4 Optimised procedure for solvent-free epoxidation of 3-chlorohimachal-7,13-ene (3).
In a flask equipped with a magnetic stirrer and a cooler, 1.15 g (4.8 mmol) of the 3-chlorohimachal-7,13-ene 3 and 16.6 mg of [MoO2(SAP)]2 (0.024 mmol, 0.005 equiv) were added. The mixture was warmed to 80 °C and aqueous TBHP (70%, 0.67 mL, 4.8 mmol, 1 equiv) was added. The mixture was stirred at 80 °C for 4 h. After cooling, the organic phase was worked up as described above for the himachalene mixture using hexane/ethylacetate (97/3) as the eluent. The epoxides 8 and 8′ were isolated in a 67/33 ratio as white solids (0.86 g, 70% yield).
8 13C NMR spectrum of the 3-chlorohimachal-7,13-ene (3)
13C NMR (δ, CDCl3): 22.3 (CH3), 24.4 (CH2), 30.3 (CH3), 31.8 (CH2), 33.1 (CH2), 34.6 (CH3), 35.8 (Cq), 41.5 (CH2), 42.9 (CH2), 43.1 (CH), 43.3 (CH2), 47.8 (CH), 73.2 (Cq), 110.3 (CH2sp2), 156.4 (Csp2).
9 Characterisation of the epoxides
9.1 From himachalenes
9.1.1 (2S,3R)-2,3 -epoxy-cis-himachal-7,13-ene (5)
Yield: 90%. 1H NMR (δ, CDCl3): 0.95 and 1,10 (6H, 2s), 1.35 (3H, s), 2.95 (1H, d), 4.78 (2H, d, J = 6.5 Hz). 13C NMR (δ, CDCl3): 22.8 (CH2), 23.3 (CH3), 24.9 (CH3), 26.1 (CH2), 28.2 (CH2), 30.9 (CH2), 32.1 (CH3), 36.9 (Cq sp3) 37.8 (CH), 39.2 (CH2), 47.5 (CH), 58.8 (Cq sp3), 62.2 (CH), 112.1 (CH2 sp2), 155.5 (C sp2).
9.1.2 (1R,6S,7R)-6,7-epoxy-3,7,11,11-tetramethylbicyclo[5,4,0]undec-2-ene (6)
Yield: 95%, 1H NMR (δ, CDCl3): 0.88 (3H, s); 0.95 (3H, s); 1.26 (3H, s); 1.73 (3H, s); 5.38 (1H, s), 13C NMR δ, CDCl3: 18.6 (CH3), 20.7 (CH2), 23.8 (CH3), 24.9 (CH2), 25.4 (CH3), 27.7 (CH2), 29.9 (CH3), 36.4 (CH2), 36.5 (CH2), 44.1 (Cq sp3), 48.8 (CH), 65.0 (Cq sp3), 65.1 (Cq sp3) 112.4 (CH sp2), 134.3 (C sp2).
9.2 From dichlorocarbenehimachal-2-ene
9.2.1 2,2-dichloro-9,10-epoxy-3,7,7,10–tetramethyltricyclo[6,4,0,01.3]-dodecane 7 and 7′
Mp = 134–136 °C, IR ν (cm−1): 615, 1050, 1230, 1410.
7: NMR 1H (δ, CDCl3): 1.04–1.09 (6H, 2s), 1.15 (3H, s), 1.30 (3H, s), 3.00 (1H, d, J = 1 Hz), NMR 13C (δ, CDCl3): 15.0 (CH3), 19.8 (CH2), 20.8 (CH2), 23.9 (CH3), 25.8 (CH2), 27.8 (CH3), 28.9 (CH3), 29.1 (CH2), 31.1 (Cq), 31.5 (Cq), 35.5 (Cq), 37.0 (Cq), 43.4 (CH), 55.0 (Cq), 61.9 (CH), 77.3 (Cq).
7′: NMR 1H (δ, CDCl3): 1.12–1.17 (6H, 2s), 1.25 (3H, s), 1.32 (3H, s), 3.10 (1H, d, J = 2.44 Hz), NMR 13C (δ, CDCl3): 17.8 (CH3), 21.8 (CH2), 22.1 (CH2), 22.9 (CH3), 24.5 (CH3), 28.8 (CH2), 30.5 (CH3), 32.1 (Cq), 35.6 (CH2), 35.9 (Cq), 39.6 (Cq), 43.6 (CH), 49.9 (Cq), 55.8 (Cq), 61.8 (CH), 77.8 (Cq).
9.3 From 3-chlorohimachal-7,13-ene
9.3.1 3-chloro-7,13-epoxy-cis-himachalane. 8 and 8′
8: 1H NMR (δ, CDCl3): 0.86 and 0.90 (CH3, 6H, 2s), 1.50 (CH3, 3H, s), 2.50 (CH2, q, J = 3.0 Hz).
8′: 1H NMR (δ, CDCl3): 0.80 and 0.90 (CH3, 6H, 2s), 1.60 (CH3, 3H, s), 2.50 (CH2,s).
8 and 8′: 13C NMR (δ, CDCl3): 19.9 (CH2), 21.8 (CH3), 27.4 (CH2), 29.4 (CH3), 29.7 (CH3), 34.4 (CH3), 35.7 (CH3), 39.1 (CH2), 40.5 (CH2), 41.7 (CH), 42.1 (CH2), 43.3 (CH2), 45.3 (CH2), 54.4 (CH), 57.0 (Cq), 72.2 (Cq).
Acknowledgements
We acknowledge the Centre National de la Recherche Scientifique (CNRS) and the Institut Universitaire de France (IUF) for financial support, the Université Paul-Sabatier and its Institut Universitaire Technologique for the facilities, and the International Associated Laboratory (LIA) “French–Moroccan Laboratory of Molecular Chemistry” (LCMMF).