

1 Introduction
The development of a novel nitrogen fixation system is one of the most important subjects in chemistry. Industrially, ammonia is produced from molecular dinitrogen and dihydrogen by the use of the iron-based heterogeneous catalysts under harsh reaction conditions, such as high temperature and high pressure in order to activate molecular dinitrogen and to produce ammonia efficiently (Scheme 1) [1]. In sharp contrast to the energy-consuming Haber–Bosch process, biological nitrogen fixation is well known to occur under ambient reaction conditions. The precise reaction mechanism still remains unclear; however, an X-ray structural model has recently been reported for the active site of nitrogenase in FeMo-cofactor (Scheme 1) [2]. It is generally believed that molecular dinitrogen is coordinated and activated at these multimetallic centers and converted into ammonia via a sequential process of protonation and reduction.

a: industrial production of ammonia (Haber–Bosch process); b: biological nitrogen fixation by nitrogenase.
Since the discovery of the first transition metal–dinitrogen complex, a ruthenium–dinitrogen complex (Scheme 2) [3], the preparation of various transition metal–dinitrogen complexes and their stoichiometric reactivities have so far been well investigated toward the goal of the achievement of novel nitrogen fixation system under mild reaction conditions [4]. Especially, molybdenum– and tungsten–dinitrogen complexes have been most intensively studied, where the coordinated molecular dinitrogen can be converted into ammonia under mild reaction conditions by treatment with inorganic acids such as sulfuric acid (Scheme 2) [5,6]. In sharp contrast to the stoichiometric formation of ammonia from the transition metal–dinitrogen complexes, reactions of these transition metal–dinitrogen complexes with molecular dihydrogen under mild reaction conditions did not yield any ammonia at all. In this reaction system, the ligand exchange of the coordinated molecular dinitrogen with molecular dihydrogen easily took place. As a result, no formation of ammonia was observed at all under these reaction conditions.

Transition metal–dinitrogen complexes and their stoichiometric reactions.
Based on these research backgrounds, we envisaged the direct reaction of the coordinated molecular dinitrogen with the coordinated molecular dihydrogen under mild reaction conditions. The terminal nitrogen atom of the coordinated dinitrogen on the transition metal complexes may attack the coordinated and electrophilic molecular dihydrogen to form nitrogen–hydrogen bonds, leading to the formation of ammonia (Scheme 3). After many trials, we evidenced the formation of ammonia through the reaction of tungsten–dinitrogen complexes with an excess amount of a ruthenium–dihydrogen complex at 55 °C for 24 h under 1 atm of molecular dihydrogen (Scheme 4) [7]. In this reaction system, only a stoichiometric amount of ammonia was produced based on the tungsten atom. However, this is the first successful example of formation of ammonia from molecular dinitrogen and dihydrogen under mild reaction conditions [8]. This work was achieved when I was an assistant professor in the group of Prof. Masanobu Hidai at The University of Tokyo (1995–2000).

Concept for direct reaction of coordinated dinitrogen with dihydrogen.
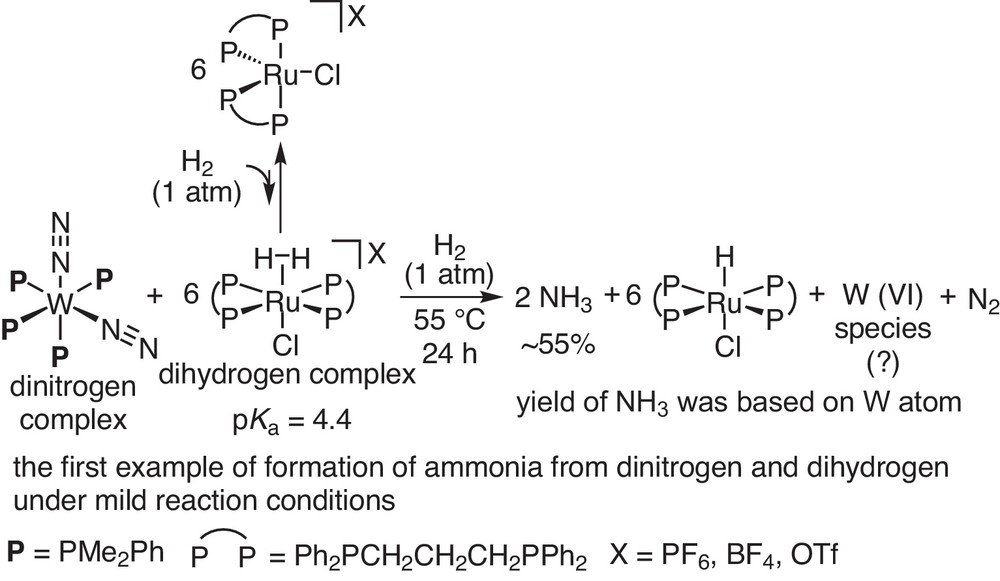
Direct reaction of dinitrogen complex with dihydrogen complex.
We also found a stoichiometric formation of ammonia by using a backminsterfullerene bearing 2 equiv of γ-cyclodextrin as ligands. The reaction of the buckminsterfullerene molecule in water at 60 °C for 1 h under 1 atm of molecular dinitrogen under visible light gave a stoichiometric amount of ammonia based on the fullerene (Scheme 5) [9]. This work was achieved as a collaborative work with the group of Kyoto University Professor emeritus Zen-ichi Yoshida at Kinki University when I was an assistant professor in the group of Professor Sakae Uemura at Kyoto University (2000–2005).
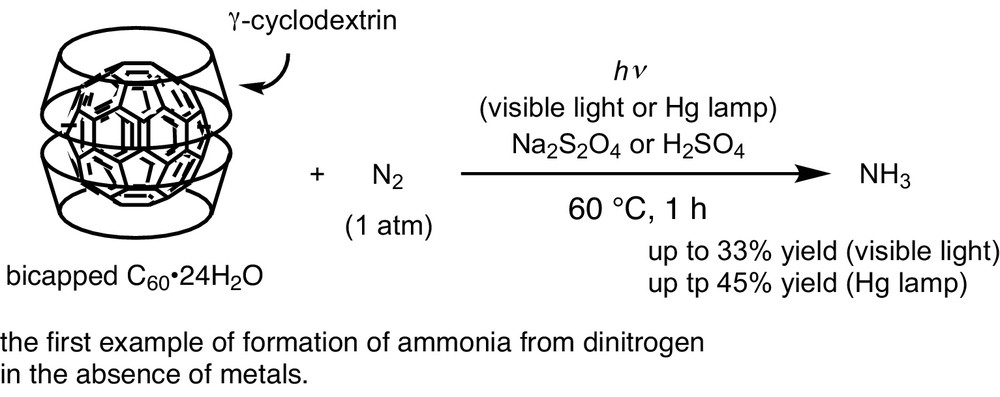
Nitrogen fixation assited by bicapped C60.
In sharp contrast to many studies on the stoichiometric formation of ammonia from transition metal–dinitrogen complexes [4], there are only a few examples of catalytic transformation of molecular dinitrogen into ammonia by using transition metal–dinitrogen complexes as catalysts. In 2003, Prof. Schrock and a co-worker reported the first successful example of the catalytic conversion of molecular dinitrogen into ammonia under mild reaction conditions by using decamethylchromocene (CrCp*2; Cp*=η5-C5Me5) as a reducing reagent and 2,6-lutidinium tetraarylborate ([LutH]BArF4; ArF=2,5-(CF3)2C6H3) as a proton source in the presence of a transition metal–dinitrogen complex as a catalyst. In their reaction system, the molybdenum–dinitrogen complex bearing a triamido- and monoamine tetradentate ligand worked as an effective catalyst, where less than 8 equiv of ammonia were produced based on the molybdenum atom in the catalyst (Scheme 6) [10], Results of the isolation of various reactive complexes as key intermediates and the theoretical study of the reaction mechanism clarified the detailed reaction pathway of the catalytic formation of ammonia. However, after the Schrock's report, there was no example of catalytic conversion of molecular dinitrogen into ammonia under mild reaction conditions by using transition metal–dinitrogen complexes as catalysts.
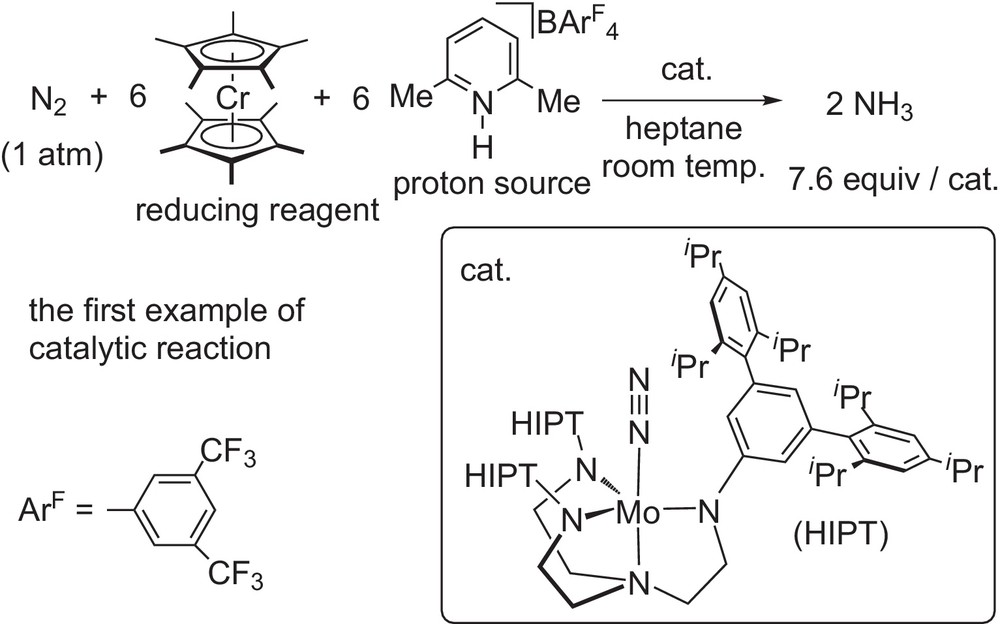
Catalytic formation of ammonia by Schrock system.
As an extension of our study on the development of novel nitrogen fixation system under mild reaction conditions [11,12], we have recently evidenced the molybdenum-catalyzed reduction of molecular dinitrogen into ammonia under ambient reaction conditions by using a dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer ligands as a catalysts (Scheme 7) [13]. In this reaction system, molecular dinitrogen can be converted into ammonia under ambient reaction conditions, where up to 52 equiv of ammonia were produced based on the catalyst (26 equiv of ammonia based on the molybdenum atom in the catalyst) by the use of slightly modified dinitrogen-bridged dimolybdenum–dinitrogen complexes [14]. This was another successful example of the direct conversion of molecular dinitrogen by using transition metal–dinitrogen complexes as catalysts when we published the original paper in 2010.
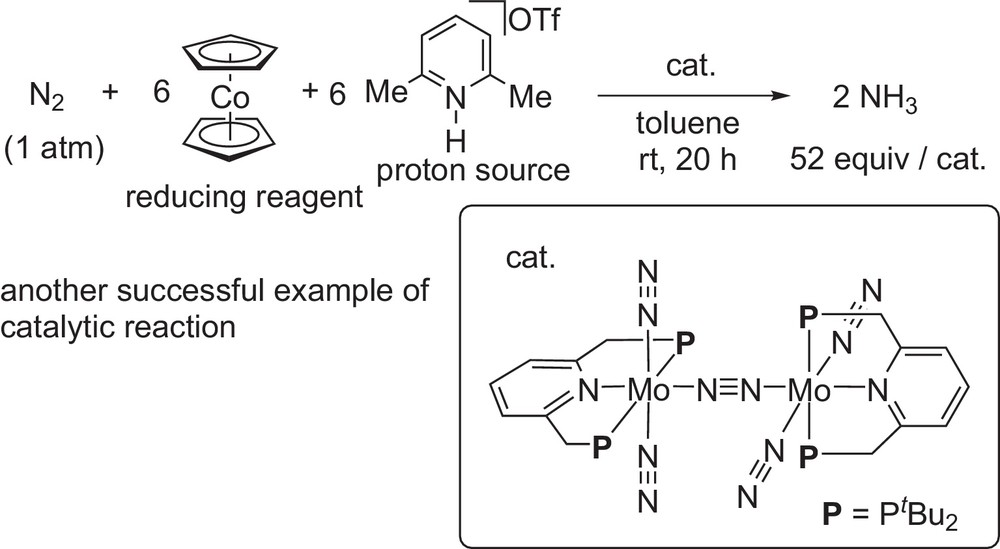
Catalytic formation of ammonia by our system.
2 Results and discussion
At first, we explained how to prepare the dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer ligands [15]. The reduction of molybdenum(III) chloride complex bearing a PNP-type pincer ligand [MoCl3(PNP)], which was prepared from the reaction of [MoCl3(thf)] complex with 1 equiv of PNP-type pincer ligand, with an excess amount of Na-Hg (6 equiv) under 1 atm of N2 in THF at room temperature for 12 h, gave the target complex in 63% isolated yield (Scheme 8). This complex can be characterized by NMR, IR, and Raman spectra. 15N{1H} NMR spectrum of the complex in THF under 1 atm of 15N2 shows three peaks, 8.5 ppm (s), −16.5 ppm (d, J = 6.1 MHz), and −29.0 (dt, J = 6.1 and 2.4 MHz) (Scheme 9). These three peaks indicate that the dinuclear structure remains even in solution.
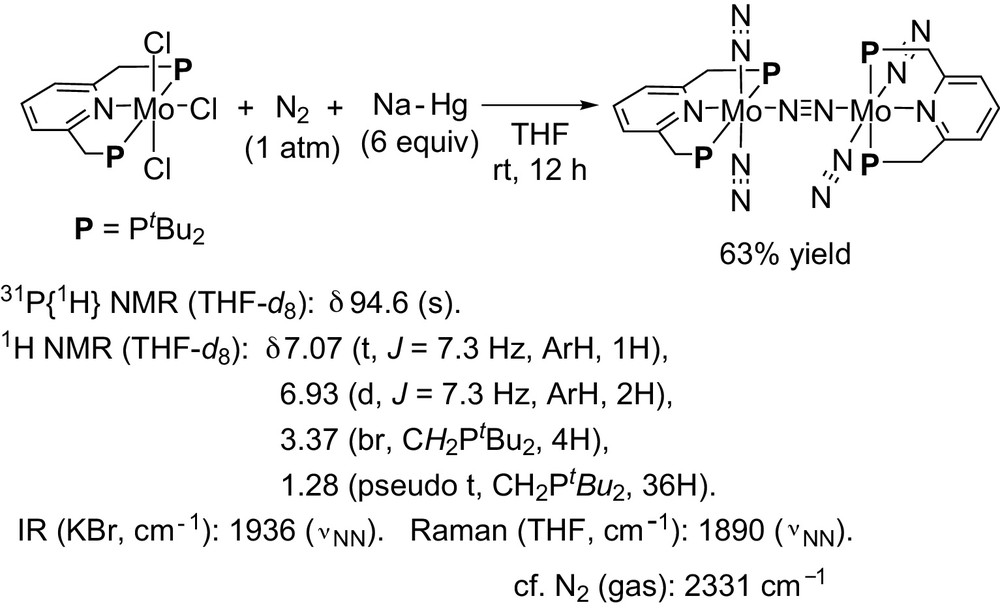
Preparation of dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer ligands.
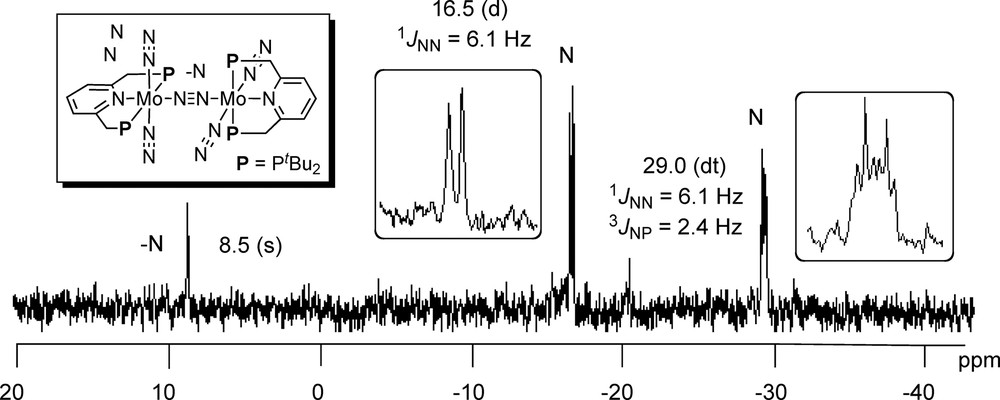
15N{1H} NMR Spectrum of dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer ligands under 15N2.
A more detailed structure of the complex is confirmed by X-ray analysis [13]. An ORTEP drawing of the complex is shown in Scheme 10. This complex contains two molybdenum moieties bearing the PNP-type pincer ligand. These two molybdenum moieties containing two dinitrogen ligands in a trans form are bridged by one molecular dinitrogen ligand in an end-on fashion with almost the liner molybdenum–dinitrogen-molybdenum bonds. These two molybdenum fragments are twisted around the molybdenum–dinitrogen-molybdenum axis with respect to each other away from the steric interaction between the two PNP-type pincer ligands. The bond distance between the two nitrogen atoms in the terminal dinitrogen ligand is slightly longer than that of the corresponding bridging dinitrogen ligand.

(Color online.) An ORTEP drawing of dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer ligands.
The reaction of molecular dinitrogen under atmospheric pressure with 72 equiv of cobaltocene (CoCp2; Cp=η5-C5H5) as a reducing reagent and 96 equiv of 2,6-lutidinium trifluoromethanesulfonate ([LutH]OTf) as a proton source in the presence of a catalytic amount of the dinitrogen-bridged dimolybdenum–dinitrogen complex bearing a PNP-type pincer ligand (0.01 mmol) in toluene at room temperature for 20 h gave 12 equiv of ammonia based on the catalyst (50% yield based on the cobaltocene) together with 13 equiv of molecular dihydrogen based on the catalyst (37% yield based on the cobaltocene) (Scheme 11) [13]. Separately, we confirmed that no formation of ammonia was observed in the absence of the catalyst and when 1 atm of Ar was used in place of 1 atm of N2. These results indicate that the use of both catalyst and N2 gas is an essential factor to promote the catalytic reaction. In all cases, no hydrazine was formed at all.

Typical result of catalytic formation of ammonia by our system.
To get a direct evidence of the conversion of molecular dinitrogen into ammonia by using the present system, we carried out the catalytic reaction by using 15N2 gas, in place of normal 14N2 gas under the same reaction conditions (Scheme 12). In this case, 10 equiv of 15NH3 were produced using this catalyst under the same reaction conditions. 15N{1H} NMR spectra in DMSO-d6 indicate the direct conversion of 15N2 gas (−68 ppm (s)) into ammonium salt
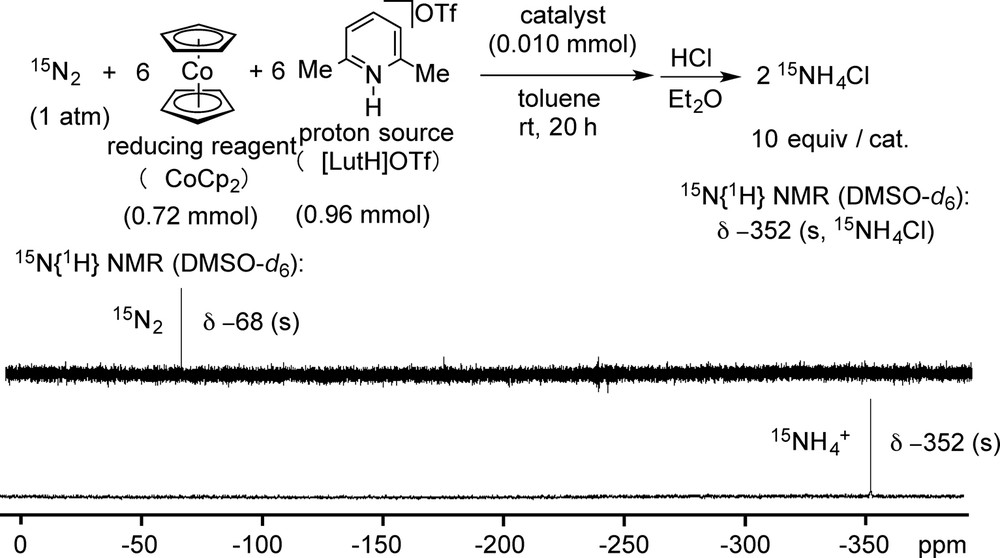
Typical result of catalytic formation of ammonia from 15N2 by our system.
Next, we investigated catalytic reactions by using other molybdenum–dinitrogen complexes as catalysts (Scheme 13) [13]. The use of dinitrogen-bridged dimolybdenum-carbonyl complex bearing PNP-type pincer ligands as a catalyst gave 0.4 equiv of ammonia based on the catalyst. The use of mononuclear molybdenum–dinitrogen complex bearing a PNP-type pincer ligand and dimethylphenylphosphine as a catalyst gave 0.2 equiv of ammonia based on the catalyst. It is noteworthy that conventional mononuclear molybdenum–dinitrogen complexes bearing four monodentate phosphines and two bidentate phosphines did not work as catalysts at all. These results indicate that only the dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer a ligands worked as an effective catalyst toward the formation of ammonia under ambient reaction conditions.
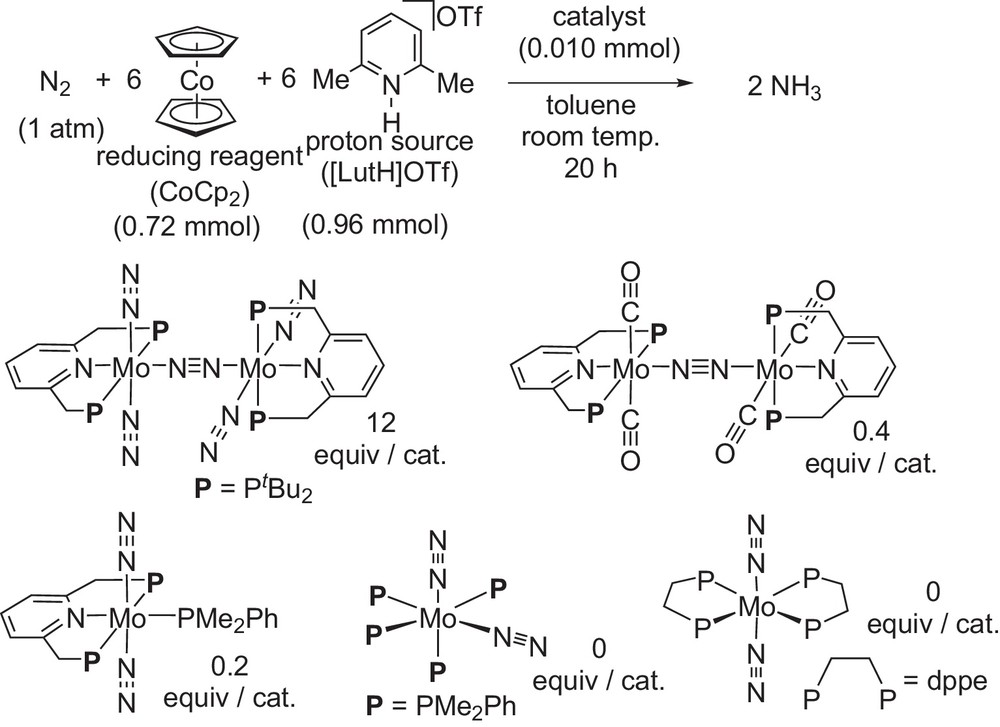
Catalytic formation of ammonia by using other molybdenum–dinitrogen complexes as catalysts.
To get more information on the reaction pathway, we carried out the following stoichiometric reactions (Scheme 14) [13]. At first, the reaction of the dinitrogen-bridged dimolybdenum–dinitrogen complex with 2 equiv of HBF4 in THF at room temperature for 24 h gave a mononuclear molybdenum–hydrazido complex bearing a PNP-type pincer ligand after the addition of pyridine as a ligand. This molybdenum–hydrazido complex can be characterized by NMR, IR, and X-ray analysis. The reaction of the isolated molybdenum–hydrazido complex with an excess amount of [LutH]OTf as a proton source under the catalytic conditions afforded ammonia in 20% yield based on the molybdenum atom. These results indicate that the coordinated molecular dinitrogen can be converted into ammonia via the molybdenum–hydrazido complex as a key reactive intermediate.
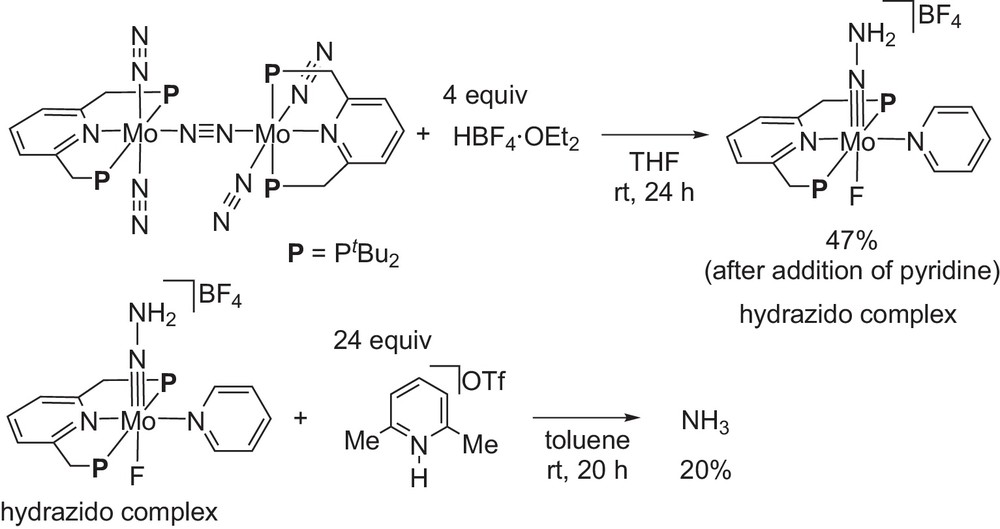
Stoichiometric reactions of dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer ligands.
The molybdenum(V)–nitride complex bearing a PNP-type pincer ligand was prepared and the molybdenum(V)–nitride complex reacted with 1 equiv of KC8 as a reducing reagent in THF at room temperature for 20 h to give the corresponding molybdenum(IV)–nitride complex bearing a PNP-type pincer ligand in 30% yield (Scheme 15) [16]. The reaction of the neutral molybdenum(IV)–nitride complex with 1 equiv of AgOTf in THF at room temperature for 14 h gave the corresponding cationic molybdenum(V)–nitride complex in 52% yield. The neutral molybdenum(IV)–nitride and the cationic molybdenum(V)–nitride complexes can be characterized by NMR and IR. Detailed molecular structures of these nitride complexes were confirmed by X-ray analysis, where both nitride complexes have a square pyramidal structure. Further reaction of the molybdenum(IV)–nitride complex with 4 equiv of CoCp2 and 4 equiv of [LutH]OTf in toluene at room temperature for 20 h gave ammonia in 82% yield based on the starting nitride complex. These results indicate that the nitride ligand coordinated to the molybdenum atom can be converted into ammonia under catalytic reaction conditions.
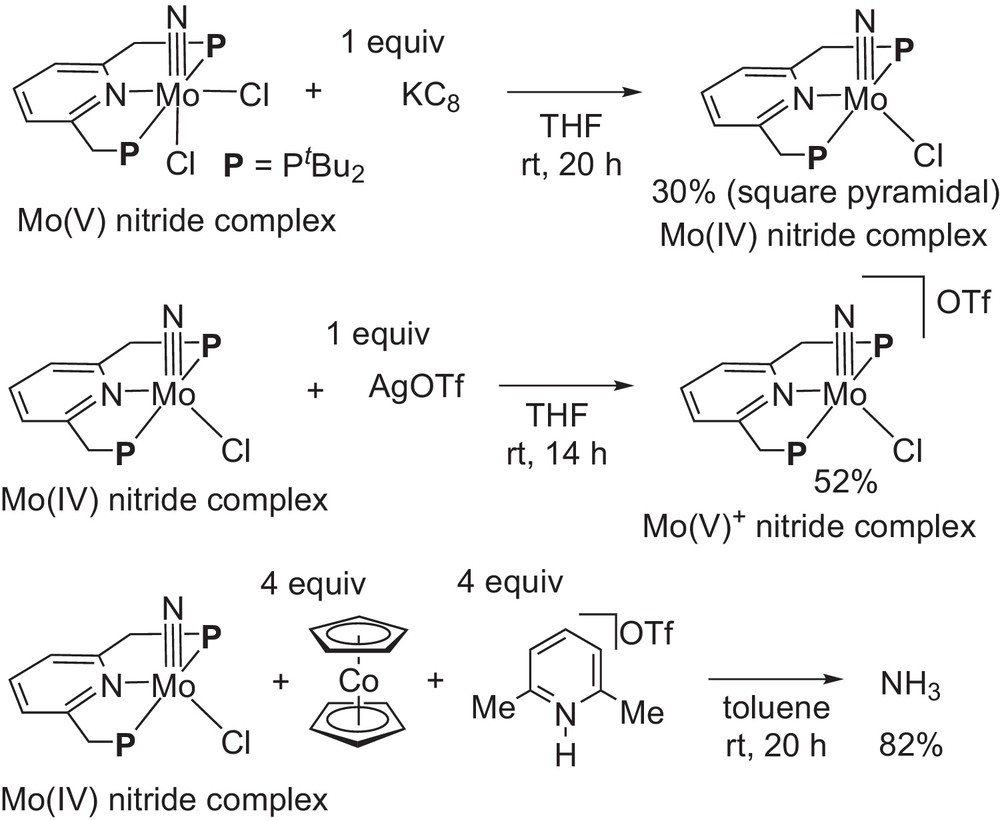
Stoichiometric reactions of molybdenum–nitride complex bearing PNP-type pincer ligand.
The catalytic activity of these molybdenum–nitride complexes was investigated under the same reaction conditions (Scheme 16) [16]. The molybdenum(V)–nitride complex did not work as an effective catalyst. In sharp contrast, the neutral molybdenum(IV)–nitride complex and the cationic molybdenum(V)–nitride complex worked as effective catalysts. Both molybdenum–nitride complexes have a similar catalytic activity toward the formation of ammonia under ambient reaction conditions. These results shown in Schemes 15 and 16 indicate that some mononuclear molybdenum(IV)–nitride complexes are considered to be key reactive intermediates of the catalytic reaction.
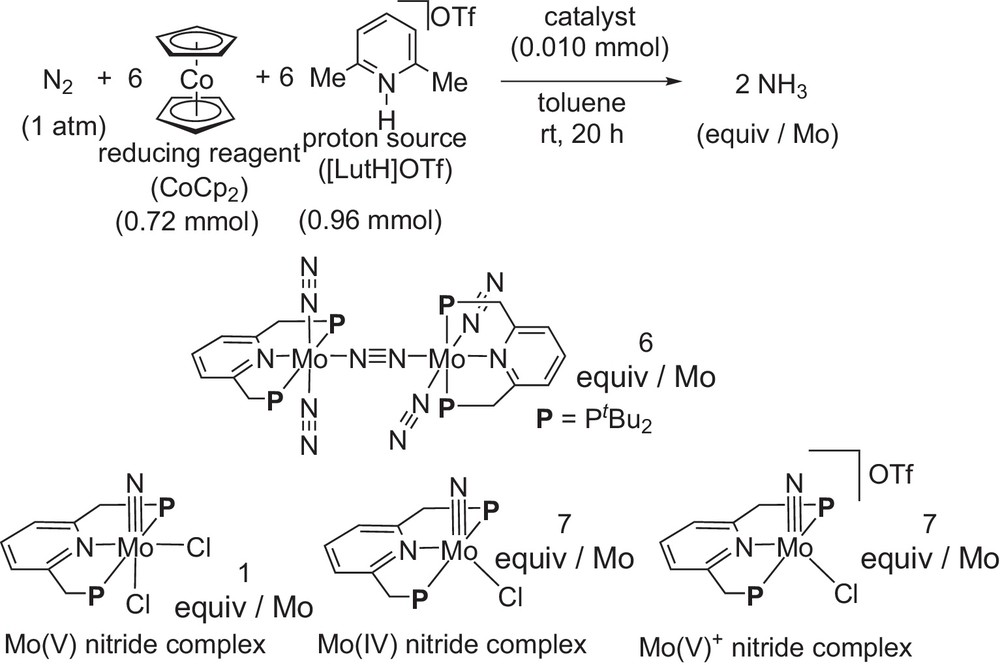
CAtalytic formation of ammonia by using molybdenum–nitride complexes as catalysts.
Based on the results of catalytic and stoichiometric reactions, we have proposed the reaction pathway shown in Scheme 17 [13]. At first, a mononuclear molybdenum–dinitrogen complex is formed from the dinitrogen-bridged dimolybdenum–dinitrogen complex. Then, protonation and reduction of the mononuclear molybdenum–dinitrogen complex give a mononuclear molybdenum-hydrazidium complex via a molybdenum–hydrazido complex as a reactive intermediate. The cleavage of a nitrogen–nitrogen single bond of the hydrazidium ligand occurs to give a mononuclear molybdenum–nitride complex together with the formation of ammonia. Further protonation and reduction of the molybdenum–nitride complex give a molybdenum-ammonia complex, which liberates the formation of ammonia by ligand exchange with another molecular dinitrogen together with the formation of the starting mononuclear molybdenum–dinitrogen complex. Separately, molecular dihydrogen is formed by the reductive elimination from the molybdenum–dihydride complex, which is generated by the protonation of the mononuclear molybdenum–dinitrogen complex after the elimination of the dinitrogen ligands [17].
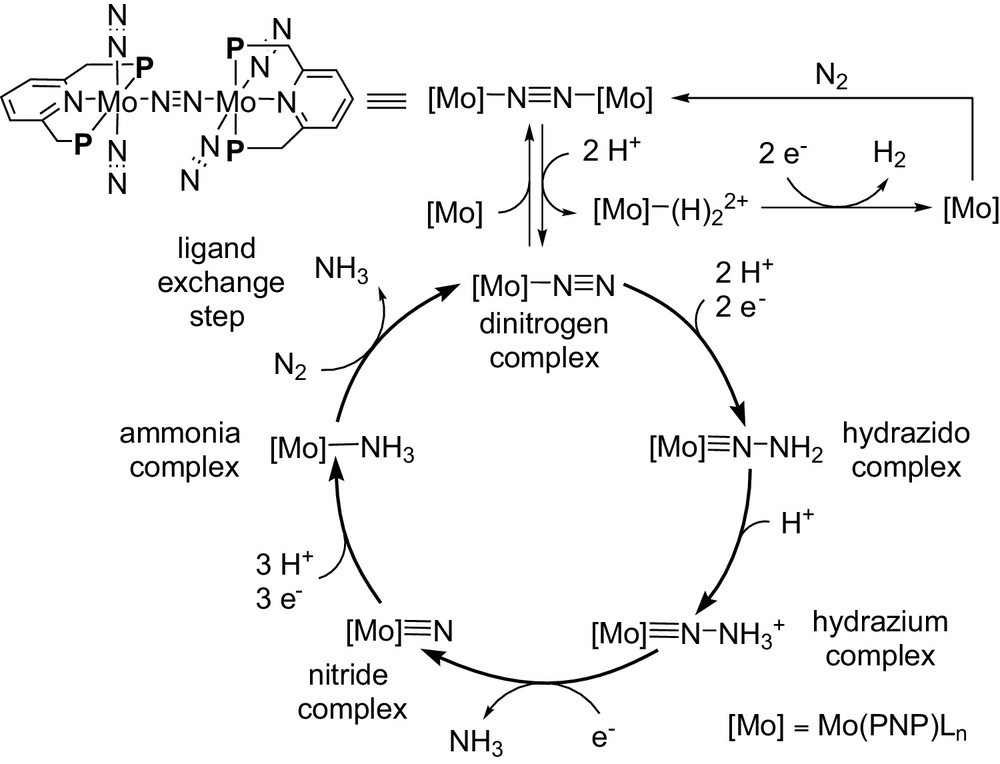
Proposed reaction pathway, where mononuclear molybdenum complexes may play an important role as key intern ediates.
To get more information on the proposed reaction pathway, we carried out the DFT calculation on the first protonation step of the dinitrogen-bridged dimolybdenum–dinitrogen complex (Scheme 18) [16]. At first, the formation of mononuclear molybdenum–dinitrogen complexes hardly occurred from the corresponding dinitrogen-bridged dimolybdenum–dinitrogen complex at room temperature. In addition, no protonation of these mononuclear molybdenum–dinitrogen complexes occurred at all under the same reaction conditions. These results indicate that mononuclear molybdenum–dinitrogen complexes did not work as real reactive intermediates. On the other hand, the protonation of dinitrogen-bridged dimolybdenum–dinitrogen complex easily occurred and the formation of a dimolybdenum–hydrazido complex was observed at room temperature. This result indicates that a dinitrogen-bridged dimolybdenum core may play an important role to promote the catalytic reaction.
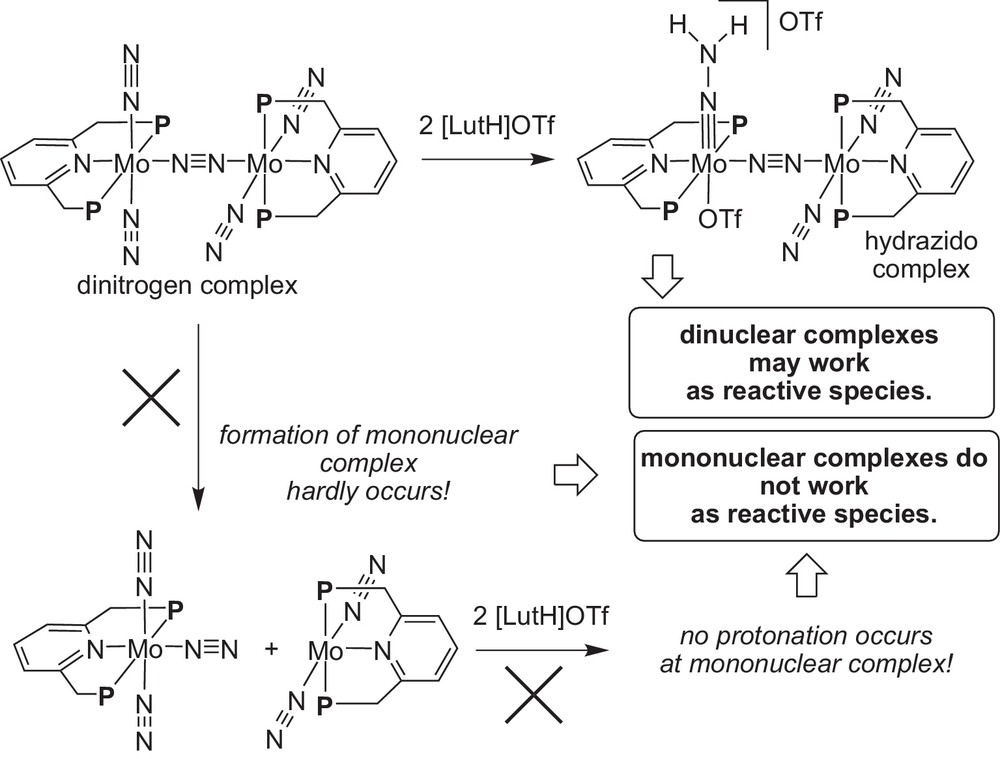
DFT calculation on the dinitrogen-bridged dimolybdenum–dinitrogen complex.
Based on the results of the DFT calculation, we have proposed a revised reaction pathway shown in Scheme 19 [16]. In this revised reaction pathway, the dinitrogen-bridged dimolybdenum core may play an important role to promote the catalytic reaction, where the dinuclear molybdenum core remains during the catalytic reaction. Direct protonation and reduction of the dinitrogen-bridged dimolybdenum–dinitrogen complex give a dinuclear molybdenum–hydrazido complex. Then, protonation and reduction of the molybdenum–hydrazido complex afford a dinuclear molybdenum–nitride complex together with formation of ammonia. Further protonation and reduction of the dinuclear molybdenum–nitride complex give a dinuclear molybdenum-ammonia complex, which liberates the formation of ammonia by ligand exchange with another molecular dinitrogen, together with the formation of the starting dinuclear molybdenum–dinitrogen complex. As shown in the proposed reaction pathway, only the left molybdenum moiety in the dinuclear molybdenum complexes works as a reactive site to convert the coordinated molecular dinitrogen into ammonia. On the other hand, the right molybdenum moiety in the dinuclear molybdenum complexes may work as an electron-pool or mobile ligand to the left molybdenum moiety in the dinuclear molybdenum complexes. Especially, in the first protonation step and the ligand exchange step of the coordinated ammonia with molecular dinitrogen, the dinuclear core of the two molybdenum moieties bridged by the coordinated molecular dinitrogen is necessary. We consider that a synergistic effect of the two molybdenum moieties is one of the most important factors to promote the catalytic formation of ammonia from molecular dinitrogen.
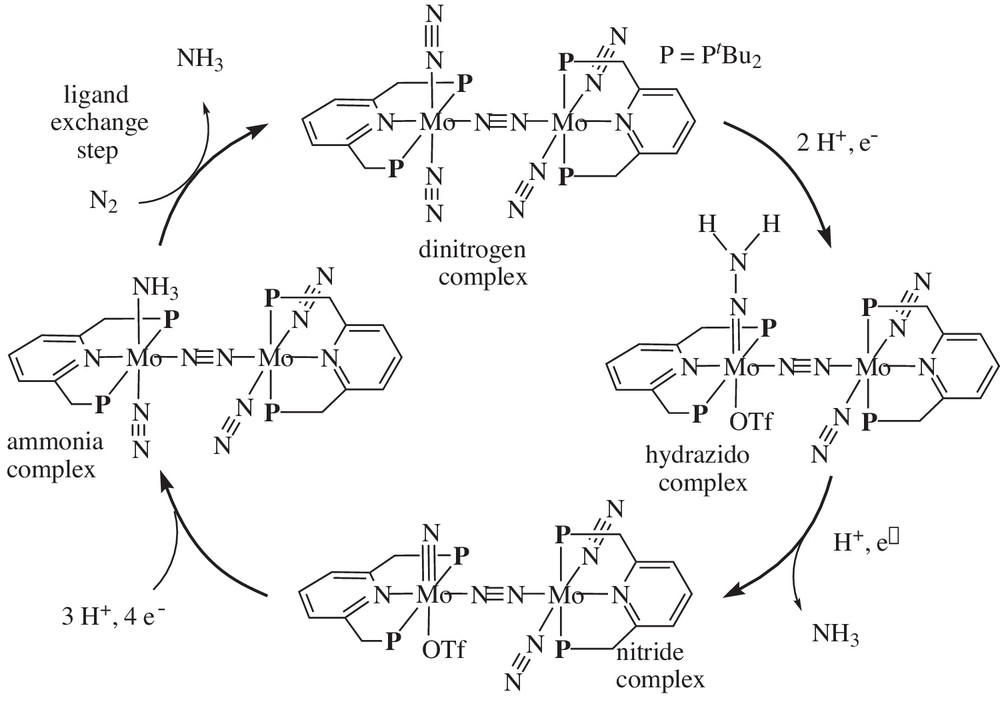
Revised proposed reaction pathway, where dinitrogen-bridged dimolybdenum complexes may play important role as key intermediates.
As shown in the previous section, we have proposed a reaction pathway for the catalytic formation of ammonia from molecular dinitrogen catalyzed by the dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-type pincer ligands. As a next step, we have focused on the development of more efficient catalysts in the catalytic formation of ammonia from molecular dinitrogen under ambient reaction conditions. According to the DFT calculation shown in the previous section, the first protonation step is considered to be one of the rate-determining steps of the catalytic reaction. We carried out the DFT calculation on the reactivity of dinitrogen-bridged dimolybdenum complexes bearing substituted PNP-type pincer ligands toward the first protonation step. As shown in Scheme 20, the introduction of electron-donating groups such as methyl and methoxy groups to the pyridine ring of the PNP-type pincer ligand dramatically decreased the activation energy of the first protonation step [14]. This result prompts us to prepare the corresponding substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes and investigate their catalytic activity toward the formation of ammonia from molecular dinitrogen in details.

DFT calculation on the first protonation step by using substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes.
According to a similar experimental procedure of the original dinitrogen-bridged dimolybdenum–dinitrogen complex, a variety of substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes can be prepared in good to high yields from the corresponding molybdenum(III) chloride complexes bearing substituted PNP-type pincer ligands (Scheme 21) [14]. The molecular structures of these substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes are confirmed by X-ray analysis and are almost the same as that of the original dinitrogen-bridged dimolybdenum–dinitrogen complex. An ORTEP drawing of methyl-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex is shown in Scheme 21.

(Color online.) Preparation of substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes.
We investigated catalytic reactions by using substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes as catalysts [14]. As shown in Scheme 22, the use of phenyl- and trimethylsilyl-substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes as catalysts gave almost the same amount of ammonia based on the catalyst as that of the original dinitrogen-bridged dimolybdenum–dinitrogen complex. In contrast, the use of alkyl-substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes as catalysts gave substantially a larger amount of ammonia based on the catalysts. As a result, the methoxy-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex worked as the most effective catalyst under the same reaction conditions.

Catalytic formation of ammonia by using substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes as catalysts.
Next, we carried out the catalytic reaction by using a methoxy-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex as a catalyst [14]. The use of larger amounts of CoCp2 as a reducing reagent and [LutH]OTf as a proton source gave the largest amount of ammonia based on the catalyst (Scheme 23). The reaction with 360 equiv of CoCp2 and 480 equiv of [LutH]OTf gave 52 equiv of ammonia based on the catalyst (26 equiv of ammonia based on the Mo atom in the catalyst). This is so far the most effective catalytic system in the catalytic formation of ammonia from molecular dinitrogen by using transition metal–dinitrogen complexes as catalysts.
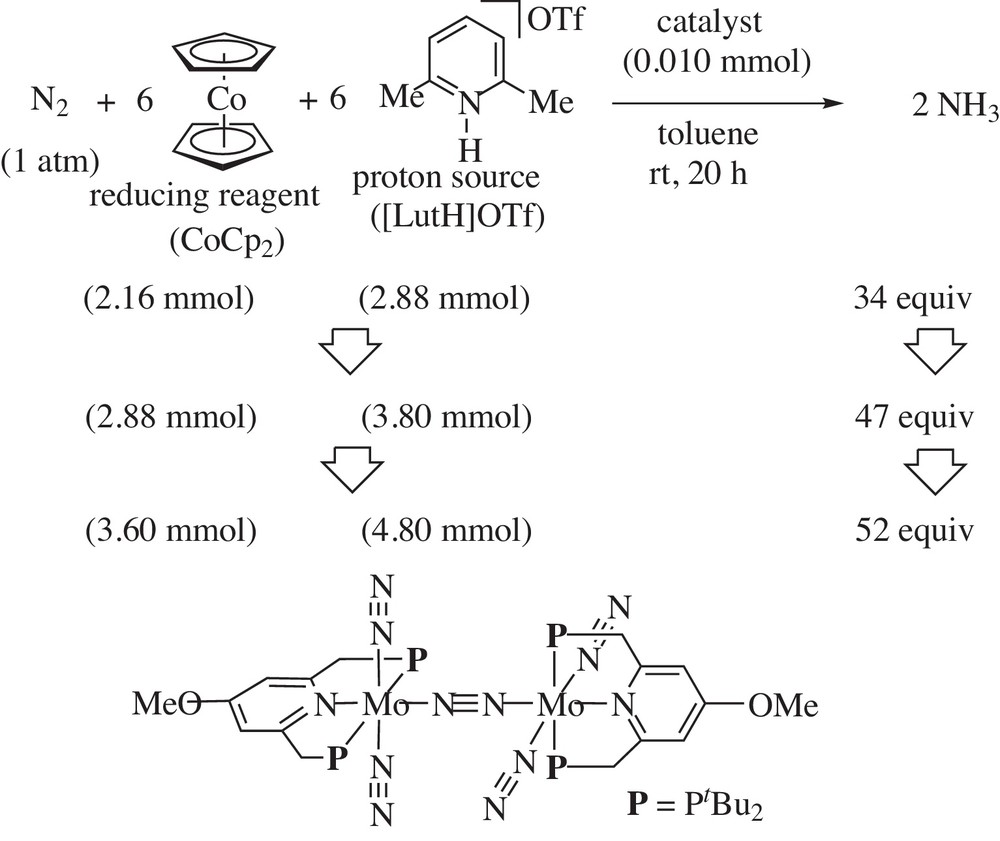
Catalytic formation of ammonia by using methoxy-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex as a catalyst.
To get information on the nature of the substituent to the PNP-type pincer ligand, we monitored the formation of ammonia by using three types of dinitrogen-bridged dimolybdenum–dinitrogen complexes as catalysts [14]. The time profiles are shown in Scheme 24. The black line indicates a time profile for the formation of ammonia by using the original dinitrogen-bridged dimolybdenum–dinitrogen complex. The green line indicates a time profile for the formation of ammonia by using the methyl-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex. In this case, the formation of ammonia proceeded more rapidly than that by using the original dinitrogen-bridged dimolybdenum–dinitrogen complex, where the formation of ammonia was almost completed within 1–2 h. On the other hand, the red line indicates a time profile for the formation of ammonia by using the methoxy-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex. In this case, the catalytic reaction did not proceed more rapidly, although the largest amount of ammonia was produced based on the catalyst.
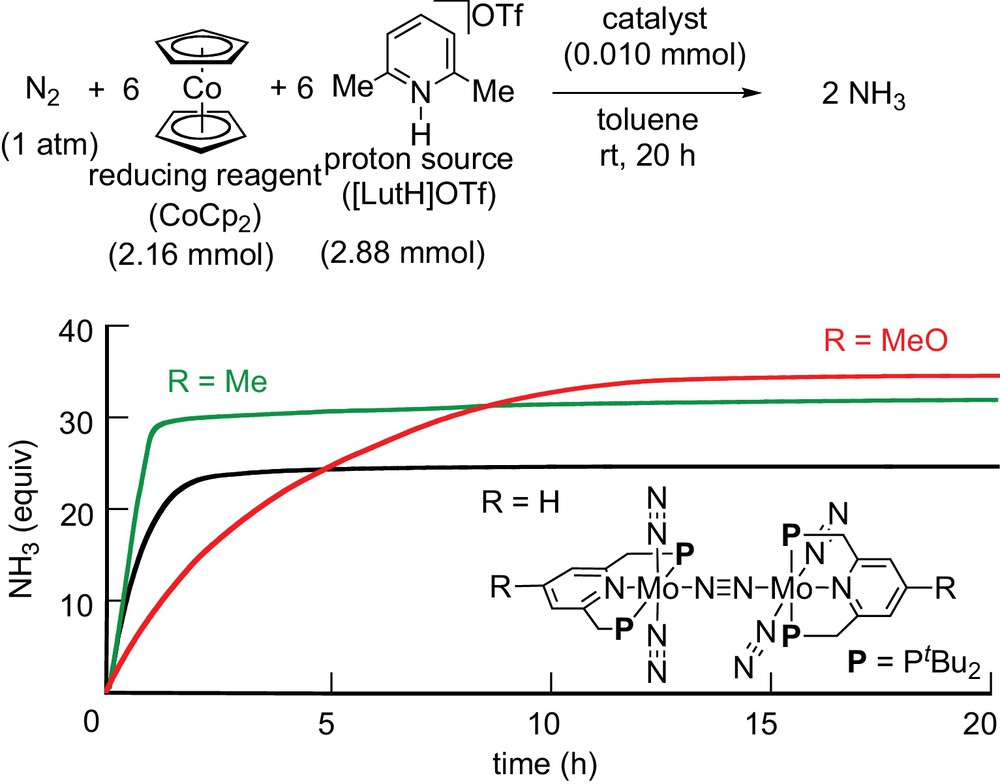
(Color online.) Time profiles of catalytic formation of ammonia by using substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes as catalysts.
A relationship between the electronic and electrochemical properties and the catalytic activity of the substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes is summarized in Scheme 25. The introduction of the more electron-donating group lowered the IR wavelength of NN stretching and the oxidation potential observed as the first reversible wave in cyclic voltammetry. Our IR spectroscopy measurements indicate that molybdenum–dinitrogen complexes bearing a more electron-donating group to the pyridine ring of the PNP-type pincer ligands have a higher reactivity toward the protonation. On the other hand, the cyclic voltammetric study indicates that the molybdenum–dinitrogen complexes bearing a more electron-donating group to the pyridine ring of the PNP-type pincer ligands have a lower reactivity toward reduction. Thus, molybdenum–dinitrogen complexes bearing a more electron-donating group have more activated coordinated dinitrogen ligands, which are easily protonated, but are rather reluctant to be reduced under the same reaction conditions. To develop more effective catalysts, the control of both opposite reactivities toward protonation and reduction is one of the most important factors. We believe that the result achieved here will provide more useful information to design more effective catalysts.
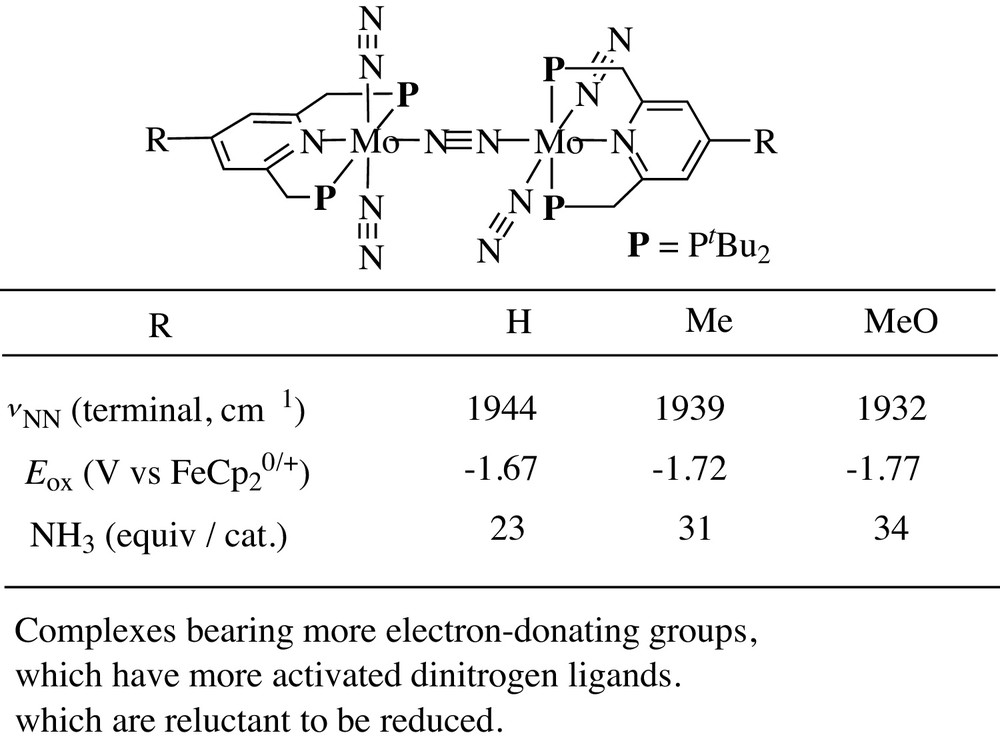
Relationship between electronic and electrochemical properties and catalytic activity of substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes.
Quite recently, Peters and co-workers have reported the first successful example of the iron-catalyzed conversion of molecular dinitrogen into ammonia by using an iron–dinitrogen complex (Scheme 26) [18]. The reaction should be carried out by using a highly acid proton source and a highly reducing reagent at a very low reaction temperature, such as −78 °C. In this reaction system, 7 equiv of ammonia were produced based on the catalyst, while its reaction mechanism has not been clarified in detail. Interestingly, the nature of coordinating atoms in the ligand dramatically affects the catalytic reactivity toward the formation of ammonia from molecular dinitrogen under the same reaction conditions [19]. In fact, the use of the iron–dinitrogen complex bearing a carbon atom in the ligand achieved the catalytic formation of ammonia; however, the use of the iron–dinitrogen complex bearing a silicon atom in the ligand gave only a stoichiometric amount of ammonia based on the catalyst.
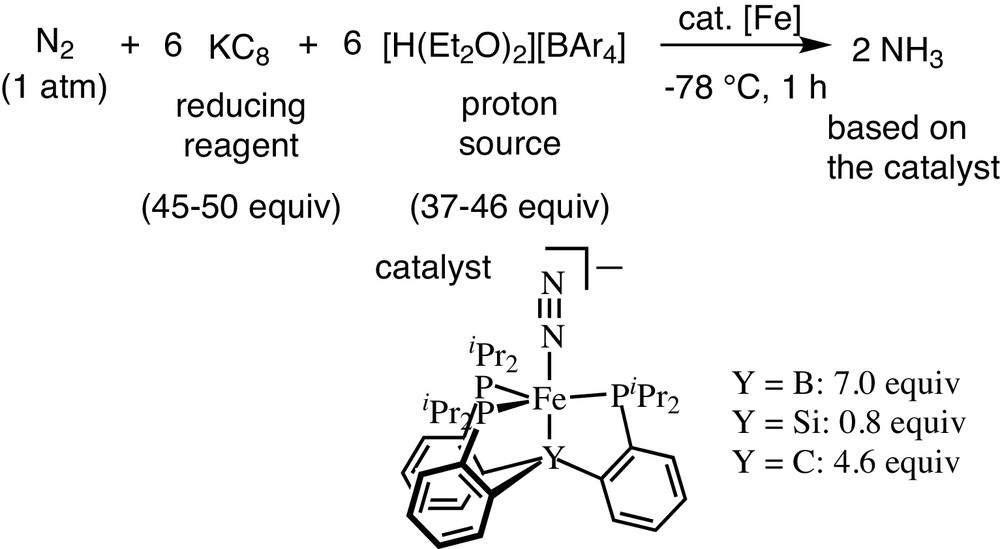
Iron-catalyzed reduction of molecular dinitrogen into ammonia under mild reaction conditions.
In summary, we have evidenced a molybdenum-catalyzed reduction of molecular dinitrogen into ammonia under ambient reaction conditions. In this reaction system, the dinitrogen-bridged dimolybdenum–dinitrogen complexes bearing PNP-type pincer ligands worked as effective catalysts, where up to 52 equiv of ammonia were produced based on the catalyst (26 equiv of ammonia were produced based on the Mo atom in the catalyst). These complexes have the highest catalytic activity toward the formation of ammonia under mild reaction conditions by using transition metal–dinitrogen complexes as catalysts. The DFT calculation has clarified the detailed reaction pathway, where dinuclear molybdenum complexes bridged by the coordinated molecular dinitrogen ligand may play an important role to promote the catalytic reaction effectively. Independently, Batista and co-workers have proposed a similar reaction pathway by the DFT calculation [20]. However, unfortunately, we have not yet obtained the exact reason why the PNP-type pincer ligand works effectively. We will report more detailed results in the near future [21].
Acknowledgement
I thank some members of my laboratory as co-workers in the present projects, especially Dr. Kazuya Arashiba, Shogo Kuriyama, Ryuji Imayoshi, Eriko Kinoshita, Aya Eizawa, and assistant professors Dr. Yoshihiro Miyake (now at Nagoya University) and Dr. Kazunari Nakajima. We also thank other members of my laboratory involved in the related projects of nitrogen fixation, Dr. Masahiro Yuki, Dr. Yoshiaki Tanabe, and Dr. Takamasa Miyazaki. Finally, I specially thank Prof. Dr. Kazunari Yoshizawa and Dr. Hiromasa Tanaka at Kyushu University for the theoretical study by DFT calculation. I thank Grants-in-Aid for Scientific Research (Nos. 26288044, 26620075, and 26105708) from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).