

1 Introduction
By virtue of the variability of their chemically different donor groups, hybrid ligands provide excellent opportunities to study the chemoselective coordination of multifunctional ligands to metals centres and to fine-tune the stereo-electronic properties of their metal complexes. This can lead to improved reactivity and catalytic properties of their metal complexes in solution [1]. For these reasons, we and others became recently interested in the synthesis and coordination chemistry of a specific family of such ligands, which possess phosphorus and N-heterocyclic carbene (NHC) donor moieties, two ubiquitous functionalities in coordination/organometallic chemistry, possibly associated with a carbon-donor function in a pincer-type system [2–4]. A transition metal of particular significance is iridium because of the unique catalytic properties of its complexes in alkane C-H activation [5]. Transfer dehydrogenation of cyclooctane (coa) has been achieved using PCP [6] and POCOP [7] iridium pincer complexes, and these systems have since served as benchmarks for this reaction (Scheme 1).
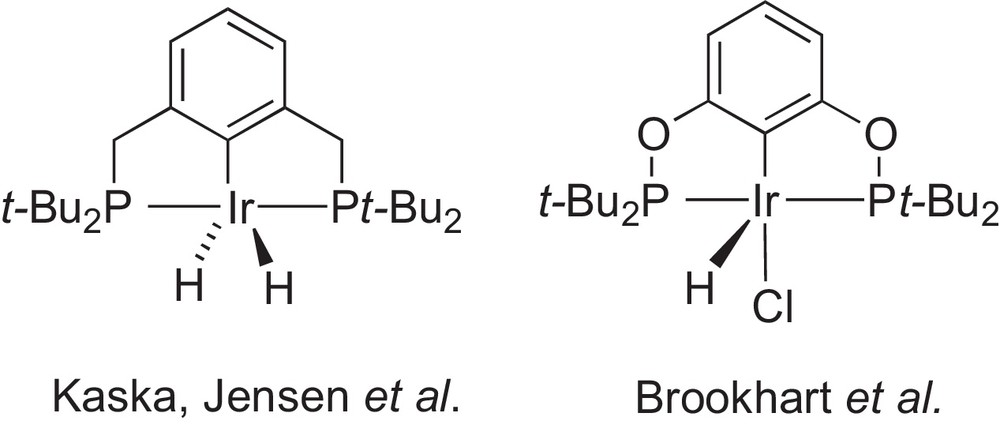
PCP [6] and POCOP [7] iridium pincer complexes used in catalytic transfer dehydrogenation of cyclooctane.
We recently reported the synthesis of Ir(I) pincer-type complexes involving phosphorus and NHC donors, [IrH(I)(PO-NHC,κP,κC,κCNHC)Me] (1a) and [IrH(I)(PO-NHC,κP,κC,κCNHC)n-Bu] (1b), with a Me or n-Bu group as N-substituent, respectively [4].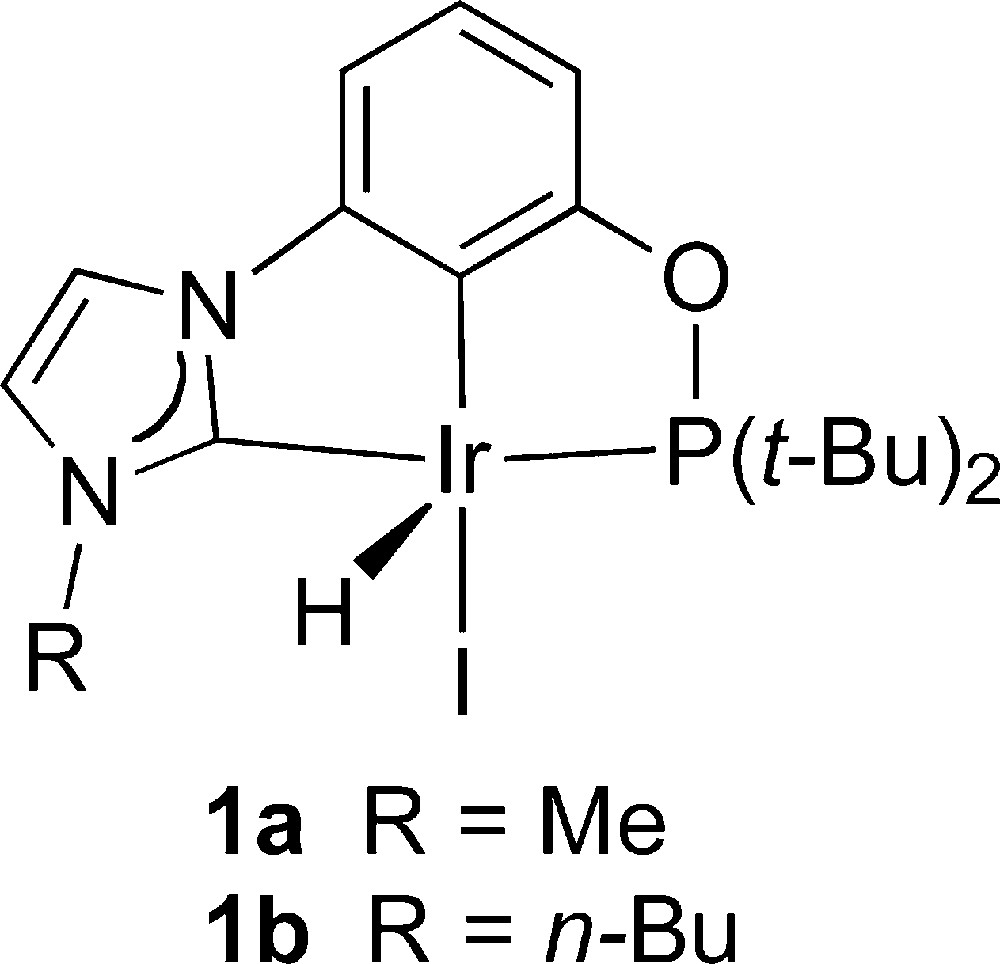
We found that slight differences in the nature of the spacers between the donor atoms may be sufficient to bring about major modifications in the structures of the corresponding metal complexes. Thus, with related NHC,P hybrid ligands, dinuclear Ir(I) complexes were obtained, also from the corresponding imidazolium salt, in which two such ligands behave as bridges rather than chelates, as in, e.g., the dicationic complex [Ir(cod)(μ-P-NHC,κP,κCNHC)]2[PF6]2 (2) (cod = 1,5-cyclooctadiene) [4].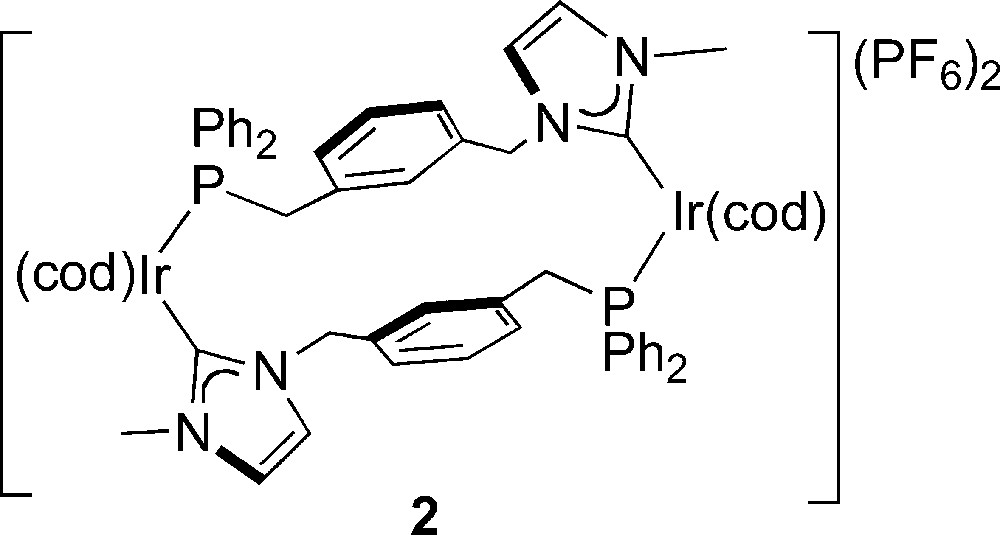
2 Results and discussion
It is highly desirable to perform the activation of alkanes by soluble transition-metal species in the absence of any competing solvent and this requires sufficient solubility of the precatalyst in the neat alkane. With the objective to prepare a neutral iridium complex that would have a better solubility in coa, and thus facilitate the activation of its C-H bonds, we envisaged replacing the neutral cod ligand of complex 2 with an anionic P,O-chelating phosphino-enolate ligand since we have previously observed that their Rh(I)/Ir(I) complexes catalysed the transfer dehydrogenation of coa [8]. The Ir(III) dihydride complex [Ir(cod)(H)2{Ph2PCH···C(···O)Ph,κP,κO}] was moderately stable in toluene solution [8]. In general, phosphino-enolate functionalities can be readily generated under basic conditions from a β-ketophosphine ligand, such as Ph2PCH2C(O)Ph, coordinated or not [9]. We thus added a THF solution of Ph2PCH2C(O)Ph to the dicationic complex 2 in the presence of a base (NaH) but we did not succeed in displacing selectively the cod ligand from the iridium centre. For comparison, we performed the same experiment with the related, dicationic complex 3 which contains two bis-NHC bridging ligands and displays a “figure 8” conformation [10].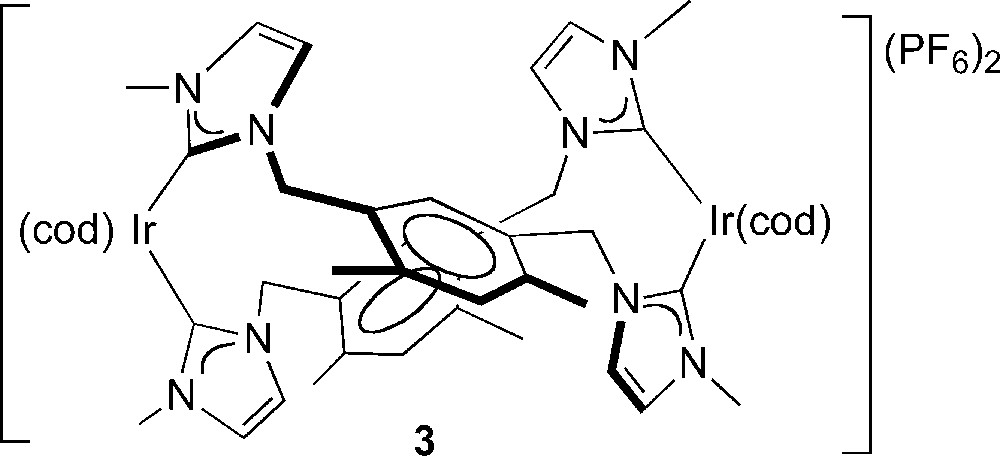
Unfortunately, this experiment was also unsuccessful. This led us to examine, for comparison, the reactivity of [Ir(cod)(μ-Cl)]2 with this β-ketophosphine with the objective to deprotonate the latter and form a chelating phosphino-enolate. First, TlPF6 was added to a THF solution of [Ir(cod)(μ-Cl)]2, in order to abstract the chloride from the metal centre and render the latter more electrophilic, and then two equivalents of Ph2PCH2C(O)Ph and NaH in slight excess were added (see Experimental Section). Analysis by 31P{1H} NMR spectroscopy indicated the presence of different species, which turned out difficult to separate and purify. Clearly, no selective displacement of the cod ligand from [Ir(cod)(μ-Cl)]2 occurs in the presence of Ph2PCH2C(O)Ph or its corresponding phosphine-enolate. In one instance however, a few yellow/orange crystals of a product could be isolated and characterized by X-ray diffraction as [Ir(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}] (5) (see below).
Because of the difficulties encountered in trying to reproduce these results, we decided to operate in two consecutive steps and isolate the intermediate product(s): first by preparing a cationic β-ketophosphine Ir(I) complex by using a chloride abstractor and secondly by reacting this complex with a base, usually a facile reaction because of the increased acidity of the PCH2 protons upon phosphorus coordination to a metal [9]. Targeting a cationic, square-planar complex of the type [Ir{Ph2PCH2C(O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}(THF)]+ or [Ir{Ph2PCH2C(O)Ph,κP,κO}2]+, we reacted [Ir(cod)(μ-Cl)]2 with 2 equivalents of Ph2PCH2C(O)Ph in the presence of TlPF6 at room temperature (equation 1), although it is often observed that the displacement by phosphines of the cod ligand from [Ir(cod)(μ-Cl)]2 requires harsher experimental conditions or tridentate ligands (e.g., CNHCCCNHC pincers) [11].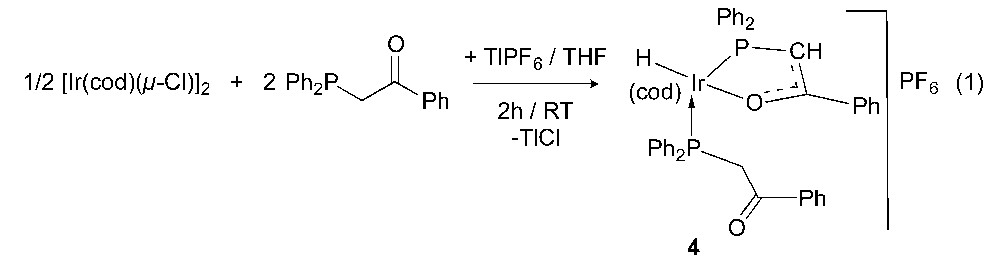
The 31P{1H} NMR spectrum of the isolated product (see Experimental section) confirmed the presence of the PF6 anion (δ −144.6 ppm) and contained two doublets, at δ 16.2 and –1.3 ppm, with 2J(P,P) = 16.6 Hz. This is consistent with the presence of two chemically different P atoms in a mutually cis-position. A 2D NMR experiment (HMQC 1H/31P) established that the more downfield-shifted resonance (16.2 ppm) corresponded to the P atom belonging to the chelating ligand. The 1H NMR spectrum contained resonances at δ 2.49 and 4.61 ppm corresponding to an ABX spin system (A = B = H, X = P) for the two diastereotopic protons of a PCH2 group (2J(H,H) = 16.6 Hz, 2J(P,H) = 10.9 and 6.8 Hz, respectively). A doublet at δ 5.91 ppm integrating for one proton is typical of the resonance for the enolate proton of a P,O-coordinated Ph2PCH···C(···O)Ph group, with a 2J(H,P) coupling of 4.6 Hz [8]. Accordingly, the 1H{31P} NMR spectrum showed only one singlet for this proton. Furthermore, the 1H NMR spectrum revealed the unexpected presence of a triplet resonance at δ −16.65 ppm (2J(P,H) = 9.0 Hz), corresponding to a hydride coupled to two P nuclei in a cis-position (the triplet pattern formally corresponds to overlapping doublets of doublets since the two P nuclei are chemically different). The IR absorptions at 1672 and 1510 cm−1 also suggested the presence of an uncoordinated ketone group of Ph2PCH2C(O)Ph and of an O-coordinated enolate function in a P,O-chelating Ph2PCH···C(···O)Ph system, respectively [12]. Furthermore, the 13C{1H} NMR data also confirmed the presence of two types of C–O carbons with resonances at 192.6 and 186.5 ppm for the uncoordinated ketone group and the O-coordinated enolate function, respectively. The former resonance is very close to that of the free ketophosphine ligand (196.9 ppm) [9].
Fortunately, plate-like colourless single crystals of this complex were obtained by slow diffusion of octane into a CH2Cl2 solution at room temperature. The X-ray diffraction analysis of 4·CH2Cl2 allowed to confirm the interpretation based on spectroscopic data and to establish its formula as [IrH(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}]PF6 (4) (Fig. 1).
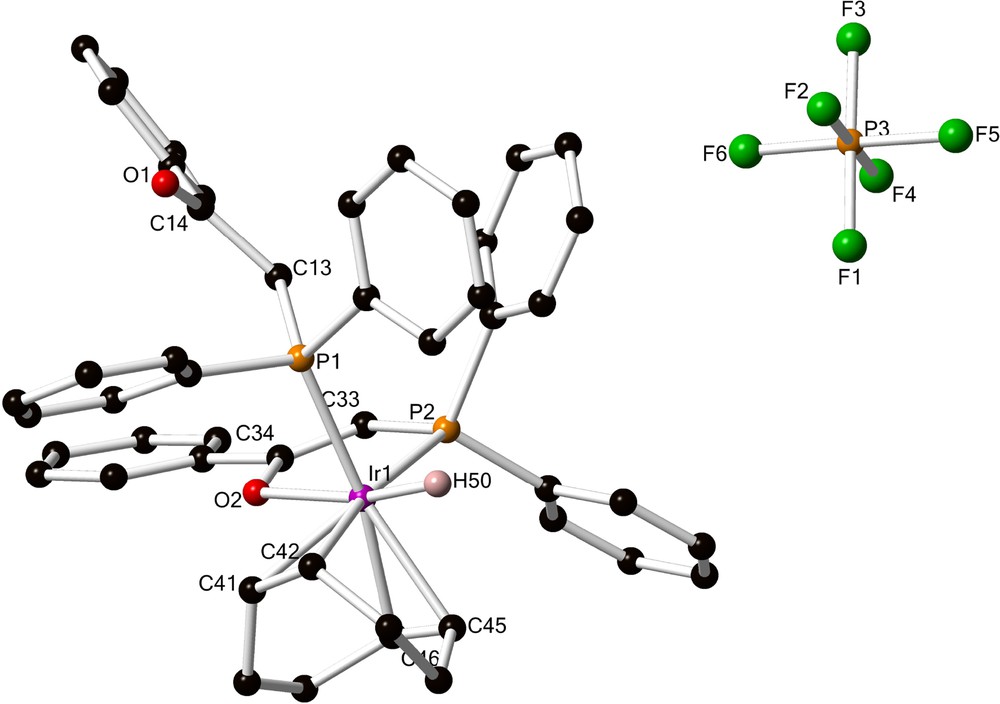
(Colour online.) View of the structure of the Ir(III) complex [IrH(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}]PF6 (4) in 4·CH2Cl2. Selected bond lengths [Å] and angles (deg): Ir–P1 2.362(2), Ir–P2 2.302(2), Ir–O2 2.152(5), Ir–H50 1.49(7), P1–C13 1.843(7), C13–C14 1.521(10), C14–C15 1.486(10), O1–C14 1.219(9), P2–C33 1.757(8), C33–C34 1.345(10), O2–C34 1.332(8); P1–Ir–P2 93.82(6), O2–Ir–P1 85.55(14).
The distorted octahedral coordination environment around the metal centre contains the cod ligand, the P atom of a monodentate ketophosphine and a P,O chelate acting as a 3-electron donor. The hydride ligand was located on the difference Fourier map and is trans to the enolate oxygen and cis to the two, mutually cis P atoms. As expected, the C-O bond length of the ketone group (O1–C14 1.219(9) Å) Å is shorter than that in the enolate moiety (O2–C34 1.332(8) Å) whereas the reverse applies to the corresponding C–C distances, of 1.521(10) Å for C13–C14 and 1.345(10) Å for C33–C34. The P–CH2 bond length is slightly longer (P1–C13 1.843(7) Å) than that of the P–CH bond (P2–C33 1.757(8) Å). All these metrical data are consistent with values reported for related complexes containing chelating phosphino-enolates or P-bound ketophosphines, respectively [8,9].
Having established the nature of the Ir(III) hydride complex 4, we can now interpret the reaction of equation (1) as having involved splitting of the chloride bridges of the precursor and formal oxidative-addition of a C–H bond of one of the two ketophosphine ligands introduced, resulting in the formation of a metal-hydride and an enolate moiety. Whether this oxidative-addition resulted from direct interaction between the P–CH2 unit and the Ir(I) centre or from the transient P,O chelation of the minor, tautomeric enol form of the ligand, Ph2PCHC(OH)Ph [9,13], cannot be stated at this stage.
Since our original objective was to prepare a neutral iridium(I) complex, we then reacted isolated 4 with NaH in THF for 2 h at room temperature (equation 2). The 31P{1H} NMR spectrum of the yellow product 5 (see Experimental section) confirmed the absence of PF6− and contained two doublets, at δ −3.5 and 14.3 ppm with 2J(P,P) = 21.5 Hz.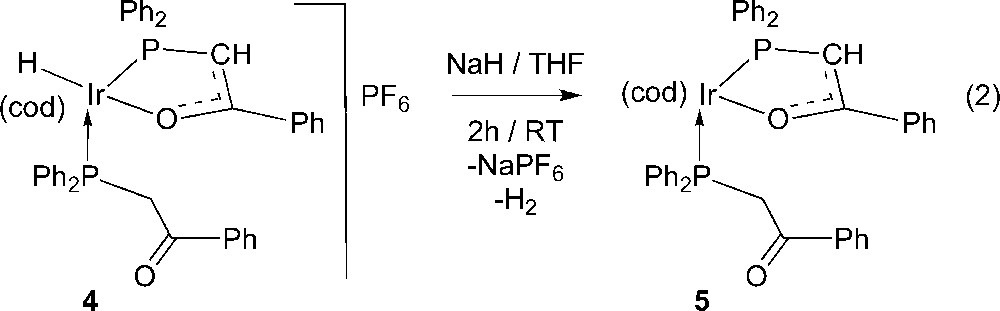
Considering the data for 4 and literature values, the doublet at 14.3 ppm was assigned to the phosphorus of a chelating Ph2PCH···C(···O)Ph ligand and that at -3.5 ppm to a P-bound Ph2PCH2C(O)Ph ligand. For comparison, the 31P{1H} NMR singlet resonance of the Ir(III) complex [Ir(cod)(H)2{Ph2PCH···C(···O)Ph,κP,κO}] was observed at 27.6 ppm [8]. The 1H NMR spectrum of 5, recorded at 203 K for a better resolution, indicated the disappearance of the hydride resonance and the presence of two PCH2 protons at 2.61 and 3.61 ppm (ABX spin system (A = B = H, X = P), partly overlapping with cod signals, with 2J(H,H) = 14.2 Hz and 2J(P,H) = 6.4 Hz) and of one PCH proton corresponding to the chelating Ph2PCH···C(···O)Ph ligand at 5.68 ppm. Consistently, the 13C{1H} NMR data included resonances at 31.2 ppm (s, PCH2) and 83.6 ppm (d, PCH, 1J(P,C) = 57.7 Hz). The IR data confirmed the presence of both a ketone group (1670 cm−1, ν(C = O)) and of an enolate moiety (1525 cm−1, [ν(C···O) + ν(C···C)]) [9a].
This complex was identical to the single crystals of 5·C7H8 obtained from the one-pot experiment (see above) by slow vapour diffusion of Et2O into a d8-toluene solution of the complex in a NMR tube. The X-ray diffraction study established the nature of the complex as [Ir(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}] (5) (Fig. 2).
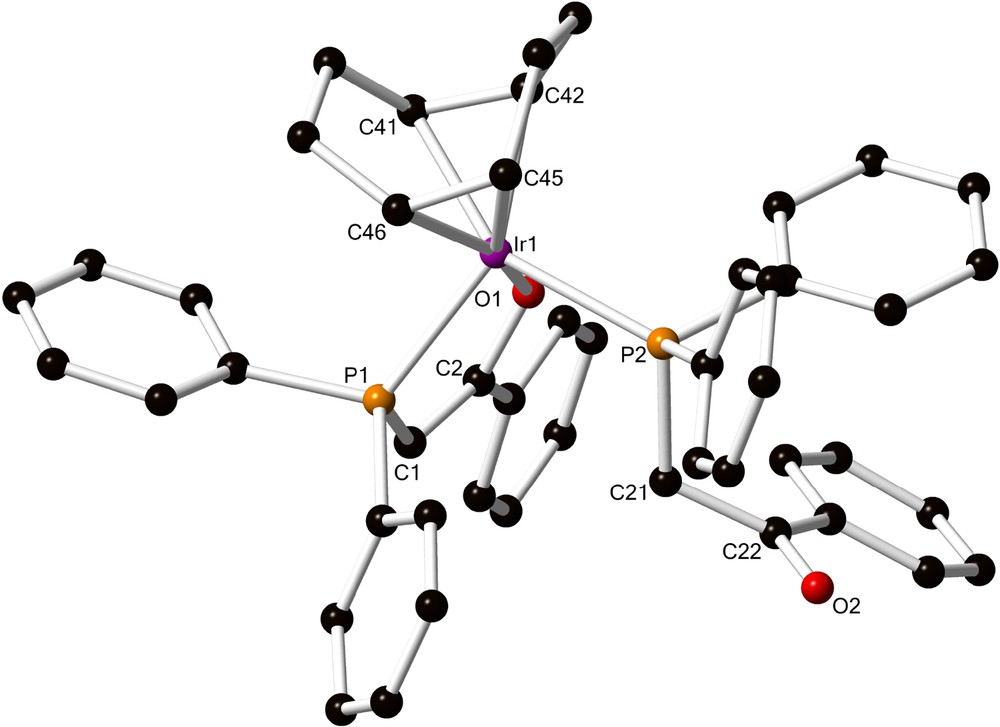
(Colour online.) View of the structure of the Ir(I) complex [Ir(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}] (5) in 5·C7H8. Hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles (deg): Ir1–P1 2.324(2), Ir1–P2 2.380(2), Ir1–O1 2.104(4), C1–C2 1.331(8), C21–C22 1.513(9), O1–C2 1.347(7), O2–C22 1.220(7); P1–Ir1–P2 100.21(6), P1–Ir1–O1 81.74(11), P2–Ir1–O1 84.85(12).
The penta-coordination environment around the metal centre includes the cod ligand, one P,O chelating ligand acting as a 3-electron donor (O1–C2 1.347(7) Å), and a P–bound ligand containing an uncoordinated ketone function (O2–C22 1.220(7) Å). The C21–C22 bond (1.513(9) Å) is longer than C1–C2 (1.331(8) Å), which is consistent with a neutral phosphine ligand and the presence of two hydrogen atoms on C21, in α position to P2. These values are similar to those in 4 for similar bonds and are typical for chelating phosphino-enolates or P-bound ketophosphines, respectively [8,9].
The reaction of 4 with NaH has thus led to the formation of H2 (not evidenced), of NaPF6 and of the Ir(I) complex 5. Although the latter had been isolated once as a result of an in situ experiment where [Ir(cod)(μ-Cl)]2 was reacted with TlPF6, the ketophosphine and NaH in THF, the sequential approach described above is much more satisfactory in terms of yields and of understanding the nature of the reaction intermediates.
3 Conclusion
It was found more difficult than anticipated to cleanly displace the Ir-coordinated cod ligand in complexes 2 and 3 by a chelating phosphine-enolate ligand. The reaction of [Ir(cod)(μ-Cl)]2 with 2 equivalents of the ketophosphine Ph2PCH2C(O)Ph in the presence of TlPF6 afforded the octahedral phosphino-enolate Ir(III) hydrido complex 4 as a result of the room temperature activation of a C-H bond from the PCH2 moiety (equation 1). The subsequent reaction of 4 with NaH in THF did not lead to the deprotonation of the P-coordinated ketophosphine but to the removal of the hydride ligand, which resulted in the formation of the pentacoordinated Ir(I) complex 5 (equation 2). The structure of both 4 and 5 was established by X-ray diffraction. It is interesting to compare the facile C-H bond activation reaction observed here with observations made previously with Ru complexes (Scheme 2). Only modest yields of the dinuclear, phosphine-enolate complex 6 were obtained when a CH2Cl2 solution of [Ru3(CO)10{Ph2PCH2C(O)Ph}2] was left standing for 1 week [14]. It required refluxing THF to convert [Ru3(CO)11{Ph2PCH2C(O)Ph}] in the phosphine-enolate Ru3 cluster 7 [15].
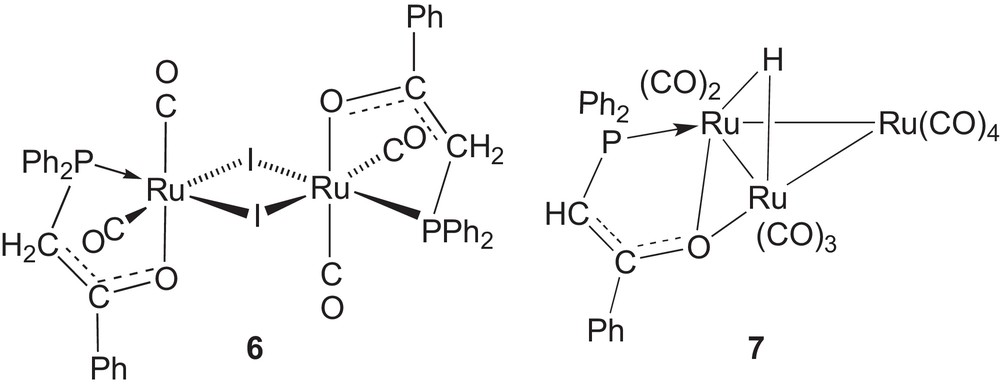
Previous examples of C-H activation of Ph2PCH2C(O)Ph by Ru complexes [14,15] were less facile than reported in this work with Ir.
It is also interesting to note that an oxidation of the metal centre somewhat related to that observed here during the formation of 4 was observed during attempts to deprotonate a Co(II) complex of the same ketophosphine ligand, [CoCl2{Ph2PCH2C(O)Ph,κP,κO}2]. Instead, the fac- and mer-isomers of the octahedral Co(III) complex [Co{Ph2PCH···C(···O)Ph,κP,κO}3] were isolated [16]. The possible occurrence of such transformations should be remembered when studying the reactivity or catalytic properties of metal complexes bearing Ph2PCH2C(O)Ph or related ligands since the corresponding phosphino-enolate ligand is known to play a key role in SHOP-type Ni(II) catalysts for ethylene oligomerization [17].
4 Experimental
General considerations: all reactions were performed under a dry argon atmosphere using standard Schlenk techniques. All solvents were distilled under argon from the appropriate drying agents and stored under argon. 1H, 13C{1H}, and 31P{1H} NMR spectra were recorded on a Bruker AVANCE 300, 400 or 600 spectrometers and internally referenced using the residual proton solvent (1H), solvent resonance (13C) or externally (31P) using H3PO4, with downfield shifts reported as positive. All NMR spectra were measured at 298 K, unless otherwise specified. Elemental analyses were performed by the “Service de microanalyses”, Université de Strasbourg. Electrospray mass spectra (ESI-MS) were recorded on a microTOF (Bruker Daltonics, Bremen, Germany) instrument using acetonitrile as solvent, nitrogen as drying agent and nebulising gas.
4.1 Synthesis of [IrH(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}]PF6 (4)
To a solution of [Ir(cod)(μ-Cl)]2 (0.5 equiv, 0.202 g, 0.301 mmol) in THF (20 mL) was added 1 equiv of TlPF6 (0.210 g, 0.601 mmol) at room temperature. The solution was stirred for 30 min, then 2 equiv of Ph2PCH2C(O)Ph (0.365 g, 1.20 mmol) was added at room temperature and the reaction mixture was stirred for 1 h, its colour changed from red to orange and a white precipitate formed. After stirring was maintained for 1 h, volatiles were evaporated and the residue was dissolved in CH2Cl2. The orange solution was filtered with a cannula equipped with a filter cap and then concentrated. Addition of hexane to the CH2Cl2 solution led to precipitation of a white product (in case this precipitate is slightly coloured, washing with 1 mL cold THF will afford a white solid). After filtration, the solid was washed with hexane and dried under vacuum. Yield: 0.232 g, 0.220 mmol, 73%. 1H NMR (400 MHz, CD2Cl2): δ −16.65 (t, 1H, Ir–H, 2J(P,H) = 9.0 Hz), 1.77–1.91 (m, 2H, CH2 COD), 2.31–2.38 (m, 1H, CHCOD), 2.49 (dd, 1H, PCHH, 2J(P,H) = 10.9 Hz, 2J(H,H) = 16.6 Hz), 2.54–2.64 (m, 2H, CH2 COD), 2.66–2.76 (m, 1H, CHCOD), 2.99–3.09 (m, 1H, CHCOD), 3.33–3.39 (m, 1H, CHCOD), 4.26–4.31 (m, 1H, CHCOD), 4.45–4.54 (m, 2H, CH2 COD), 4.61 (dd, 1H, PCHH, 2J(P,H) = 6.8 Hz, 2J(H,H) = 16.6 Hz), 5.15 (b, 1H, CHCOD), 5.91(d, 1H, CHenol, 2J(P,H) = 4.6 Hz), 6.97–8.39 (m, 30H, CHAr). 13C{1H} NMR (100 MHz, CD2Cl2): δ 24.6 (s, CH2 COD), 27.7 (s, CH2 COD), 28.8 (d, PCH2, 1J(P,C) = 30 Hz), 30.7 (s, CH2 COD), 37.5 (s, CH2 COD), 72.8 (d, PCHenol, 1J(P,C) = 73 Hz), 93.3 (d, CH COD, 2J(P,C) = 12 Hz), 96.4 (d, CH COD, 2J(P,C) = 9 Hz), 97.0 (d, CH COD, 2J(P,C) = 9 Hz), 101.6 (d, CH COD, 2J(P,C) = 12 Hz), 125.4–136.6 (CHAr), 186.5 (d, Cenol(O)Ph, 2J(P,C) = 15 Hz), 192.6 (d, C(O)Ph, 2J(P,C) = 10 Hz). 31P{1H} NMR (162 MHz, CD2Cl2): δ −144.6 (sept., 1J(PF) = 711 Hz, PF6), 16.2 (d, PCH, 2J(P,P) = 16.6 Hz), −1.3 (d, PCH2, 2J(P,P) = 16.6 Hz). Anal. calcd for C48H46IrF6O2P3: C, 54.70; H, 4.40. Found: C, 54.6; H, 4.51. MS (ESI): m/z 909.27 [M–PF6]+. IR (selected): 1672 (ν(C = O), 1510 cm−1 (ν(C···O) + ν(C···C)).
4.2 Synthesis of [Ir(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}] (5)
To a suspension of 4 (1 equiv, 0.100 g, 0.095 mmol) in THF (10 mL) was added excess NaH (5 equiv, 0.011 g, 0.46 mmol) at room temperature. The solution was stirred for 2 h and the solvent was removed under reduced pressure. The yellow residue was dissolved in toluene (5 mL) and the solution was filtered via cannula to remove NaPF6. The orange solution was evaporated under reduced pressure and the yellow residue was washed with a little cold acetone to obtain the product as a yellow powder. Yield: 0.061 g, 0.067 mmol, 70%. 1H NMR (600 MHz, CD2Cl2, 203 K): δ 0.88 (b, 3H, CH2 COD), 1.65 (b, 2H, CH2 COD), 1.77 (m, 2H, CH2 COD), 2.25 (b, 1H, CH2 COD), 2.43 (b, 1H, CHCOD), 2.61 (dd, 1H, PCHH, 2J(P,H) = 6.4 Hz, 2J(H,H) = 14.2 Hz), 3.02 (b, 1H, CH COD), 3.23 (b, 1H, CHCOD), 3.61 (dd, 1H, PCHH, 2J(P,H) = 6.4 Hz, 2J(H,H) = 14.2 Hz), 3.70 (b, 1H, CHCOD), 5.68 (s, 1H, PCHenol), 5.75–7.99 (m, 30H, CHAr). 13C{1H} NMR (150 MHz, CD2Cl2, 203 K): δ 26.4 (s, CH2 COD), 29.3 (s, CH2 COD), 31.2 (s, PCH2), 33.99 (s, CH2 COD), 36.3 (d, CH2 COD, 1J(P,C) = 6.3 Hz), 57.4 (d, CHCOD, 1J(P,C) = 32.7 Hz), 59.4 (s, CHCOD), 61.2 (s, CHCOD), 61.4 (d, CHCOD, 1J(P,C) = 6.6 Hz), 83.6 (d, CHenol, 1J(P,C) = 57.7 Hz), 126.9–142.3 (m, CAr), 181.7 (d, Cenol(O)Ph, 1J(P,C) = 26.4 Hz), 196.3 (d, C(O)Ph, 1J(P,C) = 11 Hz). 31P{1H} NMR (162 MHz, CD2Cl2): δ 14.3 (d, PCH, 2J(P,P) = 21.5 Hz), –3.5 (d, PCH2, 2J(P,P) = 21.5 Hz). Anal. calcd for C48H45IrO2P2: C, 63.49; H, 5.00. Found: C, 63.05; H, 4.83. MS (ESI): m/z 605.17 [M–L]+, 799.16 [M–COD]+, 909.27 [M–PF6]+. IR (selected): 1670 (ν(C = O)), 1525 cm−1 ((ν(C···O) + ν(C···C)).
4.3 Reaction of [Ir(cod)(μ-Cl)]2 with Ph2PCH2C(O)Ph and NaH and in situ synthesis of [Ir(cod){Ph2PCH···C(···O)Ph,κP,κO}{Ph2PCH2C(O)Ph,κP}] (5)
Freshly distilled THF (5 mL) was added to a Schlenk tube containing [Ir(cod)(μ-Cl)]2 (0.034 g, 0.051 mmol) and TlPF6 (0.035 g, 0.10 mmol). The mixture was stirred at room temperature for 3 h until a white solid precipitated (TlCl). The suspension was filtered and Ph2PCH2C(O)Ph (0.061 g, 0.20 mmol) and NaH (60% dispersion in oil, 0.009 g, 0.22 mmol) were added to the filtrate under an argon flow. When no more gas was formed (H2), the mixture was further stirred at room temperature for 30 min. After filtration, the 31P{1H} NMR spectrum of the solution contained two major doublets at 1.30 and 27.9 ppm (2J(P,P) = 9.0 Hz) (unidentified complex, most likely containing a Ph2PCH···C(···O)Ph and a Ph2PCH2C(O)Ph ligand in mutually cis position) and resonances corresponding to complexes 4 and 5 (estimated NMR yields: 80%, 10% and 10%, respectively). Addition of Et2O (10 mL) to this solution precipitated an orange solid. A few single crystals of 5·C7H8 suitable for X-ray diffraction studies were obtained by slow vapour diffusion of Et2O into a toluene solution of the complex in a NMR tube. 31P{1H} NMR (d8-toluene) for 5: δ –3.55 (d), 14.32 (d, 2J(PP) = 23.1 Hz).
4.4 X-ray data collection and structure refinement for 4·CH2Cl2 and 5·C7H8
Suitable crystals for the X-ray analysis were obtained as described above. The intensity data were collected at 173(2) K on a Kappa CCD diffractometer (graphite monochromated Mo Kα radiation, λ = 0.71073 Å). Crystallographic and experimental details for the structures are summarized in Table 1. The structures were solved by direct methods (SHELXS-97) and refined by full-matrix least-squares procedures (based on F2, SHELXL-97) [18] with anisotropic thermal parameters for all the non-hydrogen atoms.
X-ray data collection and structure refinement parameters.
| Compound reference | 4·CH2Cl2 | 5·C7H8 |
| Chemical formula CCDC ref | C49H48Cl2F6IrO2P3 1,023,389 | C48H45IrO2P2•C7H8 852,592 |
| Formula Mass | 1138.88 | 1000.11 |
| Crystal system | Orthorhombic | Monoclinic |
| Space Group | Pbca | C2/c |
| a/Å | 18.0680 (2) | 43.3764 (18) |
| b/Å | 19.9530 (4) | 10.0952 (3) |
| c/Å | 25.3600 (5) | 22.0750 (8) |
| β/° | 90.00 | 105.089 (2) |
| Unit cell volume/Å3 | 9142.6 (3) | 9333.2 (6) |
| Temperature/K | 173 (2) | 173 (2) |
| Z | 8 | 8 |
| Absorption coefficient, μ/mm−1 | 3.207 | 2.970 |
| No. of reflections measured | 81661 | 16722 |
| No. of independent reflections | 8894 | 10212 |
| Rint | 0.1581 | 0.0646 |
| Final R1 values (I > 2σ(I)) | 0.0772 | 0.0509 |
| Final wR(F2) values (I > 2σ(I)) | 0.0968 | 0.0979 |
| Final R1 values (all data) | 0.1119 | 0.1239 |
| Final wR(F2) values (all data) | 0.1040 | 0.1148 |
| Goodness of fit on F2 | 1.233 | 0.928 |
Acknowledgment
This work was supported by the “Centre national de la recherche scientifique”, the “Ministère de l’Enseignement supérieur et de la Recherche” (Paris), the International Center for Frontier Research in Chemistry, Strasbourg (icFRC, www.icfrc.fr), the China Scholarship Council and the French Embassy in Beijing (grant to X. L). We are very grateful to Johnson Matthey PLC for the loan of iridium salts. We are very grateful to Corinne Bailly for the resolution of the structure of 5·C7H8 by X-ray diffraction.