

1 Introduction
The generation of free radicals under very soft irradiation conditions (light emitting diodes (LEDs), halogen lamp, sunlight …) through the photoredox catalysis approach has been proposed in the late 2000. This approach is now characterized by many different uses in organic synthesis [1–5] and, in the last few years, has been introduced into the polymer area to initiate a free radical polymerization (FRP), a cationic polymerization (CP), a free radical promoted cationic polymerization (FRPCP) and more recently a controlled radical photopolymerization (CRP2).[5–7] Different series of complexes, e.g., ruthenium,[8] iridium,[6] zinc,[9] copper [10] or iron [11] complexes, have been proposed as photoinitiators (PIs) and/or photoredox catalysts in photoinitiating systems (PISs) upon soft light irradiation. When introduced into the polymer photochemistry area (see a recent review in [6b]), photoredox catalysis offered the potential to expand the development of photopolymerization, thereby breaking through traditional constraints (see below) by using novel three-component PISs where the PI is now defined as a photoinitiator catalyst (PIC). In previous reviews, we have detailed the reactivity/efficiency of a lot of PISs [12–15] and, in particular, that of organometallic complexes or metal-free organo-compound based PISs [6b]. The reactivity of the organic free radicals formed in cleavable organic compounds or through electron transfer/hydrogen abstraction processes and usability for the initiation of photopolymerization have also been detailed in [15,16].
In the present paper, we will focus our interest on the design of novel organometallic compound based PISs, the mechanisms involved in the formation of the radical and cationic initiating species and finally their application to photopolymerization.
Ruthenium and iridium based organometallic complexes are widely used as radical sources in the field of photoredox catalysis (e.g. for organic synthesis) under very soft irradiation conditions, the operative routes being based on oxidation or reduction cycles (see in [1–7]). UV or visible light induced polymerization reactions in the presence of a large variety of PISs containing a metal centered structure (Ru, Ir, Fe, Cu, Co, Ti, Pt…) are well documented (see a detailed review in a recent book [15]). The use of such compounds as PIs has already been reported many years ago (see, e.g., [17] and references therein). A recent revival of interest for metallic complex based PISs is noticeable. Some examples include the photopolymerization of acrylamide in the presence of Ru catalysts, the photoactivated metathesis polymerization using metal complexes [18–21] or the FRP of hydrophobic monomers, i.e. methyl methacrylate, styrene and n-butyl acrylate (using Ru complexes) [5a]. In all these examples, the metal complex behaves as a usual PI and not as a PIC. No real higher performance (when a comparison with the use of conventional organic PIs is possible) was achieved so far in the field of photocurable high/low viscosity formulations in films under air upon exposure to visible lights. Other examples involving organometallic complexes in the photopolymer area can be seen in [7] for the control of photopolymerization (CRP2).
The successful results obtained with PIC based PISs in acrylate, vinylether or epoxide photopolymerization (see example again in [6b] and references therein; see also some other recent related papers in [7]) are undoubtedly related to a careful mixing of several components which ensures improved polymerization profiles. The PISs that have been designed are consisted in a PIC (such as a Ru or Ir complex) either combined with an iodonium salt Ph2I+ and a silane R3SiH (or eventually N-vinyl carbazole NVK) or an amine AH and an alkyl halide RX (usually a phenacyl bromide Phen–Br). The characteristics of these PISs are: i) a visible light absorption depending on the metal and the ligands, ii) the generation of the same radicals, radical cations or cations, whatever may be the absorbing compound, iii) a recovery of the PIC (more or less partial), iv) a beneficial effect of the silane on oxygen inhibition (as known in other silane containing PISs, see in [22]) and v) an interesting photoreactivity (related to excellent visible light absorptions, long lived excited states and suitable redox potentials for the complex; favorable electron or hydrogen transfer reactions between the different partners).
In the PIC/Ph2I+/R3SiH PIS operating through an oxidation cycle (Scheme 1), a phenyl radical is primarily generated; then, a silyl radical R3Si• and a silylium R3Si+ are formed through a subsequent Ph•/R3SiH hydrogen abstraction and R3Si•/PIC•+ interaction, respectively. This three-component system behaves here as an efficient dual radical/cation source. In the PIC/Ph2I+/NVK PIS, the scenario is a little different. The phenyl radical adds to the NVK double bond and the resulting electron rich NVK derived radical is easily oxidized by the iodonium salt or the PIC radical cation (Scheme 1). Both initiating radicals and cations are still formed.
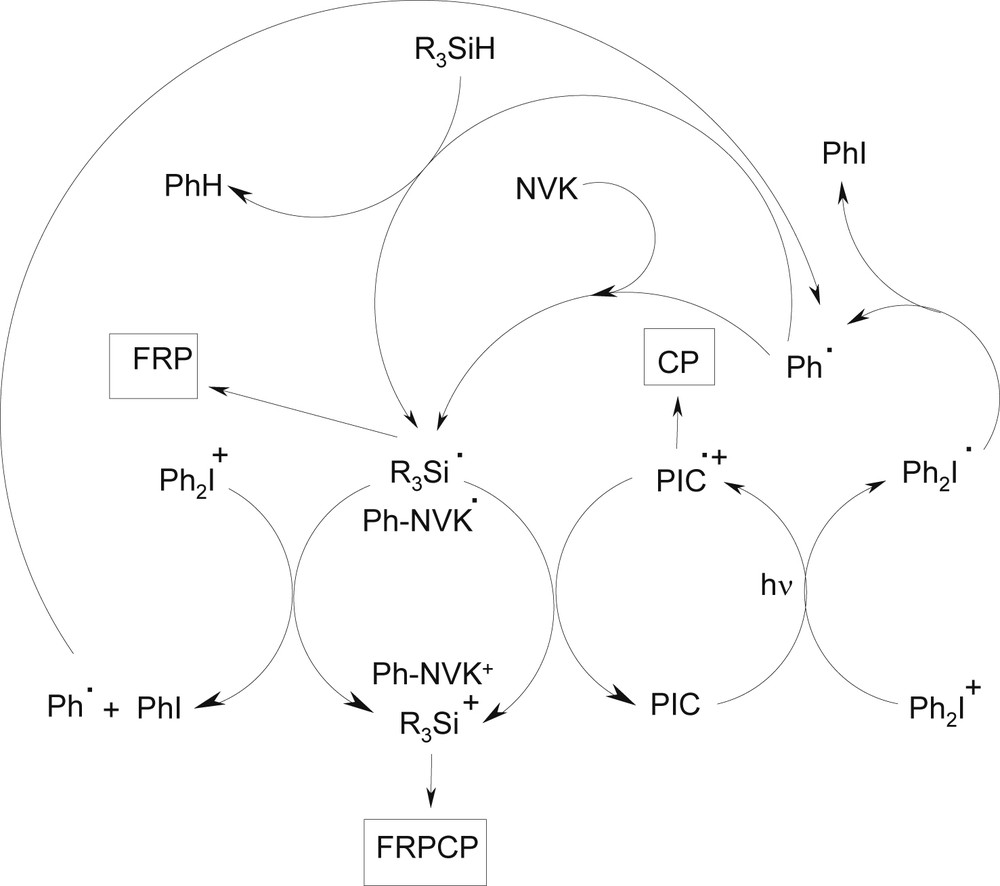
Different chemical mechanisms in photoredox catalysis to initiate polymerization processes in the presence of a silane and an iodonium salt.
On the opposite, the PIC/AH/alkyl halide (e.g. Phen–Br) PIS works through a reduction cycle (Scheme 2). A reduced form of the PIC is formed (PIC•−) through the PIC/AH electron transfer and a phenacyl radical (Phen•) is produced upon the subsequent cleavage of the phenacyl bromide radical anion (Phen–Br•−) generated in the PIC•−/Phen–Br interaction.
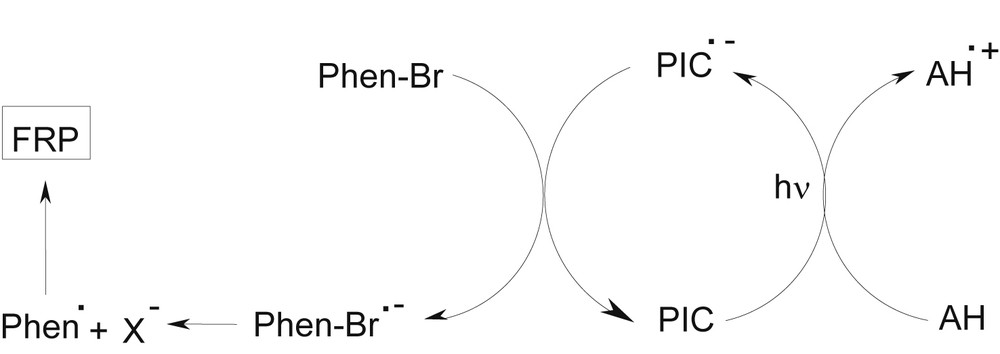
Different chemical mechanisms in photoredox catalysis to initiate polymerization processes in the presence of an amine and an alkyl halide.
Accordingly, when using appropriate metal complex based compounds, FRP, CP and FRPCP as well as the simultaneous radical/cationic polymerization of epoxide/acrylate blends leading to interpenetrating polymer networks (IPN) become feasible (Scheme 3) under exposure to polychromatic or monochromatic light ranging from the UV to the green (the development of red light sensitive PIS is still expected), even when delivered by low light intensity sources such as sun or household devices.

Dual systems to initiate both radical and cationic polymerization reactions.
Some of our recent studies have shown the FRP of acrylates, the CP of epoxides or the FRPCP of N-vinylcarbazole, epoxides or renewable cationic monomers under relatively intense (∼100 mW/cm2) or soft (∼10 mW/cm2) irradiation conditions in the visible wavelength range using Ru and Ir [5–6] complexes as PICs (like e.g. the Ir complexes shown in Scheme 4). We have also investigated other metal centered compounds as photoredox catalysts [9–11] based on Zn [9], Pt [23], Fe [11], Cu [10], Ni [24], Al [24] or Co [24]. Except the investigated Ni, Al and Co complexes which were not efficient, a quite good reactivity/efficiency was found with the other catalysts.
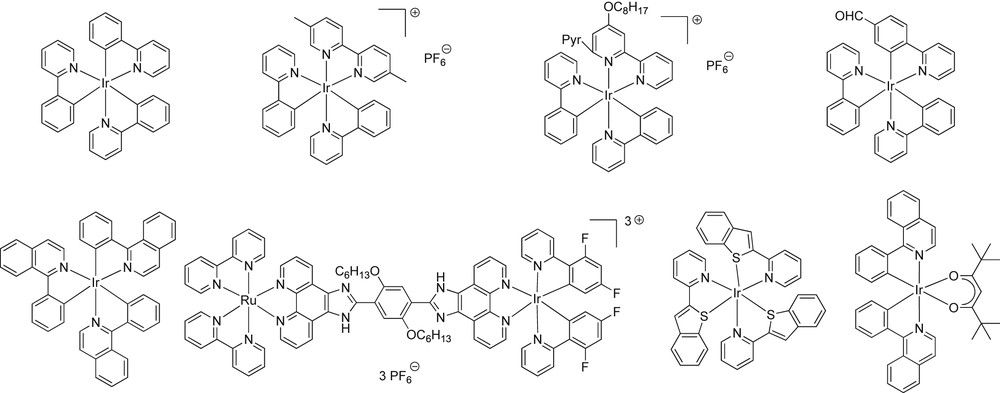
Examples of previously investigated iridium photoredox catalysts.
However, there is still a need for the development of new PICs with i) improved light absorption properties and ii) enhanced reactivity and polymerization efficiency (i.e. final conversion and polymerization rate). These new PICs should be highly worthwhile for visible light exposure; eventually, they should meet the requirements for very soft irradiation conditions. In the present paper, we are interested in Ir based complexes. Conventional Ir(III) complexes present a rather weak visible absorption. Incorporation of carefully selected ligands can lead to both a better reactivity due to longer lived transient states and longer wavelength absorptions suitable for polymerization reactions under various visible sources. We have already investigated the ability of Ir complexes to be incorporated in highly efficient PISs allowing excellent polymerization profiles (some of them are recalled in Scheme 4). For example, the commercially available tris(2-phenylpyridine)iridium Ir(ppy)3 is characterized by an intense absorption at 370 nm and exhibits only moderate absorption at λ > 400 nm, not only under an Xe–Hg lamp but also under exposure to household white LED and 462 nm blue LED or a fluorescent bulb.[5–6] Changing the ligands and/or introducing adequate conjugated substituents leads to PICs which function upon exposure to: i) Xe lamp (using tris (2-phenylpyridinato-C2,N) or (5-5′ dimethyl-2-2′-bipyridine)-bis-(2-phenylpyridine) ligand for FRP), ii) household 462 nm blue LED (using various substituted bipyridine or phenylpyridine ligands for FRPCP and FRP), and iii) halogen lamp, household 462 nm blue LED and 514 nm green LED (using a coumarin CˆN ligand moiety, tris(1-phenylisoquinoline-C2,N) or benzo[b](thiophen-2-yl) pyridinato-C,N ligands for FRPCP). The use of bis[2-(1-isoquinolinyl-κN)phenyl-κC](2,2,6,6-tetramethyl-3,5-heptanedionato-κO3,κO5)-iridium (III) (referred to as Ir(piq)2(tmd) and noted hereafter as Ir_a and known in another area [25]) has ensured, for the first time [6b], FRP and FRPCP upon exposure to a green laser diode at 532 nm.
In the following, we will explore novel structural effects of the iridium complex series ensuring enhanced and red-shifted absorptions (and even a better reactivity) by considering two novel phenylisoquinoline-based iridium complexes bearing fluorine substituents (Ir_b and Ir_c in Scheme 5) and then focus our attention on their reactivity and efficiency in the photopolymerization of a diepoxide or a triacrylate under blue, green and red lights when they are incorporated into suitable PISs. These two novel photoredox catalysts will be compared to our very recently proposed unsubstituted catalyst (Ir_a) [25]. Some comparisons with the commercially available Ir(ppy)3 will also be provided. The photochemical properties as well as the chemical mechanisms associated with these catalysts will be investigated allowing an access to their structure/reactivity relationships.
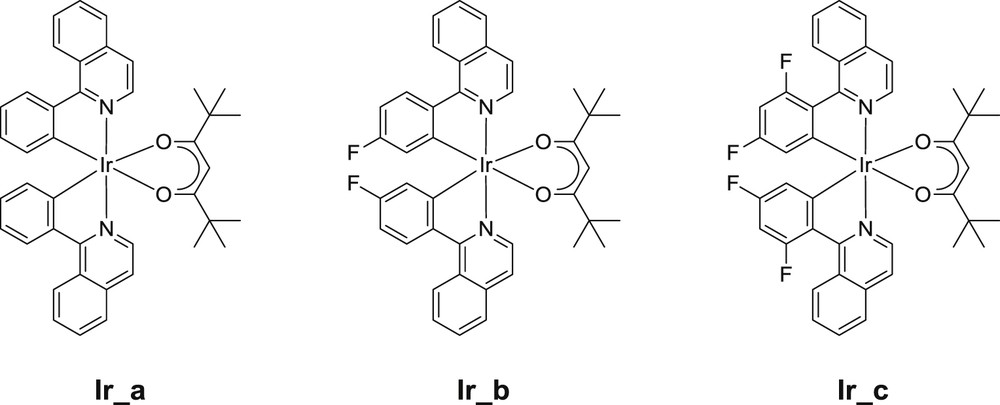
New proposed iridium complexes as photoredox catalysts.
2 Experimental part
2.1 Materials
Diphenyliodonium hexafluorophosphate (Ph2I+ or Iod), methyl diethanolamine (MDEA), the other reagents and solvents were purchased from Sigma–Aldrich or Alfa Aesar and used as received without further purification (Scheme 6). (3,4-Epoxycyclohexane)methyl-3,4-epoxycyclohexylcarboxylate (EPOX) and trimethylolpropane triacrylate (TMPTA) were obtained from Allnex (Scheme 6) and used as benchmark monomers for cationic and radical photopolymerization.
Chemical structures of additives and monomers.
2.2 Irradiation sources
Different laser diodes were used for the irradiation of the photocurable samples (@457 nm–100 mW cm−2; @532 nm–100 mW cm−2 and @635 nm–100 mW cm−2).
2.3 Photopolymerization experiments
For photopolymerization experiments, the photocurable formulations were deposited (25 μm thick) on a BaF2 pellet as a laminate (the formulation is sandwiched between two polypropylene films) for irradiation with the different lights. The evolution of the epoxy group content of EPOX and the double bond content of TMPTA was continuously monitored by real time FT-IR spectroscopy (JASCO FTIR 4100) at about 790 cm−1 and 1630 cm−1, respectively.[5–6]
2.4 Luminescence experiments
The luminescence properties of the investigated iridium complexes in acetonitrile were studied using a JASCO FP-750 spectrometer. The interaction rate constants (kq) between the studied complexes and the iodonium salt (Iod) were calculated from the classical Stern–Volmer treatment [15] (I0/I = 1 + kqτ0[additive]; where I0 and I stand for the fluorescence intensity of the studied complexes in the absence and the presence of the additives, respectively; τ0 stands for the excited state lifetime in the absence of additives).
2.5 Redox potentials
The oxidation potentials (Eox vs SCE) of the studied complexes were measured in acetonitrile by cyclic voltammetry with tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte (VoltaLab 6 Radiometer). The procedure has been presented in detail in ref. [5–6].
2.6 Laser flash photolysis
Nanosecond laser flash photolysis (LFP) experiments were carried out to determine the excited state lifetime using a Q-switched nanosecond Nd/YAG laser (λexc = 355 nm, 9 ns pulses; energy reduced down to 10 mJ) from Continuum (Minilite) and an analyzing system consisted of a ceramic xenon lamp, a monochromator, a fast photomultiplier and a transient digitizer (Luzchem LFP 212).[5–6]
2.7 ESR spin trapping (ESR-ST) experiments
ESR-ST experiments were carried out using an X-Band spectrometer (MS 400 Magnettech). The radicals were generated at room temperature upon exposure to the LED at 405 nm under N2 and trapped by phenyl-N-tert-butylnitrone (PBN) according to a procedure [5–6] described elsewhere in detail. The ESR spectra simulations were carried out with the WINSIM software.
3 Results and discussion
3.1 1/Synthesis of Ir_a, Ir_b and Ir_c
All reagents and solvents were purchased from Aldrich or Alfa Aesar and used as received without further purification. Mass spectroscopy was performed by the Spectropole of Aix-Marseille University. ESI mass spectral analyses were recorded with a 3200 QTRAP (Applied Biosystems SCIEX) mass spectrometer. The HRMS mass spectral analysis was performed with a QStar Elite (Applied Biosystems SCIEX) mass spectrometer. 1H and 13C NMR spectra were determined at room temperature in 5 mm o.d. tubes on a Bruker Avance 400 spectrometer of the Spectropole: 1H (400 MHz) and 13C (100 MHz). The 1H chemical shifts were referenced to the solvent peaks CDCl3 (7.26 ppm), and DMSO (2.49 ppm) and the 13C chemical shifts were referenced to the solvent peaks CDCl3 (77 ppm), and DMSO (49.5 ppm). All these dyes were prepared with analytical purity up to accepted standards for new organic compounds (>98%) which was checked by high field NMR analysis. 1-Phenylisoquinoline, 1-(4′-fluorophenyl)isoquinoline (4F-piq), 1-(2,4-difluorophenyl)isoquinoline (2,4diF-piq) and the corresponding dichloride-bridged iridium dimers [Ir2(μ-Cl)2(piq)4], [Ir2(μ-Cl)2(4F-piq)4] and [Ir2(μ-Cl)2(2,4diF-piq)4] were synthesized as previously reported in the literature.[26] The synthesis of the three complexes Ir_a, Ir_b and Ir_c was realized using the conventional bridge-splitting and substitution reaction of the dimer with four equivalents of 2,2,6,6-tetramethyl-3,5-heptanedione.
3.1.1 Synthesis of bis[2-(1-isoquinolinyl-κN)phenyl-κC](2,2,6,6-tetramethyl-3,5-heptanedionato-κO3,κO5)-iridium (III) (Ir_a)
In a 50 mL flask, the μ-chloride-bridged dimer Ir(III) complex [Ir2(μ-Cl)2(piq)4] (0.5 g, 0.393 mmol), 1,1,7,7-tetramethylheptan-3,5-trione (217 mg, 3 eq.) and K2CO3 (380 mg, 7 eq.) were mixed with 2-ethoxyethanol (30 mL) and the mixture was stirred under reflux overnight. After cooling to room temperature, 2-ethoxyethanol was removed under reduced pressure. The crude product was dissolved in dichloromethane (20 mL) and the obtained solid was filtered off. The dichloromethane solution (20 mL) was concentrated under reduced pressure. The residue was purified by column chromatography (SiO2) using CH2Cl2:pentane at 1:1 as the eluent. 542 mg of complex were obtained in 88% yield. 1H NMR (CDCl3) δ (ppm): 0.81 (s, 18H), 5.42 (s, 1H), 6.52 (dd, 2H, J = 1.0 Hz, J = 7.6 Hz), 6.66 (td, 2H, J = 1.1 Hz, J = 7.1 Hz), 6.90 (td, 2H, J = 1.2 Hz, J = 8.2 Hz), 7.38 (d, 2H, J = 6.3 Hz), 7.65–7.69 (m, 4H), 7.87–7.89 (m, 2H), 8.21 (d, 2H, J = 7.9 Hz), 8.34 (d, 2H, J = 6.3 Hz), and 8.98–9.01 (m, 2H); 13C NMR (CDCl3) δ (ppm): 28.1, 41.1, 89.5, 119.2, 119.8, 126.2, 126.7, 127.1, 127.3, 128.4, 129.4, 130.3, 134.3, 137.0, 140.7, 146.5, 153.9, 169.2, and 194.5; HRMS (ESI MS) m/z: theor: 784.2641 found: 784.2646 (M+. detected).
3.1.2 Synthesis of bis[5-fluoro-2-(1-isoquinolinyl-κN)phenyl-κC](2,2,6,6-tetramethyl-3,5-heptanedionato-κO3,κO5)-iridium (III) (Ir_b)
In a 50 mL flask, the μ-chloride-bridged dimer Ir(III) complex [Ir2(μ-Cl)2(4F-piq)4] (0.3 g, 0.223 mmol), 1,1,7,7-tetramethylheptan-3,5-trione (123 mg, 3 eq.) and K2CO3 (215 mg, 7 eq.) were mixed with 2-ethoxyethanol (30 mL) and the mixture was stirred under reflux for 48 h. After cooling to room temperature, 2-ethoxyethanol was removed under reduced pressure. The crude product was dissolved in dichloromethane (20 mL) and the obtained solid was filtered off. The dichloromethane solution (20 mL) was concentrated under reduced pressure. The residue was purified by column chromatography (SiO2) using CH2Cl2:pentane at 1:1 as the eluent. 307 mg of complex were obtained in 84% yield. 1H NMR (CDCl3) δ (ppm): 0.82 (s, 18H), 5.44 (s, 1H), 6.13 (dd, 2H, J = 2.7 Hz, J = 9.6 Hz), 6.64 (td, 2H, J = 2.7 Hz, J = 8.8 Hz), 7.41 (d, 2H, J = 6.4 Hz), 7.66–7.70 (m, 4H), 7.88–7.90 (m, 1H), 8.19 (dd, 2H, J = 8.9 Hz, J = 5.8 Hz), 8.26 (d, 2H, J = 6.4 Hz), and 8.87–8.92 (m, 1H); 13C NMR (CDCl3) δ (ppm): 28.0, 41.1, 89.7, 107.4 (d, J = 23.0 Hz), 119.3, 119.8 (d, J = 16.2 Hz), 125.9, 126.4, 127.4 (d, J = 43.5 Hz), 130.6, 130.9 (d, J = 9.3 Hz), 137.1, 140.4, 142.7 (d, J = 1.8 Hz), 156.8 (d, J = 6.5 Hz), 162.3 (d, J = 254.2 Hz), 168.2, and 194.7; 19F NMR (CDCl3) δ (ppm): −111.82; HRMS (ESI MS) m/z: theory: 820.2452 found: 820.2455 (M+ detected).
3.1.3 Synthesis of bis[3,5-difluoro-2-(1-isoquinolinyl-κN)phenyl-κC](2,2,6,6-tetramethyl-3,5-heptanedionato-κO3,κO5)-iridium (III) (Ir_c)
In a 50 mL flask, the μ-chloride-bridged dimer Ir(III) complex [Ir2(μ-Cl)2(2,4diF-piq)4] (0.35 g, 0.176 mmol), 1,1,7,7-tetramethylheptan-3,5-trione (137 mg, 3 eq.) and K2CO3 (240 mg, 7 eq.) were mixed with 2-ethoxyethanol (30 mL) and the mixture was stirred under reflux for 3 h. After cooling to room temperature, 2-ethoxyethanol was removed under reduced pressure. The crude product was dissolved in dichloromethane (20 mL) and the obtained solid was filtered off. The dichloromethane solution (20 mL) was concentrated under reduced pressure. The residue was purified by column chromatography (SiO2) using CH2Cl2:pentane at 1:1 as the eluent. 262 mg of complex were obtained in 87% yield. 1H NMR (CDCl3) δ (ppm): 0.68 (s, 18H), 6.40–6.46 (m, 2H), 6.59–6.66 (m, 2H), 7.42 (d, 2H, J = 6.3 Hz), 7.57–7.68 (m, 4H), 7.81 (d, 2H, J = 8.0 Hz), 8.20 (d, 2H, J = 6.3 Hz), and 8.44 (t, 2H, J = 9.2 Hz); 13C NMR (CDCl3) δ (ppm): 27.9, 41.1, 89.6, 109.2 (dd, J = 8.3 Hz, J = 27.9 Hz), 116.3 (d, J = 9.7 Hz, J = 30.8 Hz), 119.6, 125.9, 126.1 (d, J = 2.6 Hz), 126.4 (d, J = 1.5 Hz), 129.1 (d, J = 20.5 Hz), 130.6, 132.4 (d, J = 41.6 Hz), 136.6–136.9 (m), 140.6, 155.9 (dd, J = 251.5 Hz, J = 1.8 Hz), 165.7 (d, J = 229.9 Hz), 167.4 (d, J = 5.2 Hz), and 194.4; 19F NMR (CDCl3) δ (ppm): −110.8 (d, J = 21.0 Hz), −106.1 (d, J = 21.0 Hz); HRMS (ESI MS) m/z: theory: 856.2264 found: 856.2266 (M+ detected).
3.2 2/Light absorption and photochemistry of Ir_a, Ir_b and Ir_c
Very interestingly, Ir_a, Ir_b and Ir_c exhibit excellent light absorption properties. These complexes are characterized by a panchromatic behavior and can absorb light in the 300–650 nm spectral range (Fig. 1). This is a great advantage for these complexes (Ir_a, Ir_b and Ir_c) as, for more classical iridium complexes, no significant absorption at λ > 500 nm is usually found, e.g., the absorption of Ir(ppy)3 [5–6] at λ > 480 nm is quite low. Ir_a, Ir_b and Ir_c are also characterized by luminescence properties with an intense emission at about 600–620 nm. This luminescence is ascribed to the emission from their excited triplet state (phosphorescence). These triplet states can be quenched by reducing or oxidizing agents thereby opening the way for photochemical reactions. For example, *Ir_a and *Ir_b are easily oxidized by iodonium salts (r1), i.e. both phosphorescence quenching (Fig. 2A) and photolysis of the solutions (Fig. 2B) in the presence of the iodonium salt are found. This is also in agreement with the formation of phenyl radicals as observed by ESR-spin trapping experiments (the hyperfine coupling constants for the PBN adduct in a Ir_a/Iod solution being aN = 14.2 G and aH = 2.2 G in agreement with previous data for such radicals [8]). These Ir complexes operate as described in Schemes 4 and 5.
| (r1) |
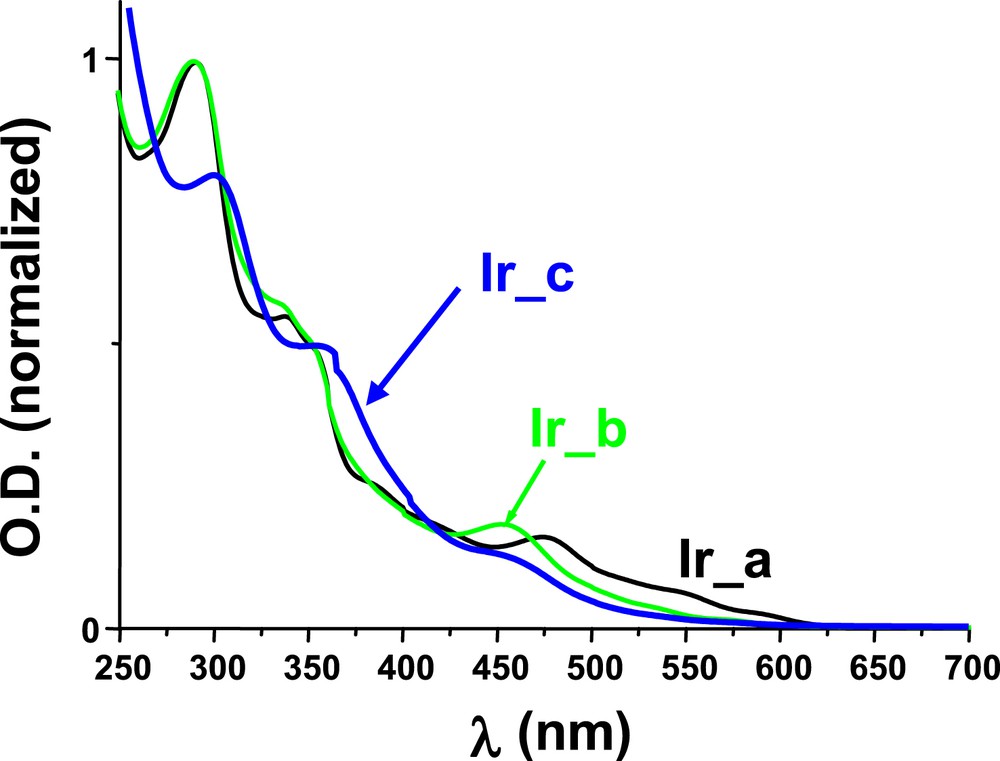
UV–visible absorption spectra of Ir_a, Ir_b and Ir_c in acetonitrile.
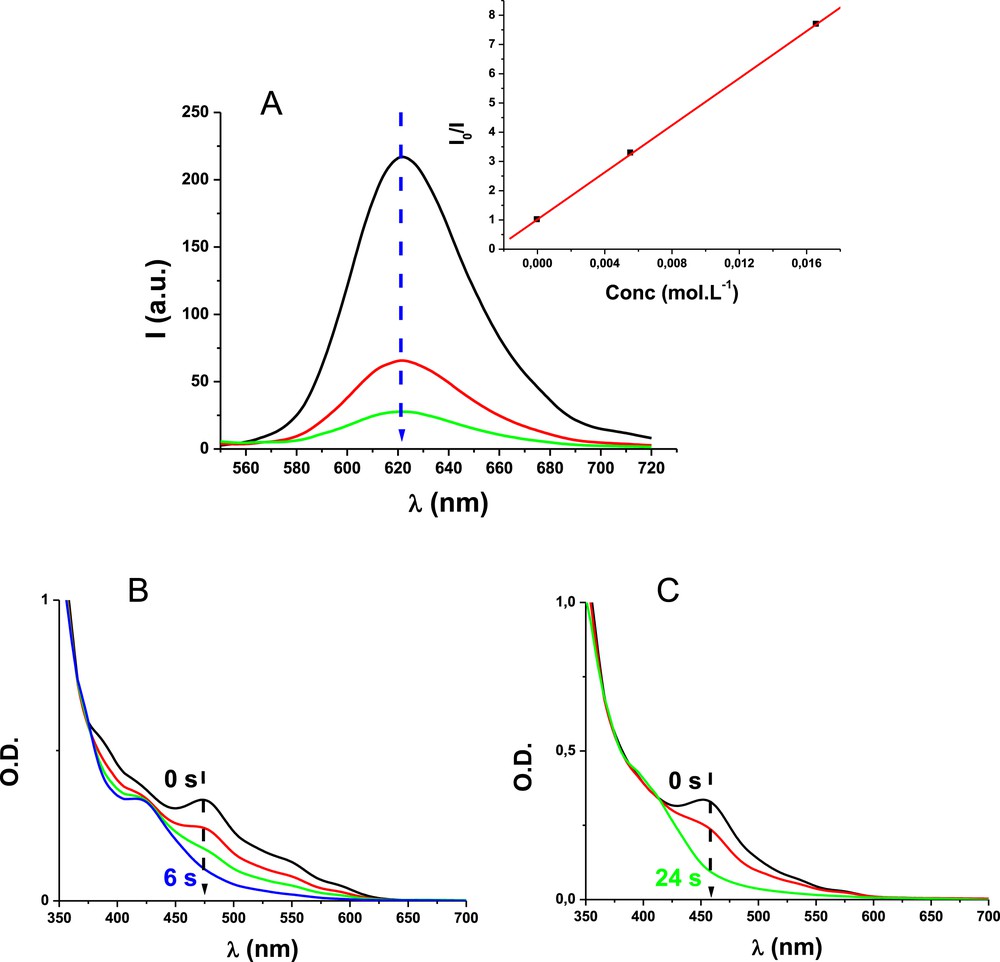
(A) Phosphorescence quenching of Ir_a/Iod in acetonitrile (inset: the Stern–Volmer plot), (B) photolysis of Ir_a/Iod in acetonitrile and (C) photolysis of Ir_b/Iod in acetonitrile. Upon a LED@455 nm exposure.
The free energy changes (ΔG) for the electron transfer between the studied Ir complexes and Iod can be calculated from the classical Rehm–Weller equation:[27] ΔG = Eox−Ered−ES (or ET) + C; where Eox, Ered, ES (or ET), and C are the oxidation potential of the electron donor, the reduction potential of the electron acceptor, the excited triplet state energies of the studied iridium complexes, and the electrostatic interaction energy for the initially formed ion pair, generally considered as negligible in polar solvents. All these parameters extracted from this work are gathered in Table 1. Reaction r1 is also in agreement with the favorable ΔG for this process (ΔG < 0). The electron transfer quantum yields (ΦeT) are also close to unity showing a quantitative process (Table 1).
Photochemical parameters governing the reactivity of Ir_a, and Ir_b.
| Eoxa (V vs. SCE) | ET (eV)b | ΔGT (eV)c | ΦeTd | |
| Ir_a | 0.67 | 2.16 | −1.29 | 0.95 |
| Ir_b | 0.86 | 2.29 | −1.23 | 0.93 |
a Eox values measured by cyclic voltammetry for NDP (this work).
b Triplet state energies (ET) extracted from the luminescence spectra.
c ΔG = free energy change for the *Ir/Iod triplet state (ΔGT) interaction; reduction potential Ered = −0.2 V for Iod; [15]
d ΦeT = Ir/Iod electron transfer quantum yields in the triplet state calculated according to ΦeT = kqτ0 [additive]/(1 + kqτ0 [additive])[15], ([Iod] = 4.7 × 10−2 M).
The presence of fluorine substituents (as the electron withdrawing group) in Ir_b not only increases the oxidation potential by almost 0.2 V (0.86 V vs. 0.67 V) but also increases the triplet state energy level (accordingly, the free energy change is weakly affected by this substitution). The good reversibility of the oxidation process for Ir_a and Ir_b found by cyclic voltammetry ensures a catalytic cycle.
3.3 3/Ir_a, Ir_b and Ir_c in new polymerization photoinitiating systems: structure/reactivity relationship
The use of Ir_a, Ir_b and Ir_c in three-component photoinitiating systems according to an oxidative cycle (Scheme 1 for Ir_a (or Ir_b, Ir_c)/Iod/NVK) or to a reductive cycle (Scheme 2 for Ir_a (or Ir_b, Ir_c)/amine/alkyl halide) has been investigated. These systems generate radicals and cations that can initiate radical and/or cationic polymerization (Figs. 3, 4 and 5). Ir_a, Ir_b and Ir_c are much better than Ir(ppy)3 in FRP in line with their excellent light absorption properties (Fig. 3, curves 2–3 vs. curve 1). In radical and cationic polymerization, Ir_a, Ir_b and Ir_c exhibit an almost similar efficiency. Ir_a and Ir_b which have a broader absorption spectrum than Ir_c are of particular interest for CP under blue, green and red light (Figs. 4 and 5) giving a panchromatic behavior to these PISs.
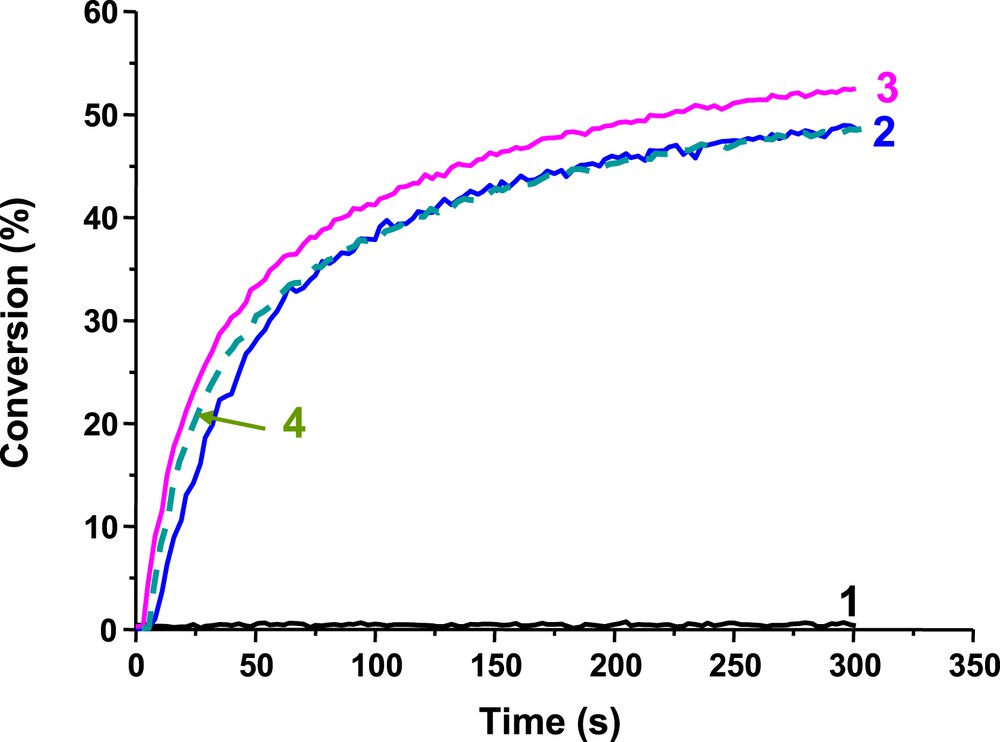
Photopolymerization profiles of TMPTA in laminate in the presence of: (1) Ir(ppy)3/MDEA/R–Br (1%/4%/3% w/w), (2) Ir_a/MDEA/R–Br (1%/4%/3% w/w); (3) Ir_b/MDEA/R–Br (1%/4%/3% w/w) and (4) Ir_c/MDEA/R–Br (1%/4%/3% w/w) upon the diode laser @457 nm exposure.
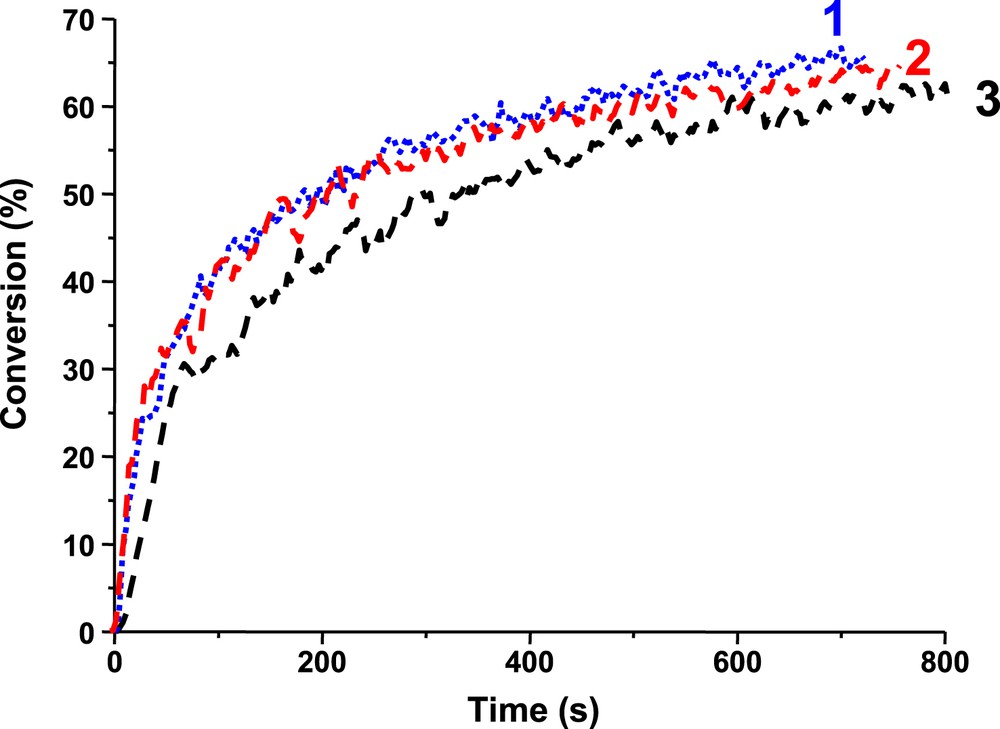
Photopolymerization profiles of EPOX under air in the presence of: (1) Ir_b/Iod/NVK (1%/2%/3% w/w), (2) Ir_c/Iod/NVK (1%/2%/3% w/w) and (3) Ir_a/Iod/NVK (1%/2%/3% w/w) upon the diode laser @457 nm exposure.
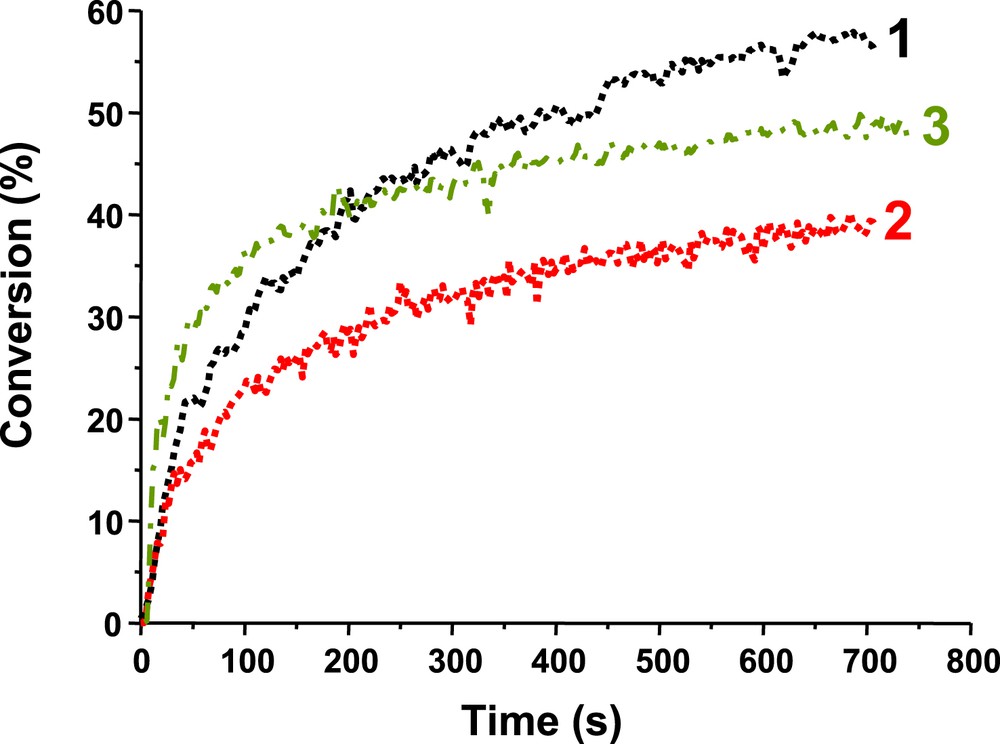
Photopolymerization profiles of EPOX under air in the presence of Ir_a/Iod/NVK (1%/2%/3% w/w) (1) upon the diode laser @532 nm exposure and (2) upon the diode laser @635 nm exposure and (3) in the presence of Ir_b/Iod/NVK (1%/2%/3% w/w) upon the diode laser @532 nm exposure.
4 Conclusion
This paper has briefly reviewed the use of the photoredox catalysis in the photopolymerization area. It also gave the opportunity to recall some interesting data obtained with a recently proposed Ir complex as PIC (Ir_a) and to present two newly synthesized related derivatives (Ir_b and Ir_c). Albeit a visible improvement of the achieved performance in the photopolymerization of a diepoxide or a triacrylate is noted, the presence of the fluorine substituents in Ir_b and Ir_c does not drastically affect their photochemical properties as well as their reactivity/efficiency. Ir_a (unsubstituted compound) and Ir_b (that contains fluorine at the ortho position on the phenyl rings) are characterized by a UV–vis spectrum broader than that of Ir_c that bears two fluorine atoms (meta and para position). These structures present a full panchromatic behavior with photosensitivity to blue-green-red laser light, e.g. i) the Ir_a based PIS is shown here as working quite well at 635 nm, ii) Ir_b/MDEA/R–Br is better than Ir_a/MDEA/R–Br at 457 nm (FRP), iii) Ir_b/Iod/NVK and Ir_c/Iod/NVK are also better than Ir_a/Iod/NVK at this wavelength (CP) and iv) Ir_b/Iod/NVK, compared to Ir_a/Iod/NVK, leads to a higher rate of polymerization at 532 nm (CP). Other Ir complexes are under development. The use of Ir_a, Ir_b and Ir_c in controlled radical photopolymerization (CRPP) like photoATRP will be presented in a forthcoming paper.