

1 Introduction
Polydentate ligands play a crucial role in transition metal chemistry, as they increase the stability of their complexes and enable a fine control of the reactivity at the metal center through their steric and electronic properties. For these reasons, mixed hard/soft donor pincer ligands have been considerably studied for their ability to stabilize metal cations in a variety of oxidation states [1] and for their role in stoichiometric and catalytic reactions [2]. A plethora of pincer ligands, coordinated to a number of d and f transition metals, has now been reported in the literature. Recently, a series of neutral mixed hard/soft donor pincer ligands has attracted considerable attention, in particular the PNP-type 2,6-(R2PX)(C5H3N) (XO, CH2, NH) ligands containing a central pyridine connected to two PR2 fragments. Considerable work, by the groups of Milstein, Jones, Brookhart and others, has demonstrated the importance of such ligands and the non-innocent role of X in hydrogen transfer catalysis [2,3]. With the reactive XNH2 or CH2 moiety replaced by an oxygen atom (XO), the R2PO(py)OPR2 (PONOP) ligands offer the advantage of a convenient and facile synthesis, but the potential of the corresponding complexes remains quite unexplored. Indeed, only a handful of PONOP complexes have been reported and structurally characterized, mostly with noble metals of the second and third rows (Ru, Rh, Pd, Ir, and Pt) [4–9] and recently with Ni [6,9] and Co [10] for metals of the first row. Here we report the synthesis and structural characterization of a series of RPONOP complexes with late first row divalent metals (Fe, Co, Ni, and Zn) and their electrochemical behavior in organic solvents.
2 Results and discussion
2.1 Syntheses and crystal structures of the complexes
The RPONOP ligands were prepared following literature protocols by mixing the 1,6-dihydroxy pyridine hydrochloride salt in THF with di(tert-butyl)-chlorophosphine or di(isopropyl)-chlorophosphine in the presence of triethylamine and tetramethylethylenediamine [4,8,9]. The series of M(tBuPONOP)X2 (MX2 = FeBr2 (1), CoCl2 (2), NiI2 (4), ZnI2 (5)) and M(iPrPONOP)X2 (MX2 = FeBr2 (1′), NiI2 (4′), ZnI2 (5′)) complexes was then readily synthesized by the addition of tBuPONOP or iPrPONOP to the anhydrous MX2 halides (Scheme 1). Because of the poor solubilities of the metal halide starting materials, a polar solvent such as THF is preferred over toluene in order to avoid extended reaction time. The reaction procedure is similar for all the compounds with initial heating at 40 °C for 1 h and then stirring overnight at room temperature. All the compounds proved to be soluble in THF except Ni(tBuPONOP)I2 (4) which deposited as a dark-red solid from the crude mixture. Removal of the solvent and washings with pentane of the solid residue afforded pure M(RPONOP)X2 compounds with yields ranging from 77 to 83%.
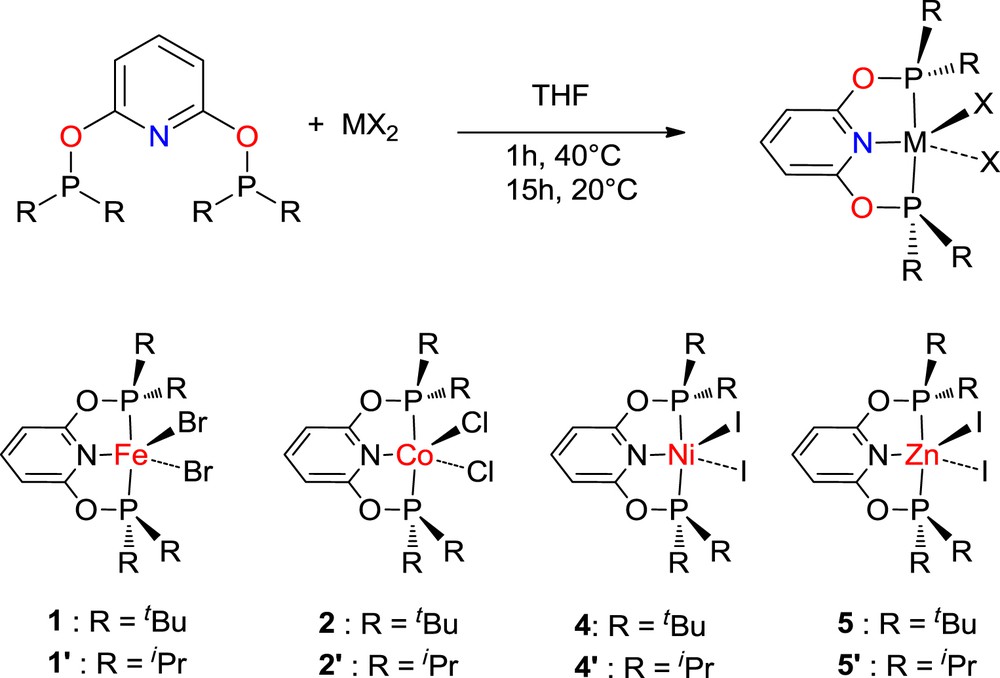
Synthesis of the M(RPONOP)X2 complexes (R = tBu and iPr).
All the compounds have been characterized by elemental analysis and 1H, 31P and 13C{1H} NMR spectra except Co(tBuPONOP)Cl2 (2), for which paramagnetism of the 3d7 Co2+ ion impedes any NMR observation. While the 1H NMR signals of the 3d8 nickel complex 4 are relatively narrow and in the diamagnetic zone (δ 1.65 (tBu) and 7.12 and 8.22 for the pyridine moiety), the corresponding signals for the 3d6 paramagnetic iron analog 1 are large and strongly shifted at δ 16.89 (tBu) and −20.39 and 54.01 for the pyridine signals, respectively, with full width at half maximum in the range of 70–150 Hz.
In contrast to 1, 2, and 5, the nickel complex 4 is almost insoluble in THF. While [Ni(tBuPONOP)Cl][Cl] is cationic in THF [9], the insolubility of 4 would suggest the formation of a similar species, e.g. [Ni(tBuPONOP)I][I]. Formation of discrete ion pairs for 4 and 4’ would also be in agreement with the lesser coordinating properties of iodide compared to the chloride anion. Surprisingly, 4′ crystallized in the neutral form [Ni(tBuPONOP)I2] (vide infra), suggesting that an equilibrium exists in solution between the cationic and neutral forms.
Crystallization of compounds 1, 2, 4, and 5 was attempted by slow diffusion of pentane into a THF solution of the complex. With the tBuPONOP ligand, only the iron complex 1 and the cobalt analog 2 crystallized as large yellow and blue crystals, respectively. Changing the solid tBuPONOP with the liquid iPrPONOP ligand clearly favored crystallization and the iron and nickel compounds 1′ and 4′ were obtained under conditions similar to those giving 1 and 2 whereas the zinc complex 5′ was obtained by cooling a hot toluene solution to room temperature. Compounds 1′, 4′ and 5′ were obtained as large yellow, dark red and colorless crystals, respectively, which were suitable for X-ray diffraction studies, as those of 1 and 2.
While the synthesis of the M(RPONOP)X2 complexes is readily achieved, the MX2:ligand ratio seems important. Thus, when a THF solution of iPrPONOP was treated with an excess of CoCl2, crystals of the dinuclear complex Co(iPrPONOP)Cl(μ-Cl)CoCl2(THF) (3) were obtained by slow diffusion of pentane to the mixture (Scheme 2). Obviously, the dihalide MX2 species is prone to intermolecular chloride interactions with the formation of bridged species, even in a polar solvent.
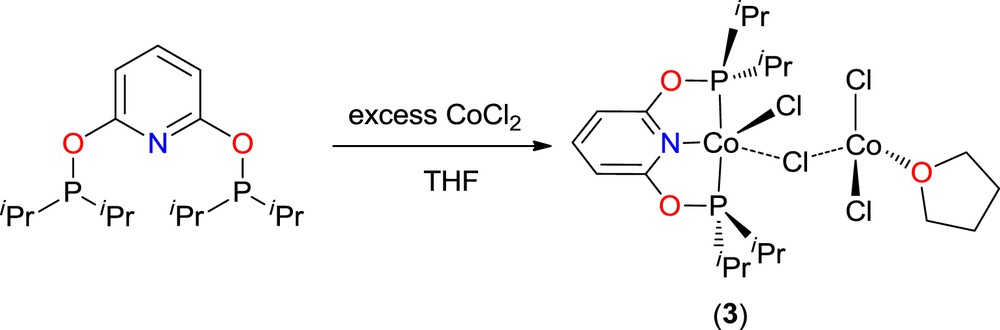
Formation of the bimetallic complex Co(iPrPONOP)Cl(μ-Cl)CoCl2(THF) 3.
Manipulations must also be carried out under exclusion of traces of air and water. In an attempt at crystallization of 5, a few colorless crystals of the unexpected THF adduct [Zn(tBuP(O)ONOP(O))l2]·0.25THF (6·0.25THF) were obtained (Scheme 3). Formation of 6 certainly resulted from accidental oxidation with traces of moist air. A similar formation of the europium species [EuCl2(H2O)3(κ2–{(O)PtBuCNCP(O)}][Cl] (tBuP(O)CNCP(O) = (2,6-bis(tert-butylphosphineoxide)CH2)-pyridine), from the reaction of EuCl3(H2O)6 and tBuPCNCP (tBuPCNCP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) in hot THF, was previously reported [11]. Sensitivity of the phosphine ligands to oxygen and water with formation of the corresponding phoshine oxide derivatives has been extensively studied [12]. In these two cases, oxidation of the phosphine pendant arms of the RPONOP and RPCNCP ligands led to derivatives coordinated by the two phosphine oxide fragments, with an uncoordinated central pyridyl moiety.
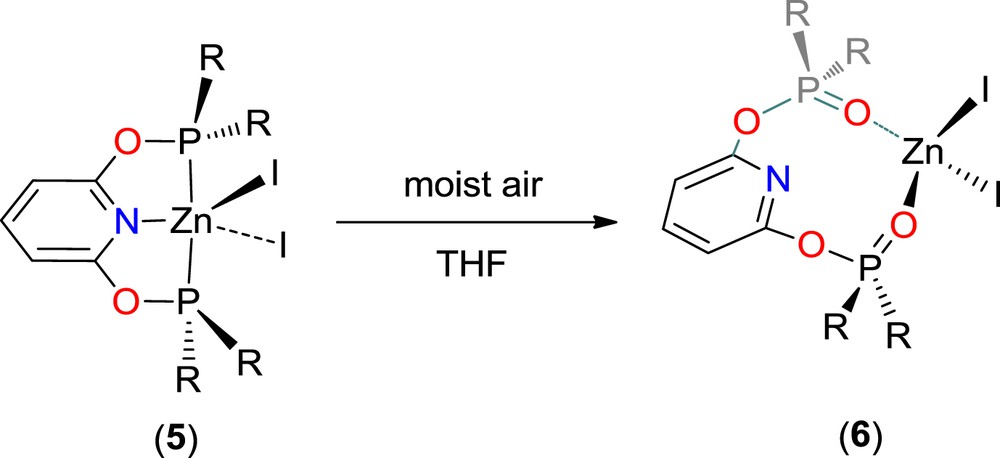
Formation of Zn(tBu(O)PONOP(O)I2 from 5.
Views of complexes 1, 1′, 2, 3, 4′, 5′ and 6·0.25THF are given in Figs. 1–7, respectively, and selected bond lengths and angles are listed in Tables 1 and 2. In all the pentacoordinate complexes 1, 1′, 2, 4′, and 5′ the metal ion can be viewed in a very distorted trigonal bipyramidal environment and best described in a distorted square pyramidal environment. In the latter case, the N1, P1, P2 and the X1 halide atoms (XCl, Br or I) form the basal plane with the halide X2 atom at the axial position. Complex 5′ is a twenty electron species which is quite common for the Zn2+ ion.
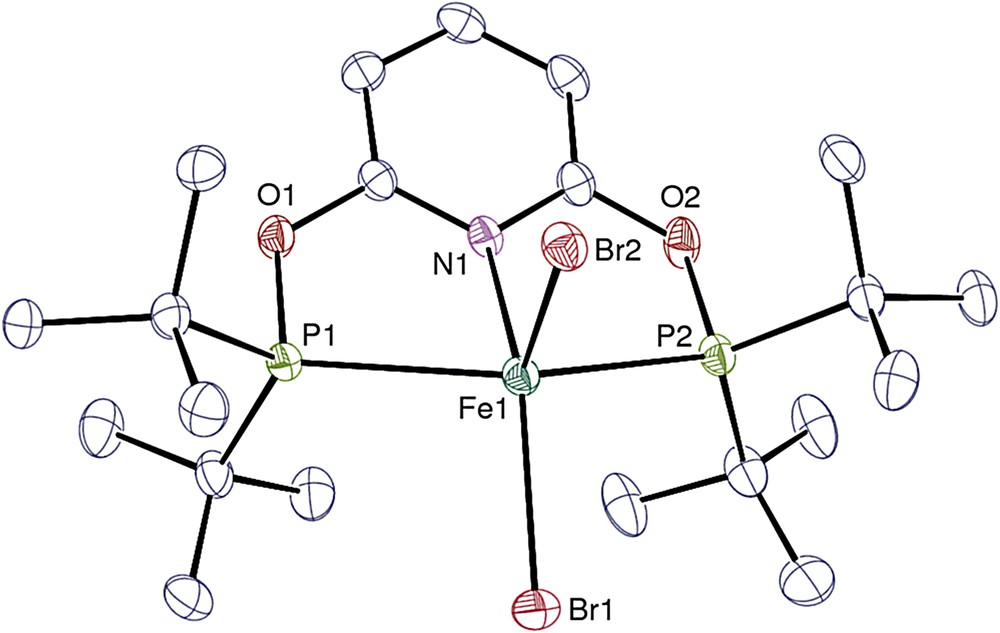
View of one of the two independent molecules in 1. Hydrogen atoms are omitted. Displacement ellipsoids are drawn at the 30% probability level.
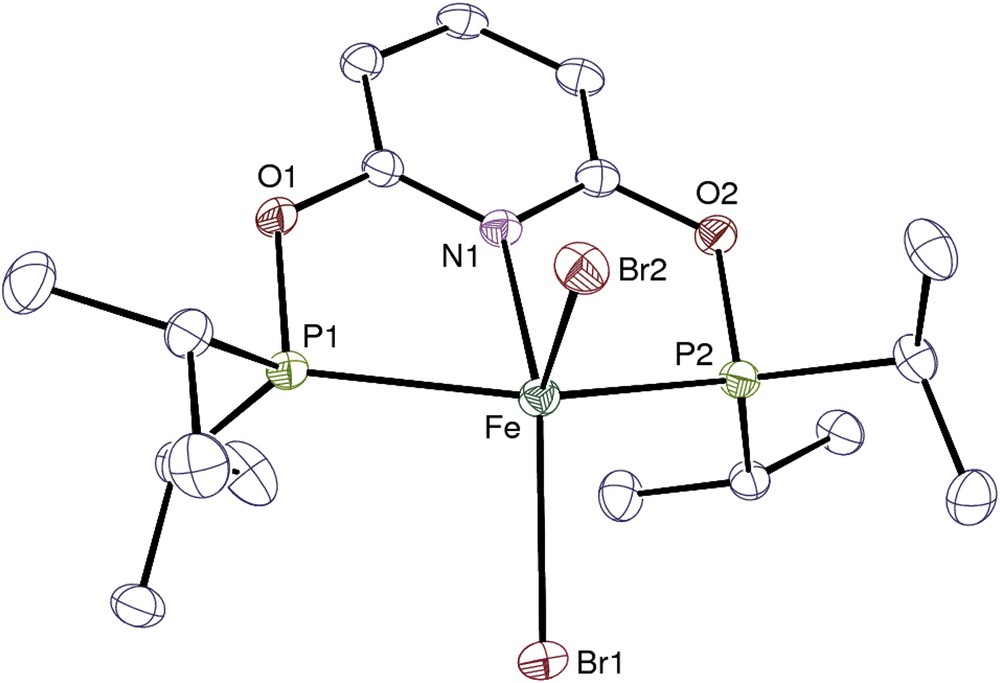
View of complex 1′. Hydrogen atoms are omitted. Displacement ellipsoids are drawn at the 30% probability level.
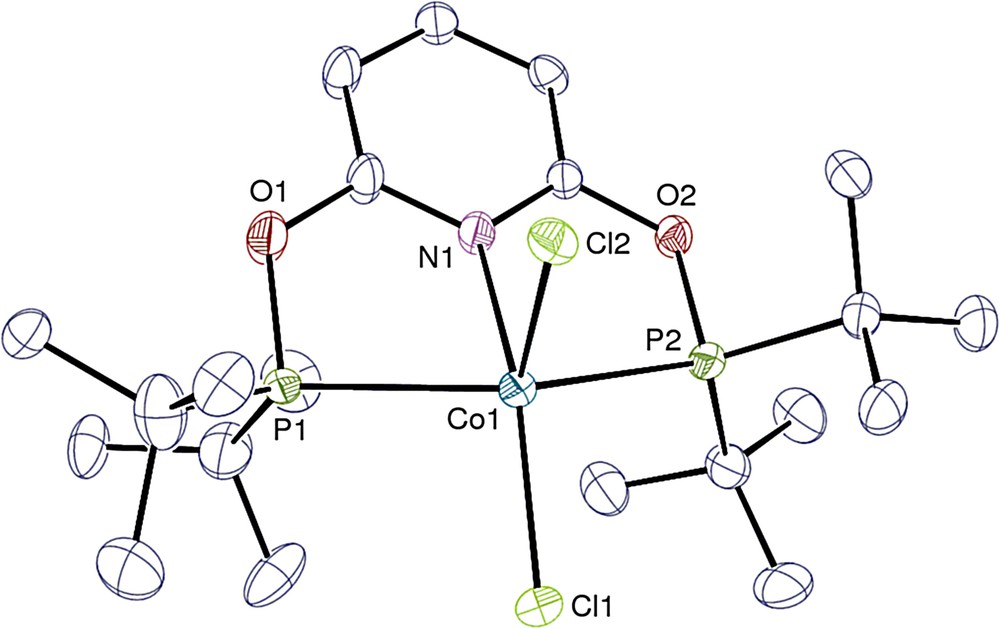
View of one of the two independent molecules in 2. Hydrogen atoms are omitted. Displacement ellipsoids are drawn at the 30% probability level.
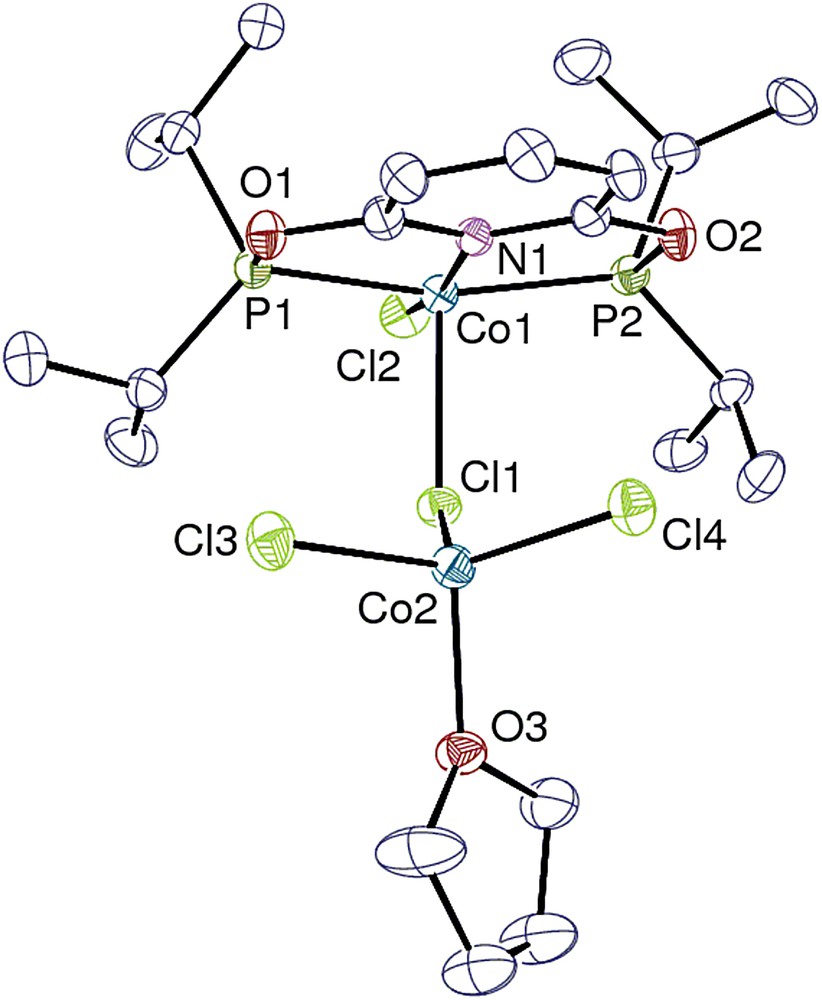
View of the bimetallic complex 3. Hydrogen atoms are omitted. Displacement ellipsoids are drawn at the 40% probability level.
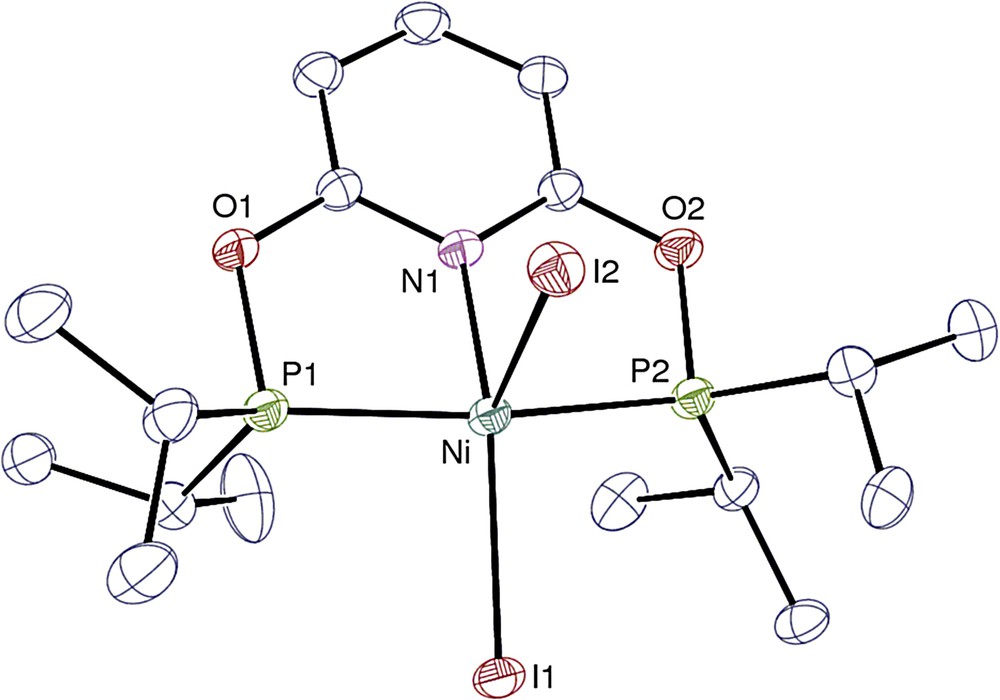
View of complex 4′. Hydrogen atoms are omitted. Displacement ellipsoids are drawn at the 30% probability level.
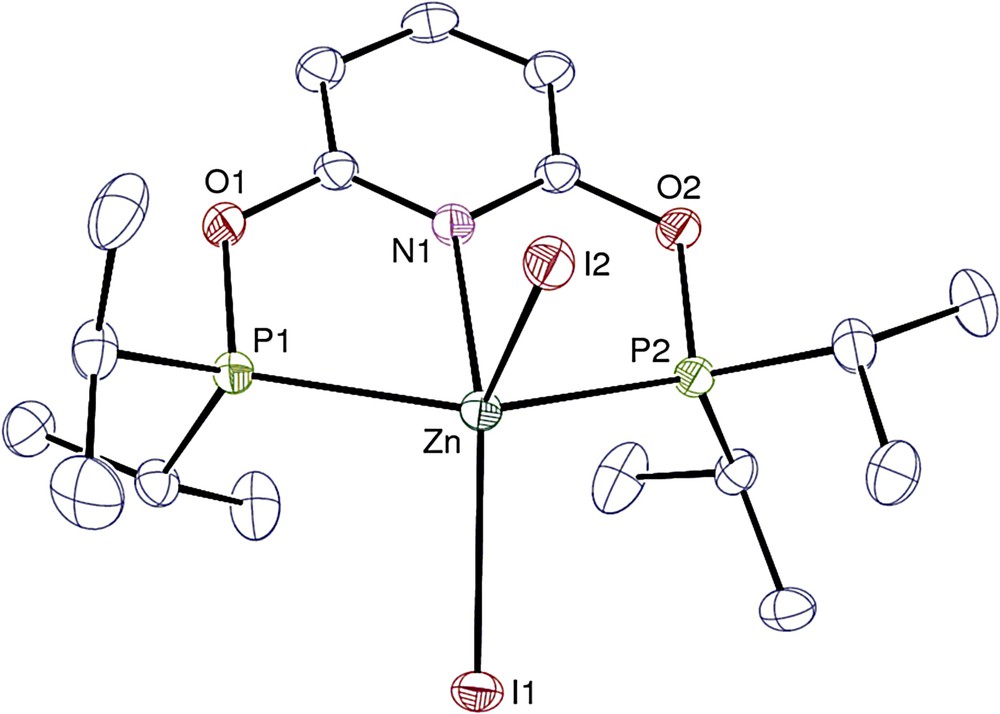
View of complex 5′. Hydrogen atoms are omitted. Displacement ellipsoids are drawn at the 30% probability level.
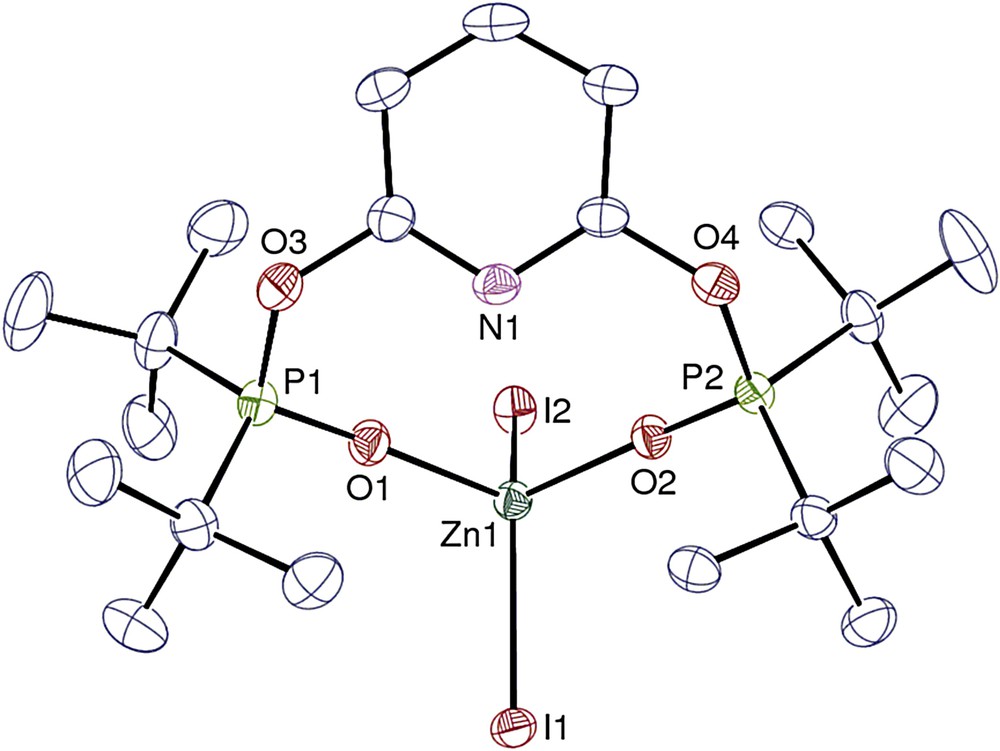
View of complex 6·0.25THF. The solvent molecule and hydrogen atoms are omitted. Displacement ellipsoids are drawn at the 30% probability level.
Selected bond lengths (Å) and angles (°) in the complexes 1, 1′ and 2.
| 1 | 1′ | 2 | |||||||
| Fe1–N1 | 2.2912(17) | Fe2–N2 | 2.2630(15) | Fe–N1 | 2.247(2) | Co1–N1 | 2.217(3) | Co2–N2 | 2.174(3) |
| Fe1–Br1 | 2.4842(4) | Fe2–Br3 | 2.4918(3) | Fe–Br1 | 2.5243(5) | Co1–Cl1 | 2.2869(12) | Co2–Cl3 | 2.3051(11) |
| Fe1–Br2 | 2.4505(3) | Fe2–Br4 | 2.4165(4) | Fe–Br2 | 2.4086(5) | Co1–Cl2 | 2.2985(12) | Co2–Cl4 | 2.2693(11) |
| Fe1–P1 | 2.5309(5) | Fe2–P3 | 2.5448(6) | Fe–P1 | 2.4492(8) | Co1–P1 | 2.4692(11) | Co2–P3 | 2.4994(12) |
| Fe1–P2 | 2.5302(6) | Fe2–P4 | 2.5362(6) | Fe–P2 | 2.4583(8) | Co1–P2 | 2.4434(12) | Co2–P4 | 2.4866(11) |
| P1–O1 | 1.6612(15) | P3–O3 | 1.6717(14) | P1–O1 | 1.6773(19) | P1–O1 | 1.624(3) | P3–O3 | 1.677(3) |
| P2–O2 | 1.6695(16) | P4–O4 | 1.6719(14) | P2–O2 | 1.6773(19) | P2–O2 | 1.666(3) | P4–O4 | 1.675(3) |
| P1–Fe1–P2 | 141.49(2) | P3–Fe2–P4 | 143.94(2) | P1–Fe–P2 | 143.67(3) | P1–Co1–P2 | 139.50(4) | P3–Co2–P4 | 150.38(4) |
| N1–Fe1–Br1 | 147.28(4) | N2–Fe2–Br3 | 145.99(4) | N1–Fe–Br1 | 142.45(6) | N1–Co1–Cl1 | 155.32(11) | N2–Co2–Cl3 | 144.31(9) |
| N1–Fe1–Br2 | 106.43(4) | N2–Fe2–Br4 | 107.48(4) | N1–Fe–Br2 | 101.66(5) | N1–Co1–Cl2 | 91.79(11) | N2–Co2–Cl4 | 106.71(9) |
| N1–Fe1–P1 | 73.16(4) | N2–Fe2–P3 | 74.64(4) | N1–Fe–P1 | 75.09(6) | N1–Co1–P1 | 73.60(9) | N2–Co2–P3 | 76.79(9) |
| N1–Fe1–P2 | 73.38(4) | N2–Fe2–P4 | 73.65(4) | N1–Fe–P2 | 74.50(6) | N1–Co1–P2 | 74.98(10) | N2–Co2–P4 | 76.32(9) |
| P1–Fe1–Br1 | 98.468(15) | P3–Fe2–Br3 | 99.418(16) | P1–Fe–Br1 | 96.90(2) | P1–Co1–Cl1 | 97.90(4) | P3–Co2–Cl3 | 98.01(4) |
| P1–Fe1–Br2 | 103.482(16) | P3–Fe2–Br4 | 103.594(18) | P1–Fe–Br2 | 96.37(2) | P1–Co1–Cl2 | 103.65(4) | P3–Co2–Cl4 | 100.18(4) |
| P2–Fe1–Br1 | 99.395(17) | P4–Fe2–Br3 | 97.283(17) | P2–Fe–Br1 | 95.21(2) | P2–Co1–Cl1 | 100.08(4) | P4–Co2–Cl3 | 96.58(4) |
| P2–Fe1–Br2 | 103.785(16) | P4–Fe2–Br4 | 101.926(17) | P2–Fe–Br2 | 108.83(2) | P2–Co1–Cl2 | 102.25(4) | P4–Co2–Cl4 | 99.23(4) |
| Br1–Fe1–Br2 | 106.285(13) | Br3–Fe2–Br4 | 106.457(13) | Br1–Fe–Br2 | 115.758(18) | Cl1–Co1–Cl2 | 112.83(5) | Cl3–Co2–Cl4 | 108.95(4) |
Selected bond lengths (Å) and angles (°) in the complexes 3, 4′,5′ and 6·0.25THF.
| 3 | 4′ | 5′ | 6·0.25THF | ||||||
| Co1–N1 | 1.9341(18) | Co2–O3 | 2.0325(17) | Ni–N1 | 1.917(2) | Zn–N1 | 2.4697(18) | Zn–I1 | 2.5730(4) |
| Co1–Cl1 | 2.4737(7) | Co2–Cl1 | 2.2965(7) | Ni–I1 | 2.5264(4) | Zn–I1 | 2.6618(3) | Zn–I2 | 2.5633(4) |
| Co1–Cl2 | 2.2200(7) | Co2–Cl3 | 2.2346(7) | Ni–I2 | 2.8289(4) | Zn–I2 | 2.5996(3) | Zn–O1 | 1.964(2) |
| Co1–P1 | 2.2108(7) | Co2–Cl4 | 2.2174(8) | Ni–P1 | 2.1689(9) | Zn–P1 | 2.4609(6) | Zn–O2 | 1.967(2) |
| Co1–P2 | 2.2087(7) | Ni–P2 | 2.1765(9) | Zn–P2 | 2.4798(6) | P1–O1 | 1.478(2) | ||
| P1–O1 | 1.6704(17) | P1–O1 | 1.676(2) | P1–O1 | 1.6579(16) | P1–O3 | 1.612(3) | ||
| P2–O2 | 1.6712(17) | P2–O2 | 1.684(2) | P2–O2 | 1.6644(15) | P2–O2 | 1.484(2) | ||
| P2–O4 | 1.610(2) | ||||||||
| P1–Co1–P2 | 162.83(3) | O3–Co2–Cl4 | 108.20(6) | P1–Ni–P2 | 164.64(4) | P1–Zn–P2 | 135.97(2) | I1–Zn–I2 | 120.186(15) |
| N1–Co1–Cl1 | 94.43(6) | O3–Co2–Cl3 | 103.55(6) | N1–Ni–I1 | 158.56(7) | N1–Zn–I1 | 142.41(4) | O1–Zn–O2 | 105.42(9) |
| N1–Co1–Cl2 | 166.95(6) | Cl4–Co2–Cl3 | 117.50(3) | N1–Ni–I2 | 92.12(7) | N1–Zn–I2 | 100.57(4) | I1–Zn–O1 | 106.59(7) |
| N1–Co1–P1 | 82.69(6) | O3–Co2–Cl1 | 98.32(5) | N1–Ni–P1 | 83.55(8) | N1–Zn–P1 | 70.25(4) | I1–Zn–O2 | 108.06(7) |
| N1–Co1–P2 | 82.97(6) | Cl4–Co2–Cl1 | 113.98(3) | N1–Ni–P2 | 84.14(8) | N1–Zn–P2 | 70.63(4) | I2–Zn–O1 | 107.89(6) |
| P1–Co1–Cl1 | 93.20(3) | Cl3–Co2–Cl1 | 112.71(3) | P1–Ni–I1 | 92.41(3) | P1–Zn–I1 | 98.451(15) | I2–Zn–O2 | 107.79(6) |
| P1–Co1–Cl2 | 97.28(3) | Co1–Cl1–Co2 | 118.25(3) | P1–Ni–I2 | 94.88(3) | P1–Zn–I2 | 107.414(15) | ||
| P2–Co1–Cl1 | 97.28(3) | P2–Ni–I1 | 95.70(3) | P2–Zn–I1 | 100.253(15) | ||||
| P2–Co1–Cl2 | 94.56(3) | P2–Ni–I2 | 94.73(3) | P2–Zn–I2 | 98.851(16) | ||||
| Cl1–Co1–Cl2 | 98.60(3) | I1–Ni–I2 | 109.224(15) | I1–Zn–I2 | 116.974(10) |
The Fe–Br bond lengths differ by ∼0.03 and 0.08 Å in the two crystallographically independent molecules in complex 1 and by ∼0.12 Å in 1′. These Fe–Br distances are in the range of values [2.3960(13)−2.547(11) Å] found in a series of analogous pentacoordinate complexes with a tridentate ligand involving one nitrogen and two phosphorus coordinating atoms [3c,13]. While the two M–X distances which average 2.290(14) Å in the cobalt complex 2 are quite similar and unexceptional [14], they are distinct by 0.06 Å in the zinc analog 5′, and differ considerably in the nickel complex 4′ for which the difference reaches 0.3 Å. The larger Ni–I2 value in the latter case would reflect an almost dissociated bonding, in agreement with the poor solubility of 4 in THF which suggests the formation of a cationic species. This is in line with the discrete cation-anion pair determined for the analogous derivative [Ni(tBuPONOP)Cl][Cl] both in the solid state and in solution. To our knowledge, 4′ is the first neutral pentacoordinate “NiI2” complex containing one nitrogen and two coordinated phosphorus atoms of the type (N)(P)2NiI2, all the other being cationic [15,16]. However, some examples of neutral complexes exist with bromide [17] or chloride [18] as halide ligands. The Ni–I distances in 4′ are longer than the mean distance of 2.48(1) Å in a series of cationic compounds [({Ph2PCH2CH2}2NCH2(p-R-C5H4))NiI]I (R = H, OMe, Br) and [({Ph2PCH2CH2}2NPh))NiI]I [16].
The Fe–N bond lengths in 1 average 2.277(14) Å, a value larger by 0.03 Å than that in 1′. The corresponding Fe–P distances, which are similar and average 2.536(6) Å for the two molecules in 1, are however quite longer than those in 1′ (average 2.454(5) Å). In 4′, the Ni–N and Ni–P distances of 1.917(2) Å and 2.1689(9)/2.1765(9) Å, respectively, compare well with the values 1.871(1) Å and 2.1841(3)/2.1809(3) Å reported for [Ni(tBuPONOP)Cl][BPh4] [9].
In the bimetallic complex 3, the two cobalt ions are connected through the bridging Cl1 atom. The Co1 atom, as in complex 2, is found in a square pyramidal environment with atom Cl1 at the axial position, while Co2 adopts a distorted trigonal pyramidal configuration. The basal plane is composed of the three chloride atoms while the O3 atom is at the apex. The Co2 atom is displaced by 0.5241(5) Å from the basal plane. The Co2–Cl distances are quite similar and average 2.25(3) Å, a value close to the Co1–Cl2 bond length but differing from Co1–Cl1 by ∼0.22 Å. The dissymmetric Co1–Cl1–Co2 arrangement would suggest the formation of the anionic species [CoCl3(THF)]− which would coordinate to the cation [Co(tBuPONOP)Cl]+. In the latter, the mean Co1–P and the Co1–N1 distances of 2.2098(11) Å and 1.9341(18) Å, respectively, are notably shorter than those in the neutral complex 2 which average 2.47(2) Å and 2.20(2) Å (including the two independent molecules), but similar to those found in a series of comparable dicationic complexes [10].
The metal atom in complex 6 is in a pseudo-tetrahedral environment. The two Zn–I bond lengths are quite identical with a mean value of 2.568(5) Å, shorter than that in 5′, in agreement with the lower coordination number. The Zn–O distances are also quite identical, averaging 1.9655(15) Å and they compare well with the distances reported in tetracoordinate zinc complexes such as ZnX2(OPPh3)2 (1.964 and 1.970 Å for X = Br; 1.966 Å for X = Cl) [19], ZnI2(OPPh3)2 (1.967 and 1.977 Å) [20] or Zn(NCS)2(OPPh3)2 (1.938–1.946 Å) [21]. As in the cation [Eu(tBuP(O)CNCP(O))(H2O)3Cl2]+ (tBuP(O)CNCP(O) = (2,6-bis(tert-butylphosphineoxide)CH2)pyridine [11], coordination of the metal center by the two phosphine oxide moieties keeps the metal away from the pyridyl group which is unfit for coordination.
The mean M−P and M−N bond lengths of the analogous complexes 1, 1′, 2, 4′ and 5′ are listed in Table 3 with the ionic radii of the M2+ ions in the coordination number of 5 (value for iron not reported) [22]. As expected, the metal−ligand bond lengths do not follow the evolution of the ionic radii since different covalent interactions develop between the metal ion and the hard or soft ligands. The M−N distances increase in the order Ni < Co < Fe < Zn with a variation of 0.49 Å at most, whereas the M−P distances follow the trend Ni < Fe ∼ Co ∼ Zn with only a gap of ca 0.3 Å between the nickel and zinc complexes. Whereas the nickel ion in 4′ interacts strongly with both the N and P donors, the cobalt and iron derivatives display overall larger M−P/N distances and, in particular, greater M−P versus M−N bond lengths, which suggests a better interaction for these ions with the harder pyridyl moiety. By contrast, 5′ shows the largest M−N/P distances, which would indicate a weaker metal−PONOP covalent interaction for Zn2+ than for the other divalent ions.
Mean M−P and M−N distances (Å) in the complexes 1, 1′, 2, 4′ and 5′.
| Complex | Metal | r2+ (CN = 5) [22] | <M−P> | <M−N> |
| 1 | Fe | Unavailable | 2.536(6) | 2.277(14) |
| 1′ | Fe | – | 2.454(5) | 2.247(2) |
| 2 | Co | 0.67 | 2.47(2) | 2.20(2) |
| 4′ | Ni | 0.63 | 2.173(4) | 1.917(2) |
| 5′ | Zn | 0.68 | 2.470(9) | 2.4697(18) |
2.2 Electrochemical studies
Key elementary steps in organometallic catalysis strongly depend on the redox properties of the metal centers and the electrochemical behavior of the PONOP complexes 1, 2, 4 and 5 was thus investigated (in acetonitrile in the presence of 0.3 M nBu4NPF6 as a supporting electrolyte; the measured potentials were referenced to the ferrocenium/ferrocene couple Fc0/+).
2.2.1 Electrochemical behavior of the free tBuPONOP ligand
The ligand tBuPONOP is characterized by its successive oxidation peaks at EpO,L = +0.79 V/Fc0/+ and EpO′,L = +0.96 V/Fc0/+ (Fig. 8a), and by its 2-electron reduction peak at EpR,L = −3.21 V/Fc0/+ (Fig. 8b).
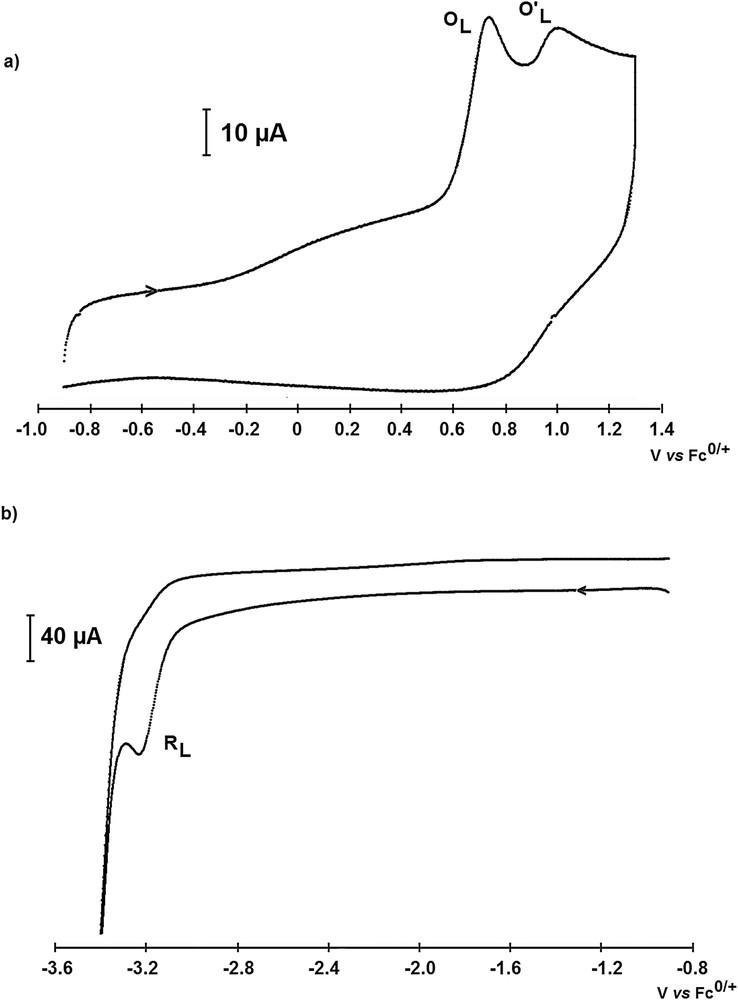
Cyclic voltammetry curves of tBuPONOP, obtained in CH3CN with nBu4NPF6 (0.3 M) as the supporting electrolyte at a vitreous C disk electrode (d = 2 mm) and at a scan rate of 0.5 mV·s−1, at 25 °C.
2.2.2 Electrochemical behavior of Fe(tBuPONOP)Br2 (1)
As depicted in Fig. 9a, the cyclovoltammogram of 1 exhibits two distinct reduction peaks, R1 (EpR1 = −2.19 V/Fc0/+) and R2 (EpR2 = −1.78 V/Fc0/+). The oxidation peaks of the free tBuPONOP ligand are also observed (Fig. 9a), suggesting that 1 dissociates in the electrolyte. This finding is confirmed by the reduction peak at R2, which is attributed to FeBr2 by comparison with an authentic sample (2-electron peak at EpR2 = −1.78 V/Fc0/+). When the first scan was performed towards negative potentials, a large adsorption peak corresponding to the oxidation of electro-generated iron(0) was observed at EpO2 = −0.49 V/Fc0/+, revealing that a low valent iron PONOP complex is unstable in solution. The electrochemical behavior of 1 therefore points to the occurrence of an equilibrium between the PONOP complex, the free ligand and FeBr2 (Scheme 4). The plateau-shaped reduction peak between R2 and R1 moreover attests to the existence of such an equilibrium, which is continuously displaced towards the formation of FeBr2 due to its consumption in the diffusion layer starting at its reduction at R2. Minor peaks at Ep,ox = +0.42 V/Fc0/+ and Ep,red = −0.15 V/Fc0/+ are attributed to the Br− anion resulting from the partial dissociation of FeBr2 or 1 in acetonitrile.
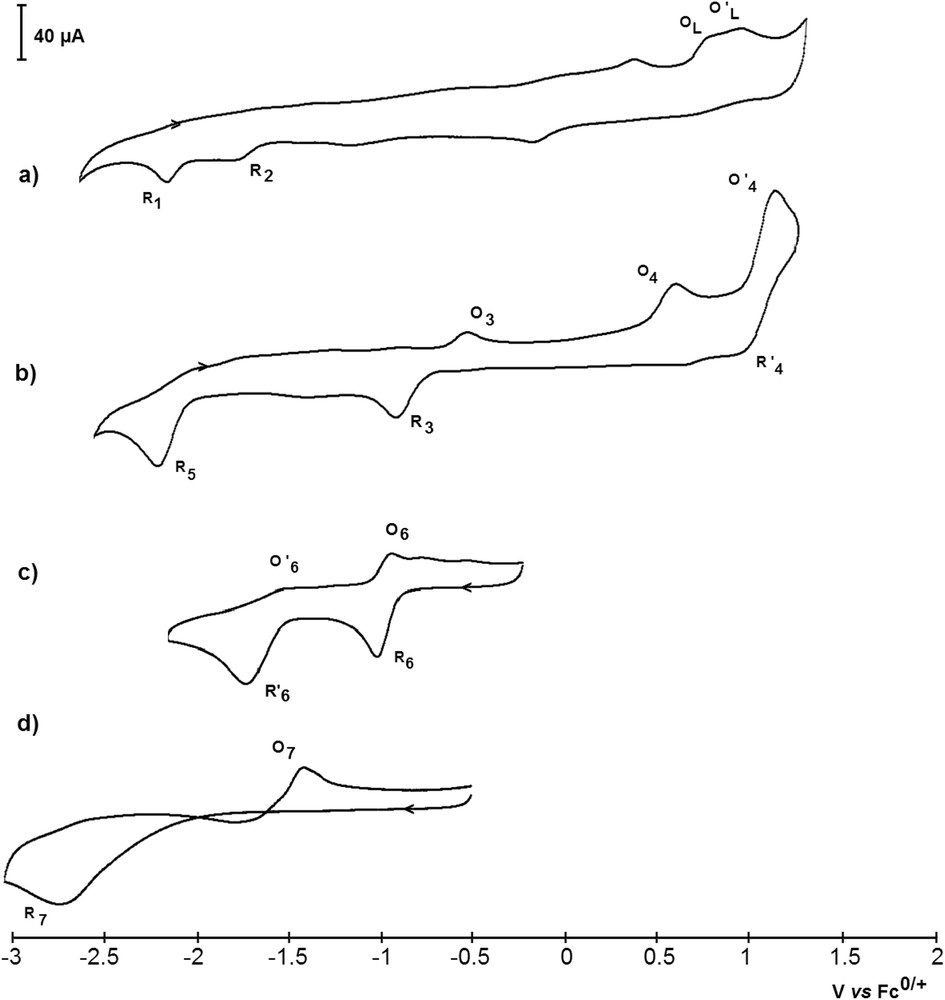
Cyclic voltammetry curves obtained in CH3CN with nBu4NPF6 (0.3 M) as the supporting electrolyte at a vitreous C disk electrode (d = 2 mm) and at a scan rate of 0.5 mV.s−1, at 25 °C. a) Fe(tBuPONOP)Br2 (1) (5.5 mM); b) Co(tBuPONOP)Cl2 (2) (5.5 mM); c) Ni(tBuPONOP)I2 (4) (5.5 mM); d) Zn(tBuPONOP)I2 (5) (5.5 mM).
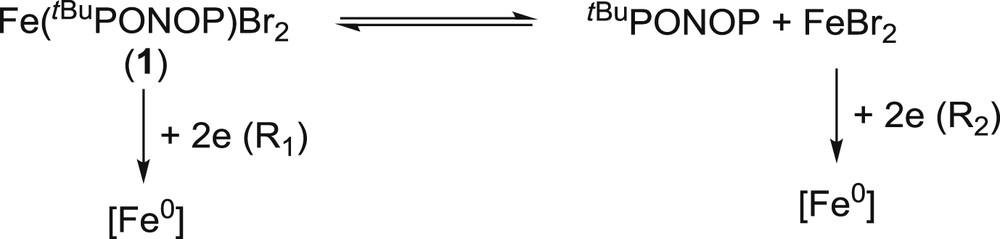
Electrochemical behavior of 1.
2.2.3 Electrochemical behavior of Co(tBuPONOP)Cl2 (2)
Examination of the cyclic voltammogram of cobalt complex 2 indicates the presence of one quasi-reversible redox system (EpR3 = −0.94 V/Fc0/+ and EpO3 = −0.54 V/Fc0/+; the resting potential being evaluated at ca. −0.7 V/Fc0/+), one quasi-reversible system (EpO′4 = +1.13 V/Fc0/+ and EpR′4 = +1.01 V/Fc0/+), and two irreversible peaks (EpO4 = +0.61 V/Fc0/+ and EpR5 = −2.30 V/Fc0/+) (Fig. 9b). No free tBuPONOP ligand was detected. The three peaks O4, O′4 and R′4 were attributed to the CoII anion by comparison with reported data [23]. This anion exhibits two distinct oxidation peaks EpO4 and EpO′4. The peak at EpO4 = +0.61 V/Fc0/+ is close to the oxidation potential of free Cl−, which accounts for a dissociation of the coordinated Cl− anions of 2 to afford the dicationic complex [(tBuPONOP)Co]2+ (22+) (vide infra). The oxidative event at EpO′4 = +1.13 V/Fc0/+ results in the formation of CoIII, which is reduced at EpR′4 = +1.01 V/Fc0/+. On the basis of the reduction current peak ratio ipR5/ipR3 = 1.5 > 1, it can be ascertained that the reduction peak R5 does not correspond to the reduction of a species electro-generated in the diffusion layer at R3 [24]. Since no free tBuPONOP ligand was detected in either the oxidation or reduction mode (see Table 5), and since both reduction peaks R5 and R3 disappeared upon the addition of an excess (30 mM) of nBu4NCl, it is likely that complex 2 first dissociates into Cl− and [(tBuPONOP)Co]2+ (22+), the latter partially leading to the formation of CoII and [(tBuPONOP)2Co]2+ via ligand scrambling (Scheme 5) [25]. The measured reduction potential of 22+ (EpR3 = −0.94 V/Fc0/+) is also consistent with that measured for the complex [Co(PONOP)][BF4]2 [10].
Measured oxido-reduction potential peaks of the investigated complexes.
| Complexes | ||
| (1) | −2.19 (R1) | |
| −1.78 (R2) | ||
| [Fe0] | −0.49 (O2) | |
| (2) | −0.94 (R3) | |
| −0.54 (O3) | ||
| +0.61 (O4) | ||
| +1.13 () | ||
| +1.01 () | ||
| [Co(tBuPONOP)2]2+ | −2.30 (R5) | |
| Cl− (from NCl) | +0.64 | |
| Ni(tBuPONOP)I2 (4) | −1.07 (R6) | |
| NiI(tBuPONOP)I | −0.99 (O6) | −1.75 () |
| Ni0(tBuPONOP) | −1.51 () | |
| Zn(tBuPONOP)I2 (5) | −2.67 (R7) | |
| [Zn0] | −1.44 (O7, broad) | |
| tBuPONOP | +0.79 (OL); +0.96 () | −3.21 (RL) |
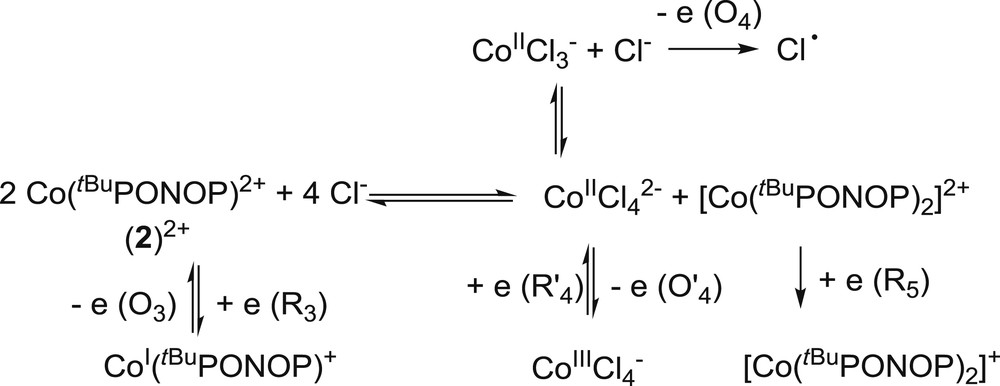
Ligand scrambling from 2 and electrochemical behavior of the subsequently formed complexes.
It is notable that, although complex 2 is blue in the solid state and in THF solution, it turns spontaneously purple in polar and coordinating media (e.g., acetonitrile, THF or acetonitrile solutions of nBu4NPF6 0.3 M). Consequently, dissociative properties and ionic strength of the medium seem to play a key role in the exchange process described above.
2.2.4 Electrochemical behavior of Ni(tBuPONOP)I2 (4)
The nickel complex 4 is characterized by two successive quasi-reversible monoelectronic reduction peaks corresponding to the reduction of the NiII center to NiI (EpR6 = −1.07 V/Fc0/+) and Ni0 (EpR′6 = −1.75 V/Fc0/+) (Fig. 9c and Scheme 6). The first reduction event is partly reversible (EpO6 = −0.99 V/Fc0/+), whereas the second one is almost irreversible (EpO′6 = −1.51 V/Fc0/+). Characteristic oxidation peaks of I− (Ep = −0.1 V/Fc0/+) and electro-generated I2 (Ep = +0.25 V/Fc0/+) are detected on the reverse scan, which can be explained by the displacement of I− with acetonitrile molecules in the nickel coordination sphere.
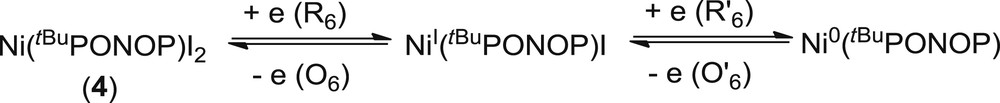
Electrochemical behavior of 4.
2.2.5 Electrochemical behavior of Zn(PONOP)I2 (5)
Complex 5 is reduced in a bi-electronic process at EpR7 = −2.67 V/Fc0/+ (Fig. 9d). A large adsorption peak EpO7 = −1.44 V/Fc0/+ is detected on the reverse scan, showing that the electro-generated zinc(0) species is unstable and led to a metallic deposit on the surface of the electrode (Scheme 7). 5 is thus the less easily reducible of the series, which is in agreement with the strong reductive power of zinc(0) complexes and the instability of zinc(I) complexes.

Electrochemical behavior of 5.
2.3 Behavior of 1, 2, 4 and 5 in catalytic dehydrogenation of formic acid
Because of its high hydrogen storage capacity (4.4 wt %), formic acid is an attractive energy carrier. It could indeed serve as the intermediate for the production of methanol and its derivatives or directly as a H2-vector. Controlling and improving the rate of its dehydrogenation to H2 and CO2 (H2 release) is therefore a current challenge to meet the requirement for this technology [26]. This reaction has been widely investigated with precious metal catalysts such as gold, iridium, and ruthenium [27] that lead to high turnover numbers (TONs). Only a few examples of catalysts involving earth abundant and cheap metals (Fe, Ni and Al) have been recently described [28–31]. With the iron catalyst ({iPr2PCH2CH2}2NH)FeH(O2CH), bearing a PNP-type pincer ligand [30], a TON close to 1,000,000 was reported by the groups of Hazari and Schneider while the aluminum and nickel catalysts show a lower TON of 5200 and 626, respectively [28,29]. We thus investigated the activity of the different M(tBuPONOP)X2 complexes in formic acid dehydrogenation.
Heating 0.16 mmol of HCOOH in THF at 150 °C for 24 h with 1 mol% of 1, 2, 4 or 5 gave no conversion. However replacing formic acid with an azeotrope solution of formic acid/triethylamine (5:2) yielded complete conversion of formic acid to H2 and CO2 after 8 h at 150 °C in THF with 1 mol% of Ni(tBuPONOP)I2 (4), while 1, 2 and 5 still proved unreactive under these conditions. No by-product was observed by 1H or 13C NMR spectroscopy (Scheme 8).
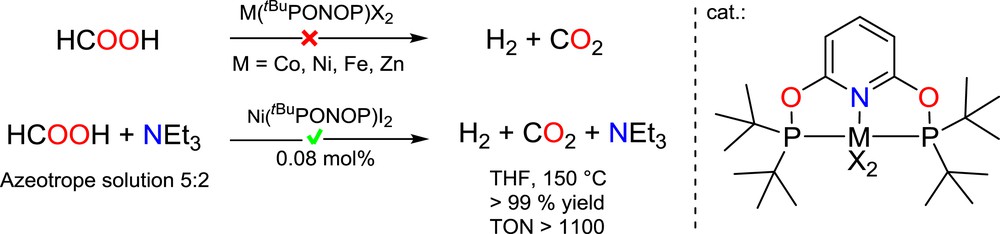
Catalytic dehydrogenation of formic acid.
If 4 was not active at temperatures below 140 °C, TONs up to 1143 could be attained after 19 h at 150 °C while the formic acid/triethylamine (5:2) mixture is not degraded under such conditions in the absence of 4. Complex 4 is only the second example of a nickel catalyst that efficiently catalyzes the dehydrogenation of formic acid, the other one being Ni(tBuPCP)H (tBuPCP = {C6H3-2,6-(PtBu2)2}), which affords a TON of 626 after 3 h at 80 °C with dimethyl-n-octylamine as the solvent [29]. Based on the experimental data and previous literature studies [27–31], a schematic mechanistic proposal is depicted in Scheme 9. Given the strong coordination ability of formate ions, the iodide anions in 4 are likely displaced in a formate medium to yield a nickel(II) bis(formate) complex (A). Decarboxylation of a formate might require the decoordination of one formate ligand to generate an unsaturated nickel(II) cationic center (B), able to promote C–H bond activation via β-hydride elimination or direct hydride abstraction from a free formate anion; in line with the ability of the PONOP ligand to stabilize a cationic nickel(II) center (vide supra). The resulting (PONOP)Ni(OCHO)(H) complex (C) then affords H2 and the catalytically active bis(formate) complex by protonolysis of the hydrido-nickel functionality.
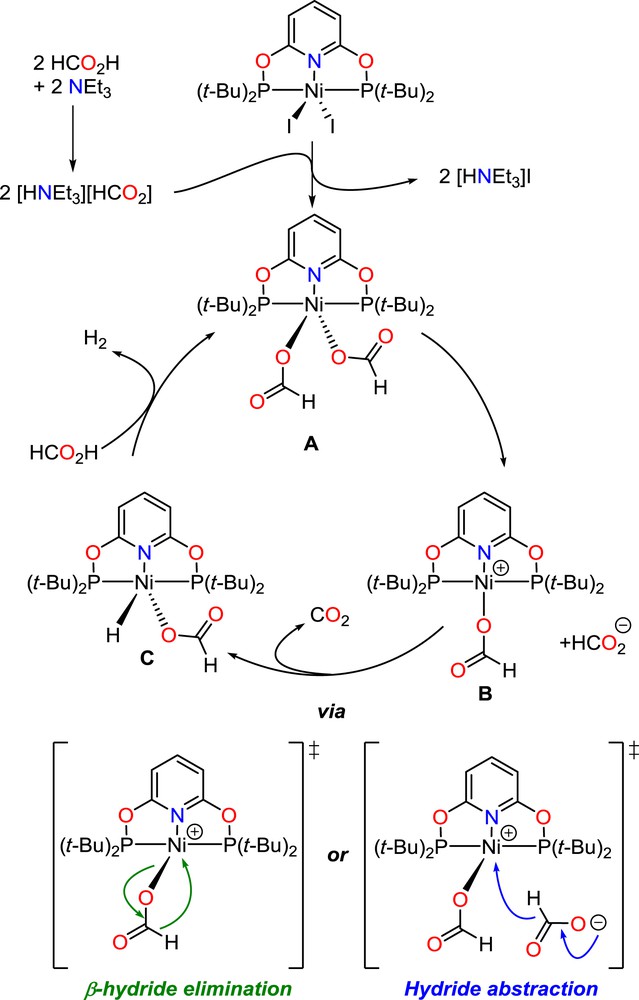
Mechanistic proposal for the catalytic dehydrogenation of formic acid with 4.
3 Conclusion
In conclusion, preparation and full characterization (crystal structure, NMR) of metal complexes of the Fe2+, Co2+, Ni2+ and Zn2+ ions have been described with the RPONOP (R = tBu, iPr) pincer ligands and their electrochemical behavior in organic solvents has been studied. In the presence of a strongly coordinating medium, the compounds are prone to dissociation of the RPONOP ligand or ligand scrambling, except the nickel compound which is stable. They are scarce examples of M(RPONOP)X2 complexes with first row elements. While the Fe2+, Co2+ and Zn2+ complexes displayed no activity in dehydrogenation of formic acid, the Ni(RPONOP)I2 proved active with TONs up to 1143 after 19 h at 150 °C. Complex 4 is a rare example of a cheap metal catalyst for this transformation. Such pincer complexes, as shown in a number of studies, might display activity for bond activation and catalysis, and they are currently tested for the activation of CO2.
4 Experimental
The complexes described below are oxygen and moisture sensitive. Syntheses and manipulations of the compounds were conducted under an ultra high purity argon atmosphere with rigorous exclusion of air and water, using Schlenk-vessel and vacuum-line techniques or in a dry glovebox.
The solvents, toluene, pentane and tetrahydrofuran were dried over a mixture of sodium-benzophenone, and over KH for acetonitrile, and were distilled immediately before use. Deuterated tetrahydrofuran and methanol (Eurisotop) were distilled over potassium and stored over 3 Å molecular sieves (for CD3OD) in a glovebox. The white solid 2,6-bis(di-tert-butylphosphinito)-pyridine) (tBuPONOP) and the colorless liquid 2,6-bis(di-isopropylphosphinito)-pyridine) (iPrPONOP) were prepared by literature methods [4,8,9]. The supporting electrolyte nBu4NPF6, for the electrochemical studies was purchased from Sigma–Aldrich and used as received. The 1H, 31P and 13C{1H} NMR spectra were recorded on a Bruker DPX 200 MHz instrument and referenced internally using the residual protio solvent resonances relative to tetramethylsilane (δ 0). The spectra were recorded at 21 °C when not otherwise specified. The 1H NMR signals of THF-d8 are referenced at δ 3.58 and 1.73 and those of methanol-d8 at 4.87 and 3.31 whereas the 13C NMR signals are at δ 67.57 and 25.37 for THF and δ 49.0 for methanol. The 31P NMR signals were referenced from H3PO4 (δ 0 ppm). Elemental analyses were performed by Medac Ltd at Chobham (Surrey, UK) or by the Service de Microanalyse of the Institut de Chimie des Substances Naturelles of the CNRS at Gif-sur-Yvette (France).
4.1 Synthesis and crystals of Fe(tBuPONOP)Br2 (1) and Fe(iPrPONOP)Br2 (1′)
- a) A 50 mL round bottom flask was charged with tBuPONOP (500 mg, 1.25 mmol) and FeBr2 (270 mg, 1.25 mmol) and THF (15 mL) was distilled in it under reduced pressure at −78 °C. After stirring at 40 °C for 1 h under argon and then at room temperature for 15 h, the solvent was evaporated off. The resulting beige solid was then washed with pentane (2 × 5 mL) and dried under vacuum for 10 h, affording a beige powder (592 mg, 77%).1H NMR (THF-d8): δH −20.39 (s, w1/2 = 42 Hz, 1H), 16.89 (s, w1/2 = 140 Hz, 36H), 54.01 (s, w1/2 = 46 Hz, 2H). 13C NMR (THF-d8): δC −174.5, −5.95, 128.8, 187.7 (s, w1/2 = 80 Hz), 274.7; 31P NMR (THF-d8): δ 73.26, 135.64 (s, w1/2 = 185 Hz), 156.82 (s, w1/2 = 180 Hz). Anal. Calcd for C21H39Br2FeNO2P2 (MW = 615.15 g mol−1): C 41.00, H 6.39, N 2.28. Found: C 41.53, H 6.85, N 2.17.
- b) FeBr2 (10 mg, 4.6 10−5 mol) and iPrPONOP (20 mg, 5.8 10−5 mol) were charged in a NMR tube with 1 mL THF. The suspension was heated at 80 °C for 15 h, affording a yellow solution. After evaporation of the THF, washing with pentane (2 × 1 mL) and drying under vacuum for 5 h, Fe(iPrPONOP)Br2 (1′) was isolated as a yellow solid in almost quantitative yield (∼28 mg, 90%). Large yellow crystals of 1′ were obtained by slow diffusion of pentane into a THF solution of the product. 1H NMR (THF-d8): δH 53.23 (s, w1/2 = 120 Hz, 2H), 12.14 (s, w1/2 = 250 Hz, 12H), 10.35 (s, w1/2 = 230 Hz, 12H), −21.69 (s, w1/2 = 100 Hz, 1H). No conclusive 13C NMR data could be obtained for this compound. 31P NMR (C6D6): δ −56.7 (s), −7.5 (s, w1/2 = 220 Hz), 144.8 (s, w1/2 = 180 Hz),
4.2 Synthesis of Co(tBuPONOP)Cl2 (2) and crystals of the binuclear complex Co(iPrPONOP)Cl(μ-Cl)CoCl2(THF) (3)
A 50-mL round bottom flask was charged with tBuPONOP (200 mg, 0.50 mmol) and CoCl2 (65 mg, 0.50 mmol) and THF (10 mL) was distilled in it under reduced pressure at −78 °C. After stirring at 40 °C for 1 h and then at room temperature for 15 h, the solvent was evaporated, affording a blue solid which was extracted with toluene (15 mL). After evaporation of the solvent, the solid was further washed with pentane (2 × 5 mL) and dried under vacuum for 10 h, and 2 was obtained as a blue-purple powder (220 mg, 83%). No NMR spectra could be obtained for this paramagnetic species. Anal. Calcd for C21H39Cl2CoNO2P2 (MW = 529.33 g·mol−1): C 47.65, H 7.43, N 2.65. Found: C 47.26, H 7.76, N 2.66. A suspension of CoCl2 (10 mg, 7.7 10−5 mol) and tBuPONOP (30.7 mg, 3.2 10−5 mol) in 0.5 mL THF was heated at 80 °C for 2 h giving a blue-purple solution. Large dark blue crystals of 2 were obtained by slow diffusion of pentane into a THF solution of the product.
In an attempt to crystallize a mixture of iPrPONOP and excess (ca 2 equiv.) of CoCl2 in THF, by slow diffusion of pentane into the solution, large dark brown crystals of the dinuclear complex Co(iPrPONOP)Cl(μ-Cl)CoCl2(THF) (3) were obtained.
4.3 Synthesis of Ni(tBuPONOP)I2 (4) and Ni(iPrPONOP)I2 (4′)
- a) A 50 mL round bottom flask was charged with tBuPONOP (500 mg, 1.25 mmol) and NiI2 (391 mg, 1.25 mmol) and THF (15 mL) was distilled in it under reduced pressure at −78 °C. After 1 h of stirring at 40 °C and then for 15 h at room temperature, an abundant dark red solid deposited. Evaporation of the solvent and washings of the solid with pentane (2 × 5 mL) led, after 10 h of drying under vacuum, to a dark red powder of 4 (703 mg, 79%). 1H NMR (CD3OD): δH 1.65 (vt, 36H), 7.12 (d, J = 8.2 Hz, 2H), 8.22 (t, J = 8.2 Hz, 1H). 31P NMR (MeOD): δ 203.84 (s). 13C NMR (CD3OD): δC 28.4, 43.5 (t, J = 6.1 Hz), 105.6, 150.0, 165.6–165.9 (m). Anal. Calcd for C21H39I2NiNO2P2 (MW = 712.00 g·mol−1): C 35.43, H 5.52, N 1.97. Found: C 35.13, H 5.32, N 2.50.
- b) NiI2 (10 mg, 3.2 10−5 mol), iPrPONOP (11 mg, 3.2 10−5 mol) and 1 mL THF were charged in a NMR tube. The suspension was heated at 90 °C for 15 h, affording a red-violet solution. After evaporation of the THF, the red solid Ni(iPrPONOP)I2 (4′) was washed with pentane (2 × 1 mL) and dried under vacuum (∼95%, 20 mg). Large red black crystals of 4′ were obtained by slow diffusion of pentane into a THF solution. 1H NMR (THF-d8): δH 7.87 (t, J = 8.1 Hz, 1H), 6.68 (d, J = 8.1 Hz, 2H), 3.52–3.23 (m, 4H), 1.60–1.48 (m, 24H); 13C NMR (THF-d8): δC 163.8 (m), 147.1, 143.8, 105.5 (m), 102.1 (m), 35.7 (m), 33.2 (m), 19.91 (dt, J = 25.9, 4.7 Hz), 17.36 (dt, J = 10.3, 5.3 Hz). 31P NMR (C6D6): δ 197.2 (s).
4.4 Synthesis of Zn(tBuPONOP)I2 (5) and Zn(iPrPONOP)I2 (5′). Crystals of Zn(tBuP(=O)ONOP(=O))l2·0.25THF (6·0.25THF)
- a) A 50-mL round bottom flask was charged with tBuPONOP (500 mg, 1.25 mmol) and ZnI2 (400 mg, 1.25 mmol) and THF (15 mL) was distilled in it under reduced pressure at −78 °C. After stirring at 40 °C for 1 h and then at room temperature for 15 h, the solvent was evaporated to give a white solid. The latter was washed with pentane (2 × 5 mL) and dried under vacuum yielding a white powder of 5 (725 mg, 82%). 1H NMR (THF-d8): δH 7.95 (s, w1/2 = 60 Hz, 1H), 7.11 (s, w1/2 = 101 Hz, 2H), 1.35 (s, 18H), 1.29 (s, 18H). 13C NMR (THF-d8): δC 27.8, 28.05, 33.6, 34.8, 106.2, 106.6, 146.9, 162.9. 31P NMR (C6D6): δ 69.1, 107.1 (s, w1/2 = 90 Hz), 172.1 (s, w1/2 = 100 Hz). Anal. Calcd for C21H39NO2P2ZnI2 (MW = 718.68 g·mol−1): C 35.10, H 5.47, N 1.95. Found: C 34.57, H 5.69, N 2.10.
In an attempt to crystallize 5 (in an NMR tube), a few colorless crystals were collected after a few weeks at room temperature. The X-ray diffraction study showed the latter to be the THF adduct [Zn(tBuP(O)ONOP(O))Cl2]·THF (6·THF) resulting from the oxidation of the phosphine ligand by adventitious traces of air and water.
- b) ZnI2 (10 mg, 3.1 10−5 mol) and iPrPONOP (11 mg, 3.2 10−5 mol) were charged in a NMR tube and 1 mL THF was condensed in it. The suspension was warmed at 90 °C for 15 h, affording a colorless solution. After evaporation of the THF, the solid was washed with pentane (2 × 1 mL) and dried under vacuum affording pure 5′ (∼95%, 20 mg). Colorless crystals of 5′ were obtained by slow diffusion of pentane into a THF solution. 1H NMR (THF-d8): δH 7.85 (t, J = 7.9 Hz, 1H), 6.70 (d, J = 7.9 Hz, 2H), 2.85–2.43 (m, 4H), 14.60–1.22 (m, 24H). 13C NMR (THF-d8): δC 160.2 (m), 146.1 (m), 129.1 (m), 106.3, 28.1, 17.6 (m). 31P NMR (TDF): δ 63.1 (s) and 95.1 (s, w1/2 = 216 Hz).
4.5 Catalytic dehydrogenation of formic acid
In a J. Young NMR tube, formic acid (6 μL, 0.16 mmol) and NEt3 (9 μL, 0.06 mmol) were added to a solution of the catalyst in THF-d8 (0.3 mL), and heated at 150 °C. The solution was periodically cooled down to room temperature and monitored by NMR spectroscopy.
4.6 Electrochemical experiments
Cyclic voltammetry experiments were carried out in a three-electrode cell under argon flow (working electrode: vitreous C, d = 2 mm; auxiliary electrode: Pt wire; quasi-reference electrode: silver wire), and cyclovoltammograms were recorded with a PAR Versastat 4 potentiostat. nBu4NPF6 was used as the supporting electrolyte (0.3 M) with CH3CN (freshly distilled on KH) as the solvent (6 mL). Half-peak potentials were referenced to the ferrocenium/ferrocene couple by adding ferrocene at the end of each experiment.
4.7 Crystallography
The data were collected at 150(2) K on a Nonius Kappa-CCD area detector diffractometer [32] using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The crystals were introduced into glass capillaries with a protective coating of Paratone-N oil (Hampton Research). The unit cell parameters were determined from ten frames, and then refined on all data. The data (combinations of φ- and ω−scans with a minimum redundancy of 4 for 90% of the reflections) were processed with HKL2000 [33]. Absorption effects were corrected with SCALEPACK [33]. The structures were solved either by direct methods with SHELXS-97 [34] or with SHELXT [35], expanded by subsequent difference Fourier synthesis and refined by full-matrix least-squares on F2 with SHELXL [34]. All non-hydrogen atoms were refined with anisotropic displacement parameters. Complexes 1 and 2 as well as complexes 1′, 4′ and 5′ are isomorphous. In complex 2, restraints had to be applied for some bond lengths and/or displacement parameters for atom O1 and the two tert-butyl groups C6–C9 and C10–C13, which seem to be affected by unresolved disorder; the highest residual electron density peaks are located near these tert-butyl groups. The crystals of 6·0.25THF desolvate very quickly when out of their native solution; the THF solvent molecule, located on a twofold rotation axis, is very badly resolved and it has been refined as a complete molecule, with an occupancy parameter of 0.25 in order to retain acceptable displacement parameters, and with restraints on bond lengths, angles and displacement parameters. In all compounds, the hydrogen atoms were introduced at calculated positions and were treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom (1.5 for CH3, with optimized geometry). The drawings were done with ORTEP-3 [36]. Crystal data, structure refinement parameters and CCDC deposition numbers are given in Table 4. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystal data and structure refinement details.
| 1 | 1′ | 2 | 3 | 4′ | 5′ | 6·0.25THF | |
| Chemical formula | C21H39Br2FeNO2P2 | C17H31Br2FeNO2P2 | C21H39Cl2CoNO2P2 | C21H39Cl4Co2NO3P2 | C17H31I2NNiO2P2 | C17H31I2NO2P2Zn | C22H41I2NO4.25P2Zn |
| M (g mol−1) | 615.14 | 559.04 | 529.30 | 675.13 | 655.88 | 662.54 | 768.67 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | Pī | P21/c | Pī | P21/n | P21/c | P21/c | C2/c |
| a (Å) | 10.9406(5) | 15.7751(6) | 10.8095(6) | 8.1410(4) | 15.4659(4) | 15.8067(4) | 29.7668(12) |
| b (Å) | 16.0526(8) | 10.2847(8) | 15.5895(8) | 36.2450(14) | 10.7081(6) | 10.6976(4) | 10.9094(4) |
| c (Å) | 16.0999(5) | 14.4404(10) | 16.0037(5) | 10.8026(6) | 14.2027(7) | 14.7256(5) | 22.4171(7) |
| α (°) | 98.946(3) | 90 | 97.083(3) | 90 | 90 | 90 | 90 |
| β (°) | 95.988(3) | 97.108(4) | 97.155(3) | 111.857(3) | 98.276(3) | 98.438(2) | 118.735(2) |
| γ (°) | 99.434(2) | 90 | 98.962(2) | 90 | 90 | 90 | 90 |
| V (Å3) | 2730.8(2) | 2324.8(3) | 2615.0(2) | 2958.4(3) | 2327.62(18) | 2463.05(14) | 6383.2(4) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 8 |
| Dcalcd (g cm−3) | 1.496 | 1.597 | 1.344 | 1.516 | 1.872 | 1.787 | 1.600 |
| μ (Mo Kα) (mm−1) | 3.612 | 4.234 | 1.000 | 1.614 | 3.635 | 3.644 | 2.829 |
| F(000) | 1256 | 1128 | 1116 | 1392 | 1280 | 1288 | 3040 |
| Reflections collected | 162,155 | 75,010 | 141,945 | 84,509 | 82,978 | 88,027 | 120,400 |
| Independent reflections | 16,642 | 5994 | 9908 | 7621 | 7090 | 7510 | 8242 |
| Observed reflections [I > 2σ(I)] | 11,934 | 3986 | 7850 | 4805 | 4625 | 5834 | 6324 |
| Rint | 0.044 | 0.044 | 0.045 | 0.054 | 0.043 | 0.036 | 0.042 |
| Parameters refined | 547 | 234 | 541 | 306 | 234 | 234 | 337 |
| R1 | 0.034 | 0.035 | 0.055 | 0.039 | 0.035 | 0.027 | 0.037 |
| wR2 (all data) | 0.080 | 0.079 | 0.159 | 0.094 | 0.085 | 0.062 | 0.103 |
| S | 0.994 | 0.958 | 1.069 | 0.961 | 0.921 | 0.996 | 1.081 |
| (e Å−3) | −0.74 | −0.63 | −0.98 | −0.69 | −1.15 | −0.98 | −0.86 |
| (e Å−3) | 0.81 | 0.58 | 2.25 | 0.65 | 0.85 | 0.50 | 1.18 |
| CCDC deposition number | 1050104 | 1050105 | 1050106 | 1050107 | 1050108 | 1050109 | 1050110 |
Acknowledgments
This work was supported financially by the CEA, CNRS, CHARMMMAT Laboratory of Excellence, and the European Research Council (ERC starting grant agreement no. 336467). T.C. thanks the Fondation Louis D.–Institut de France for its support.