

Pathological calcification can occur at multiple anatomical sites and under a variety of physiological conditions. The composition and structure of the resultant calcified materials may provide clues to the pathological causes and molecular mechanisms leading to calcification, and possibly suggest avenues for therapeutic intervention. Basic information on the composition of these materials can be obtained with simple chemical analyses such as determination of elemental composition. However, information which may be critical to identifying pathological causation, such as biomineral crystal structure, requires more sophisticated physicochemical methodologies which provide structural information to complement chemical analysis [1,2].
X-ray diffraction is a powerful tool for defining the spatial arrangement of atoms in crystalline or multi-crystalline materials. It can be used to determine the crystal structure, and by comparison with published database “fingerprints” of chemically or synthetically well-defined materials, chemical composition in favourable cases. It is widely used in characterizing the crystalline components of biominerals including sialoliths [3–5]. Fourier transform infrared spectroscopy (FTIR) can also be used to identify the structure and physical phases of the material by probing the molecular vibrational modes which are functions of molecular and crystal structures [6]. In pure organic phases or mixed organic–inorganic composites, the amide stretches due to the C–O and C–N backbone of a peptide, for instance, gives distinctive IR spectra which can be related to the backbone conformation. Common pathological biomineral phases, such as calcium oxalate solvates and polymorphs widespread in kidney stones, can be distinguished by zero-crossing-point first-derivative spectrophotometry [7], and different apatites and other calcium phosphate materials can be identified by assigning P–O stretches. Scanning electron microscopy (SEM) examines the surface of the calcified material, and is a useful method for characterizing the crystallite morphology [8]. These important physical methods, and others such as powder neutron diffraction (PND) and X-ray fluorescence (XRF), have long been widely used to study many kinds of pathological calcified materials to determine the molecular composition and establish possible links between structural and chemical properties and the etiopathogenesis of biomineralization [9].
Solid-state nuclear magnetic resonance spectroscopy (ssNMR) is widely used to study solid material structures by virtue of its sensitivity to the local environment of each NMR receptive atom [10]. Unlike X-ray scattering techniques, which rely on molecular crystallinity and long range order for interpretable data, ssNMR (like FTIR) can effectively study materials such as amorphous solids without long range order and provide information from which the local structure can be inferred. In addition NMR data are often easier to interpret in terms of molecular structure as each chemically unique atom gives rise to a single signal the frequency of which directly depends, often in an interpretable way, on its chemical environment. Considering the example of a protein, 13C atoms in amino acids containing methyl groups (Ala, Ile, Leu, Met, and Val) will give rise to signals which are distinct from each other, and from other sp3 carbon atoms, which are distinct from aromatic carbons (His, Phe, Tyr, and Trp), which in turn are distinct from backbone and sidechain amide carbons, and acidic carboxylate carbons. Similarly in phosphatic biominerals different phosphate environments, such as the groups in apatite, give rise to different 31P signals from , for instance brushite. Under favourable circumstances NMR can yield complete atomic level molecular and even crystal structures, although this is far from realization for complex mixtures such as pathological calcifications. The potential of NMR in the study of the composition of renal calculi has been demonstrated by Bak et al. [11] who distinguished from each other the common organic (calcium oxalates and uric acid) and inorganic (apatite, struvite, and brushite) constituents. Apatitic stones in particular have also been shown by ssNMR to contain polysaccharides and proteins of highly variable apparent composition [12] and citrate [13] in variable proportions. Using the ssNMR technique, REDOR atomic level (sub-nanometre) interactions between urolith phosphates, and the polysaccharides, proteins and (when present) citrate, were also demonstrated, prompting the suggestion that such interactions might reflect processes central to urinary calculus biogenesis [12].
In this paper, we apply ssNMR to extend the characterization of the composition of, and some aspects of the mineral–organic interactions present in, salivary stones (sialoliths) [14,15], to our knowledge for the first time in these pathological materials. We show that this technique constitutes a unique tool, complementary to the other techniques described above, to address many aspects of the organic constituents of pathological calcifications. The necessary background to the methods we use – cross-polarization (CP), magic angle spinning (MAS) and 13C{31P} REDOR [12], and dipolar dephasing (DD) [13] – is described in the respective references just cited. Amino acid analyses were also performed on four of the samples, and the salivary protein statherin was identified as the most likely major organic component of the stones, consistent with the signals observed in the 13C ssNMR. The function of the statherin is widely believed to be, at low concentrations, the inhibition of propagation of calcium phosphate crystallization and crystal growth, exerted by binding strongly with the nucleating mineral surface. However at high protein concentrations such proteins can solidify and actually act as crystallization promoters via heterogeneous nucleation processes. It is evidently the latter process which predominates in the stone materials we have studied.
1 Materials and methods
Eight sialoliths were from the Wharton's ducts of patients undergoing surgical sialolithotomy and they were initially examined by FTIR spectroscopy at the Hôpital Tenon, Paris. They were used in all analytical procedures with informed patient consent and institutional ethics approval.
Each one was studied using a stereomicroscope to define morphological type [16] and a Spectrospin Vector 22 Fourier transform infrared spectrometer (Bruker, Karlsruhe, Germany) to determine its mineral composition and detect the presence of proteins and lipids [17].
All solid-state NMR measurements were performed on a Bruker 400 MHz Avance II spectrometer, at frequencies of 400.42, 162.1, and 100.6 MHz for 1H, 31P, and 13C, respectively, using standard Bruker double and triple resonance magic angle spinning (MAS) probes. Samples were powdered using a pestle and mortar, packed into 4 mm zirconia rotors and spun at 12.5 kHz, except for the DD experiments where a spin rate of 6.25 kHz was used. Samples were characterized using cross-polarisation magic angle spinning (CP-MAS; 1H 90° pulse length 2.5 μs, 31P CP contact time 10 ms, 1H–13C CP contact time 2.5 ms, spin lock field strength 70 kHz, SPINAL64 broadband decoupling at 100 kHz 1H field, recycle time 2 s), DD (same 1H–13C CP parameters, 13C refocussing π-pulse at the centre of the DD period, DD time 100 μs) and REDOR (same 1H–13C CP parameters, 10 ms dephasing time, interpulse spacing in 31P π pulse train 80 μs synchronized with the MAS period). 31P and 13C chemical shifts were externally referenced using commercial macrocrystalline HAp (Fluka) at a shift of 2.6 ppm relative to 85% phosphoric acid at 0 ppm, and the methylene signal of the α-polymorph of glycine (Sigma) at 43.1 ppm relative to TMS at 0 ppm, respectively. Sample masses available for the study varied between ca. 100 and only a few milligrams; accordingly acquisition times for the most demanding experiment, the REDOR, varied from overnight to three days. Other experiments could be correspondingly shorter, typically a few hours to overnight.
2 Results
We were able to carry out ssNMR on eight sialoliths. 31P NMR confirms the apatitic nature of the stones and two spectra are overlaid in Fig. 1. Some stones showed only a single signal at the chemical shift expected for pure HAp, broadened by environmental heterogeneity on account of the nanocrystalline nature of the mineral. Other stones showed distinct shoulders at low frequency of this apatite signal, the origin of which is unclear but which may reflect substitutions of minor cations (e.g., Mg2+) and anions (e.g., ) into the calcium phosphate lattice, the presence of minor hydrogen phosphate species, and/or interactions with organic macromolecules [18]. Fig. 1 illustrates these two extremes. Fig. S1 in the online supporting information summarizes the 31P data from all eight samples.
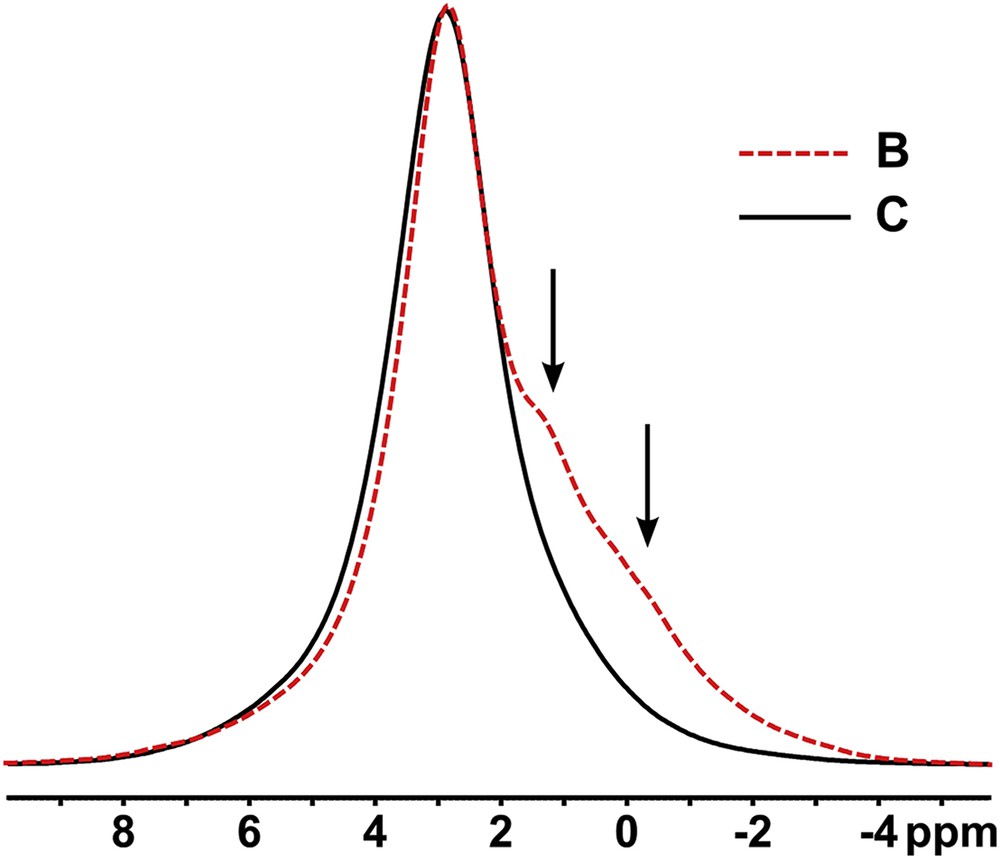
Two representative 31P sialolith spectra, one (sample C) showing no shoulders, the other (sample B) prominent shoulders (arrowed) at low frequency of the main apatite signal at ca. 2.6 ppm. All samples referred to throughout the paper are lettered (A, B, etc.) alphabetically in the order in which they were received and anonymized.
Typical 13C spectra are shown in Fig. 2, along with some assignments of relevant signals to chemical functionalities; spectra from all eight samples are overlaid for comparison in online supporting information Fig. S2. The spectral intensities suggest that organic materials are much more abundant relative to mineral than is usual in apatitic kidney stones; it is also of a rather more uniform composition. While it is impossible to identify specific proteins or other biomacromolecules precisely by our ssNMR data, the spectra do provide some compositional information. In particular the unusually intense peaks (relative to the spectrum as a whole, and to a typical protein) between 110 and 160 ppm indicate that a high proportion of the proteins in these samples have abundant His, Phe, and Tyr (and possibly Arg, signal at ca. 156 ppm) contents. There is no sharp signal from the γ-carbon of collagen hydroxyproline at ca. 70 ppm, suggesting that the proteins are not of collagenous origin.
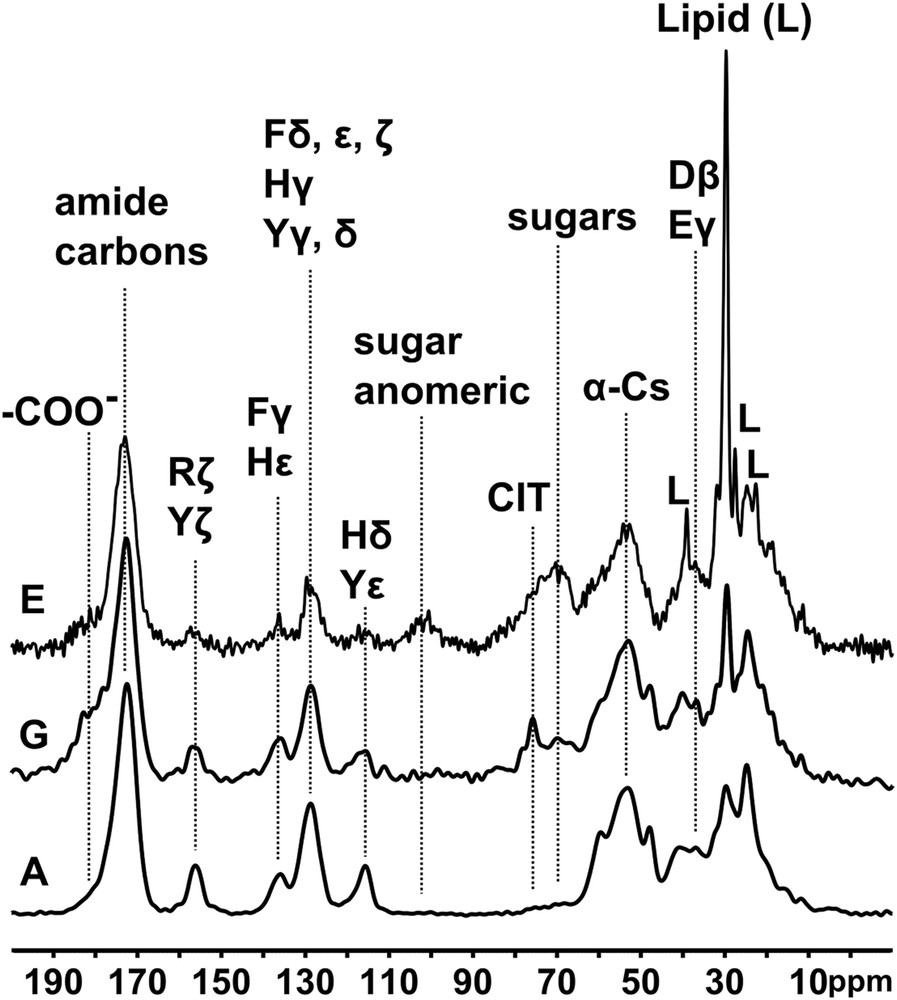
Three typical 13C spectra with key amino acids, sugars, citrate, and lipid, labelled. Note the variability of the citrate, sugar, and lipid signal relative intensities.
Varying relative amounts of sugar signals are observed in all samples. These are the broad peaks between ca. 65 and ca. 80 ppm due to the majority of the hydroxylated sugar carbons, and the signal from sugar anomeric carbons at ca. 100 ppm which is particularly diagnostic of sugars. The former, lower frequency spectral region also contains signals from citrate (the quaternary 3-carbon signal at ca. 76 ppm) when it is observable. The overlapping sugar and citrate signals can be distinguished by the DD procedure, which broadens the sugar signals beyond detection due to 13C–1H dipolar coupling with hydrogens directly attached to the sugar carbons, while the citrate quaternary carbon, at 76 ppm, experiences no significant change. The DD experiment also identifies signals from methyl groups and other mobile protonated carbons because molecular motion serves to reduce the 13C–1H dipolar coupling which leads to signal loss. In particular the DD experiment highlights signals from segmentally mobile lipids when they are present at observable levels. Finally this experiment also highlights quaternary aromatic signals (e.g., His, Phe and Tyr Cγ, and Tyr Cζ), and amide (ca. 175 ppm) and carboxylate (shoulder at ca. 180 ppm) carbons. All these effects are exemplified in Fig. 3, and summarized for all eight stones in Fig. S3 (online supporting information).
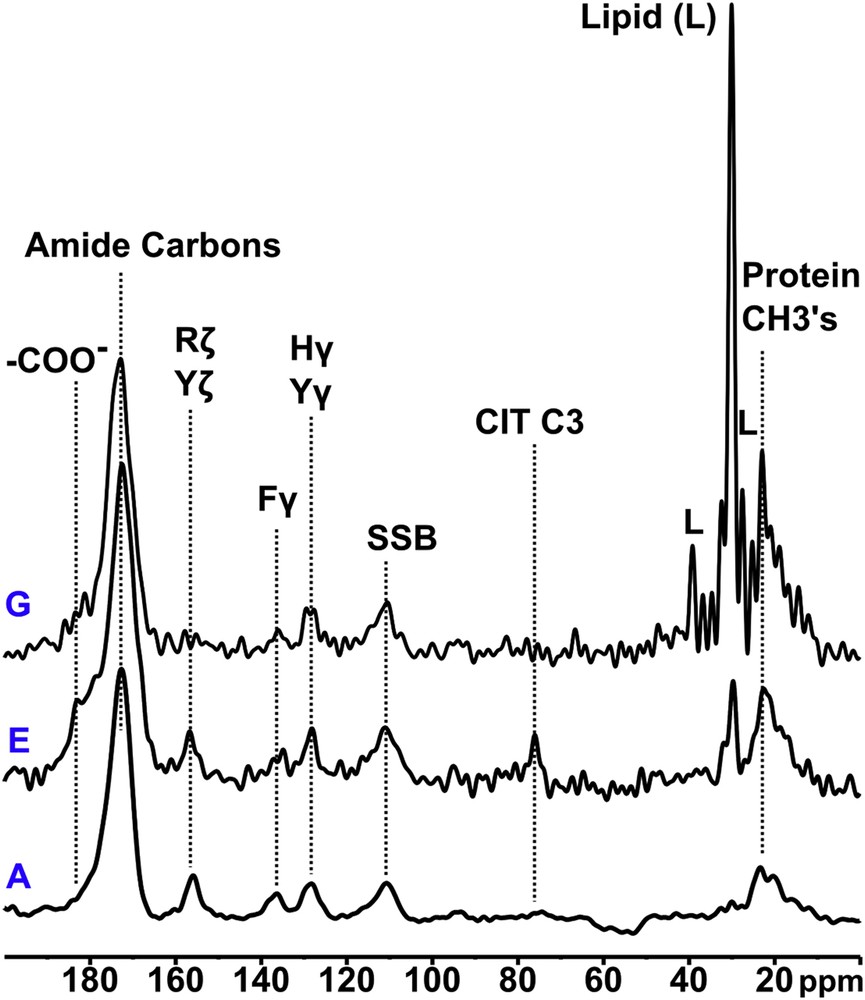
Three typical 13C dipolar dephased spectra showing protein amide, quaternary aromatic, and methyl, citrate and protein carboxylate, citrate C3, and lipid, non-dephasing signals.
The 13C{31P} REDOR experiment identifies signals from organic functionalities which are in close proximity to phosphate ions, the bulk of which may be assumed to be in mineral although there may be small contributions from organic phosphates such as phosphoserine. The technique is thus a powerful tool for mapping organic functionalities in close proximity to mineral, in effect within about 0.5 nm, and therefore likely involved in atomic length-scale interactions with mineral atoms.
A typical REDOR experiment consists of two phases; in the first phase a reference spectrum is obtained in which any through-space interaction between 13C and 31P is removed by the MAS procedure, while in the second phase this interaction is restored by a series of perturbing RF pulses applied at the 31P resonance frequency. The REDOR effect manifests as a reduction in the intensity of signals from carbon atoms close to phosphorus atoms, in practice within at most 1 nm, and is best appreciated by overlaying the reference spectrum and the REDOR spectrum; typical examples are shown in Fig. 4, and all 13C{31P} REDOR data are summarized in Fig. S4 (online supporting information). The REDOR experiment is a powerful tool for mapping the organic functionalities in close proximity to mineral, and likely involved in atomic length-scale binding with mineral atoms.
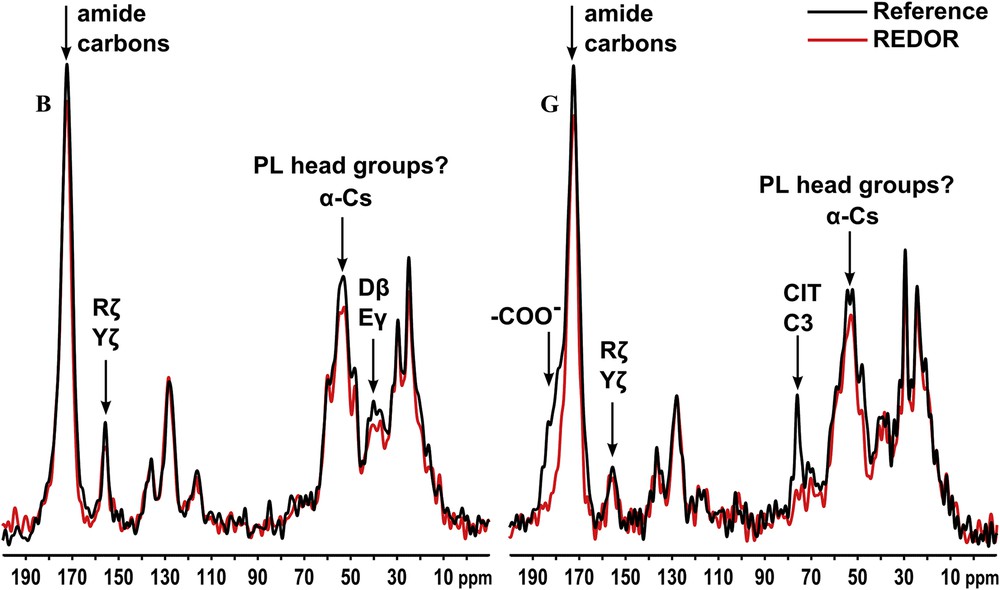
Typical 13C{31P} REDOR output from two stones. The entire 13C{31P} REDOR experiment is performed in two stages. First a reference 13C spectrum is acquired in which the through-space 13C–31P nuclear magnetic dipole coupling is removed (by the magic angle spinning procedure), then a REDOR spectrum is acquired under circumstances (application of a series of MAS period-synchronized π RF pulses at the 31P resonance frequency) which restore the 13C–31P coupling, and broaden any signals from carbons close to phosphorus to the extent that they become undetectable. These signals are best identified by overlaying reference and 13C{31P} REDOR spectra; loss of intensity in the latter relative to the former is evidence of a 13C{31P} REDOR effect.
Where citrate is observable, it undergoes a strong 13C{31P} REDOR effect involving its central C3 quaternary (76 ppm) and carboxylate (high frequency shoulder ca. 180 ppm) carbons (the methylene carbons are obscured by protein signals) implying a strong binding interaction with mineral, as observed in other calcified materials [19]. The broad sugar signal envelope between ca. 70 and 80 ppm also shows 13C{31P} REDOR, as do some protein signals, in particular the broad envelope of signals between ca. 50 and 60 ppm. In these respects the 13C{31P} REDOR behaviour of the sialoliths resembles that previously reported for apatitic uroliths [12]. Notes summarizing NMR data, as well as compositional information from standard FTIR, for all sialoliths appear in the online supporting information.
We were able to obtain amino acid content analyses for four of the samples using standard amino acid analysis; the data are summarized in Table 1 and the raw data (duplicate determinations for each sample) are presented in Table S1 (online supporting information). Table S1 shows that there is some variability in the measurements even within individual samples; this is not surprising considering that the powdered concretions are likely heterogeneous mixtures of mineral, polysaccharides, small molecules including citrate and lipids, and undoubtedly several different proteins. However there are some clear trends from the overall data. All four stones show a high content of Glu/Gln, Gly, and, significantly, Pro, and (consistent with the well resolved aromatic signals in the ssNMR), His, Phe, and Tyr. A protein database similarity search detected matches with statherin with fair confidence (score of 115), and the calculated amino acid compositions of statherin and its precursor peptide are compared with mean amino acid contents of each of the four stones analysed, in Table 1. As mentioned, our stone constituents are unlikely to represent a unique protein, but certainly proline-rich salivary proteins, of which statherin is representative, are significantly present.
Amino acid composition (in mole percent) of four of the eight sialoliths studied. Quoted values are means of two measurements; the corresponding raw data are shown in Table S1 (online supplementary information). N.B. the methodology does not quantify Cys or Trp. Calculated amino acid compositions of statherin and the statherin precursor peptide are also shown for comparison.
| Amino acid | Sample | Model protein | ||||
| A | B | C | D | Statherin | Statherin precursor | |
| Ala | 2.7 | 2.9 | 3.6 | 5.0 | 0.0 | 4.8 |
| Arg | 7.8 | 7.7 | 8.1 | 7.0 | 7.0 | 4.8 |
| Asp/Asn | 5.4 | 5.1 | 6.1 | 9.3 | 2.3 | 1.6 |
| Glu/Gln | 15.1 | 15.4 | 14.1 | 10.6 | 16.3 | 11.3 |
| Gly | 9.8 | 9.3 | 9.5 | 10.4 | 7.0 | 4.8 |
| His | 5.9 | 4.6 | 7.4 | 8.5 | 9.3 | 8.1 |
| Ile | 3.3 | 3.8 | 3.4 | 3.3 | 2.3 | 4.8 |
| Leu | 5.3 | 5.6 | 5.0 | 6.6 | 4.7 | 8.1 |
| Lys | 4.1 | 3.9 | 4.9 | 6.0 | 2.3 | 3.2 |
| Met | 0.5 | 0.7 | 0.7 | 1.4 | 0.0 | 4.8 |
| Phe | 7.6 | 7.5 | 8.3 | 5.9 | 7.0 | 9.7 |
| Pro | 13.3 | 14.5 | 11.8 | 5.7 | 16.3 | 11.3 |
| Ser | 3.0 | 2.6 | 2.5 | 4.1 | 4.7 | 4.8 |
| Thr | 2.1 | 2.1 | 1.8 | 3.6 | 2.3 | 1.6 |
| Tyr | 10.5 | 10.6 | 9.2 | 6.6 | 16.3 | 11.3 |
| Val | 3.5 | 3.7 | 3.4 | 6.0 | 2.3 | 4.8 |
3 Discussion
Many sialoliths consist of an organic-rich core surrounded by concentric laminations of alternating inorganic and organic materials [20]. The inorganic mineral phases are calcium phosphates, usually hydroxyapatite, carbonated apatite (carbapatite), and the mineral whitlockite (often containing some magnesium), and our samples are no exception. Reports of the organic components imply much less consistency in composition. An early study showed that the organic matrix forms a much higher proportion of salivary stones than it does of urinary tract calculi [21], consistent with our observation, and contains proteins and carbohydrates (galactose, glucose, mannose, rhamnose, fucose). Osuoji and Rowland report sugars, and protein rich in glycine, alanine, glutamate, and aspartate, but devoid of collagen (hydroxyproline) and keratin (cysteine) “marker” amino acids [22]. Basic proteins rich in proline, glycine, and glutamate/glutamine are abundant in saliva [23,24]. It is now clear that saliva contains groups of unique proline-rich acidic and basic proteins, which may be glycosylated [25,26], proposed to act as inhibitors of calcium phosphate precipitation. They may thus be important in maintaining calcium phosphate supersaturation and hence protecting dental enamel, and might play an antibacterial role as well [27]. They include the calcium binding seryl-phosphorylated polypeptide statherin [28]. A variety of ssNMR methods have been used to study aspects of the interaction between statherin model peptides and calcium phosphates [29], such as the orientation of the two N-terminal Phe rings of adsorbed statherin relative to the surface of HAp [30,31]. Using site specific 13C labelling of N-terminal acidic residues the importance of glutamates and aspartates in controlling statherin binding to HAp surfaces has been demonstrated, using 13C{31P} REDOR methods similar to those we report here [32]. Site-specific 13C labelling of arginine and 13C{31P} REDOR [33] and 15N labelling of lysine and 15N{31P} REDOR [34] demonstrate the importance of basic as well as acidic groups in the statherin–HAp phosphates.
The organic phase of submandibular sialoliths can contain about 10% lipids by weight [35]. On the basis of lipid content and composition it has been proposed that proteolipids, and calcium complexes with acidic phospholipids, probably derived from salivary gland constituents, play a mechanistic role in the stone mineralization process [36]. High salivary calcium, low salivary phytate (myo-inositol hexakisphosphate, a possible precipitation inhibitor) and magnesium, but not pH and citrate, correlate with propensity towards sialolithification [37]. Proteomic mass spectrometry detects peptides in submandibular sialoliths consistent with the presence of members (e.g., the A chain of HNP-3) of the defensin class of antibacterial proteins [38].
Citrate is abundant in bone and has been invoked as playing a role, not clear as yet, in orderly biomineralization [19]. It also occurs commonly but not invariably in pathological apatitic mineralizations besides sialoliths such as vascular calcifications and kidney stones [13]. Its occurrence in the latter is interesting in view of the widespread use of oral potassium citrate in urolithiasis prophylaxis [39]. The invariably strong 13C{31P} REDOR effect from citrate (central C3 carbon, and carboxylate carbons) we observe in all the sialoliths in which citrate is detectable by NMR implies a close atomic level association between citrate and mineral which may be mechanistically significant in the sialolithiasis process in at least some instances. In addition to citrate, polysaccharide species are also intimately associated with HAp in uroliths [12] and other pathological calcifications [40]. Similarly where polysaccharides are detectable in our sialoliths 13C{31P} REDOR effects again provide evidence for strong sugar–mineral association, which may be mechanistically significant.
In view of the detailed ssNMR studies published on the geometry of interactions between HAp and model statherin subsequences, and our findings that our sialoliths contain high proportions of salivary proteins typified by statherin, it is relevant to ask whether any of the 13C{31P} REDOR effects we observe are consistent with the proposed geometries. In the sialoliths the most consistent 13C{31P} REDOR effects are observed for the broad envelope of 13C signals between ca. 50 and 60 ppm comprised of numerous overlapping signals from the α-carbon atoms of most of the amino acid types, as well as certain others such as the δ-carbons of Pro, and possibly from glyceryl and head group atoms of any phospholipids which may be present which will undergo strong intramolecular 13C{31P} REDOR effects. In some but not all stones there are also 13C{31P} REDOR effects in the 35–40 ppm spectral region which contains signals from Asp and Asn β- and Glu γ-carbons. Strong 13C{31P} REDOR effects observed for carboxylate groups (spectral region around 180 ppm) are particularly indicative of the involvement of acidic groups in the mineral structure but unfortunately it is impossible to distinguish the effects from acidic amino acids (Asp, Glu) from those from citrate. There are no reliably discernible 13C{31P} REDOR effects from any of the aromatic signals between 110 and 160 ppm, including the signal at ca. 156 ppm from Tyr Cζ which may contain a contribution from Arg Cζ. Statherin, and possibly other salivary proteins, is phosphorylated on some Ser residues; phosphoseryl residues should exhibit strong intraresidue 13C{31P} REDOR effects corresponding to Cα (ca. 58 ppm) and Cβ (ca. 65 ppm) [41] but again it is impossible to distinguish these (if any) from those from other protein and sugar signals. For such reasons of spectral overlap and signal degeneracy it is difficult to confirm that the detailed geometric models proposed for the statherin–apatite interaction are reproduced in our materials. It seems unlikely however that aromatic groups are located within 0.5 nm from the mineral.
In comparison with apatitic kidney stones, the protein content of sialoliths as reflected in 13C NMR is strikingly uniform although there is greater variation between stones in respect of the less abundant sugars, citrate, and lipids (compare Fig. S2 which overlays the spectra of eight sialoliths, with an overlay of ten apatitic urolith spectra in the online supplementary figure of Reid et al. [13]). There is an emerging consensus that the causative event in urolithiasis is the occlusion of renal ducts by calcium phosphate crystals which eventually extrude into the urinary space and nucleate further deposition of the crystalline material, very commonly calcium oxalates [42]. There is no generally accepted single causative event in the genesis of sialoliths, although obstruction, low salivary flow rate, dehydration, changes in salivary pH, epithelial cell debris, and bacterial infection, are all considered significant to the aetiology [43]. The high content of proline-rich salivary proteins in apatitic sialoliths is interesting in view of the proposed role of at least some of them as inhibitors of calcium phosphate crystal propagation. Whatever proteins solidify in sialolithiasis the low intensity of the 13C{31P} REDOR effects from protein signals suggests that the bulk of protein residues are not in atomic length-scale contact with mineral. In this respect they resemble the protein material in uroliths in which 13C{31P} REDOR effects from proteins are much less apparent than those from polysaccharides and citrate [12]. In spite of intensive study of the protein content of clinically significant kidney stones, which is dominated by inflammatory proteins, [44] there is no corresponding data yet for the proteome of the tiny apatitic Randall's plaques believed to represent the causative nidus of urolithic mineralization [45]. As such there is no circumstantial or mechanistic evidence yet linking specific proteins (or other biomolecules) with urolithogenesis.
The same is even more true of sialolithogenesis. The single published proteomics study concentrated on the centre and periphery of the core, and was limited to stones in which these structures were well developed and discernible [38]. In such cases the A chain of the antibacterial defensin HNP-3 was identified using proteolytic fragment mass spectrometry. The amino acid content of this peptide showed very low correspondence with our experimental amino acid content measurements. Our studies examined the entire stone as it was not possible to isolate core material specifically, so it is possible that defensin peptides are present at low abundance and insignificant compared to the predominant proline-rich salivary proteins we overwhelmingly find. It is not surprising that salivary proteins are major constituents of sialoliths. At low concentrations these proteins are thought to inhibit crystallization processes by binding to the growing faces of the crystallite nidus. At higher concentrations they can solidify and promote calcium phosphate deposition by heterogeneous nucleation. If our stones conform to the widely accepted model of sialoliths as an amorphous mineralized core surrounded by concentric laminations of organic and inorganic material [20] we might assume that the mineralization-inhibitory salivary proteins solidify physically separated from mineral. This would be consistent with the comparatively insignificant 13C{31P} REDOR effects unambiguously assignable to protein, relative to citrate, signals. It may imply alternating episodes of deposition of mineral, and of mineralization-inhibitory salivary proteins. Testing this will require isolation and study of separate organic and mineral laminations in cases where this is possible. Another explanation may be that proline-rich proteins aggregate and solidify under sialolithogenic conditions and then accumulate solid calcium phosphate via the heterogeneous mechanisms alluded to earlier. Under such circumstances protein–protein may predominate over protein–mineral contacts, again consistent with the weak protein–mineral 13C{31P} REDOR effects. Future work should be aimed at characterizing the sialolith proteome in more detail, and studying the distribution and variation in composition of the protein content of stone core and peripheral laminations. In this way a better understanding should be attained of the detailed mechanistic relationship between mineralization and mineral–organic composite formation leading to sialolithogenesis, and of treatments for prophylaxis.
4 Conclusions
- 1. We have applied ssNMR to extend characterization of the composition of, and some aspects of the mineral–organic interactions present in, salivary stones (sialoliths), to our knowledge for the first time in these pathological materials.
- 2. This technique represents a unique and complementary tool to address details of the organic components in biocalcifications which may prove central to the mechanisms driving pathological mineralization.
- 3. Indeed, we have identified different organic compounds in addition to the abundant proteins, such as polysaccharides, lipids, and the high affinity calcium ligand citrate.
- 4. 13C{31P} rotational echo double resonance (13C{31P} REDOR) indicates a degree of binding between proteins and the mineral phase, probably related to the fact that some of them (e.g., proline-rich salivary proteins such as statherin) are specific inhibitors of the mineralization process.
Such approaches provide a unique opportunity to better understand the etiopathogenesis of these calcifications, whereby ssNMR has significant potential in the characterization of pathological biomineralization.
Acknowledgements
We thank the U.K. EPSRC (Y. L.) and MRC (D. G. R.) for funding. We thank our colleague Peter Sharratt in the Department of Biochemistry, University of Cambridge, for amino acid analyses and protein database searching, and helpful discussions.