

1 Introduction
Regular secondary structures such as α-helices are essential components of protein architectures and are frequently found at protein–protein interfaces [1]. The coiled-coil motif which consists of two or more amphiphilic α-helical segments interacting together by interdigitation of their hydrophobic side chains is also common among protein tertiary structures and protein oligomerization domains [2]. The knowledge of these protein–protein interactions (PPIs) at atomic resolution has provided the basis for diverse applications of helically folded synthetic peptides including, for example, the modulation of therapeutically relevant PPIs [3,4] or the construction of bioinspired self-assembled (one-dimensional or three-dimensional) nanostructures [5,6]. However, in the absence of additional stabilizing effects (packing, favorable electrostatic interactions, and N- and C-capping motifs) [7,8], short peptide helices are only weakly populated in water thus limiting the development of their therapeutic applications. Today, a number of synthetic strategies are available to further increase the stability of short-chain α-helices, among which are the insertion of conformationally constrained amino acids (e.g., Cα-tetrasubstituted amino acids such as α-amino isobutyric acid, Aib [9,10], and β-amino acids [11]), side chain crosslinking [12,13], and helix nucleating templates [14–16]. The latter approach is aimed at pre-organizing the first amide bonds through the use of a rigid template generally placed at the N-terminus of the peptide chain (e.g., Kemp tricyclic templates [17,18]) or a hydrogen bond surrogate (HBS) in which the N-terminal intramolecular i,i+4 hydrogen bond is substituted with a covalent linkage (e.g., Arora HBS helices [19,20]). More recently, chiral templates have also been used to control helix screw sense preference in α-peptides made of helicogenic achiral Aib residues [21,22].
Surprisingly and despite potential interest, the use of a helical foldamer (synthetic folded oligomer) [23–25] as a template to nucleate/stabilize peptide helices has hardly been investigated; (αβ + α) peptides disclosed by the Gellman group represent to our knowledge the only case in the literature of a foldamer/α-peptide chimera mimicking an α-helix [26,27]. Most of the published studies are rather related to hybrid foldamer sequences whereby α-amino acid residues alternate with non-natural monomer units [28–30]. We [31] and others [32] have recently started to explore the concept of foldamer/α-peptide chimeras (or block co-foldamers as termed by Clayden [32]) with aliphatic oligoureas, a class of foldamers that adopt well-defined and stable helical structures akin to α-helices (similar helicity, polarity and pitch but a more complex three centred H-bond network and a larger diameter, Fig. 1a) [33–38]. In a previous study, we have shown that oligourea/peptide chimeras form well-defined helical structures in polar organic solvents with the propagation of a continuous intramolecular H-bond network spanning the entire sequence and connecting two geometrically distinct helices (α-helix and oligourea 2.5-helix) [31]. Furthermore, our results in solution also suggest that short triurea sequences have the ability to induce α-helicity when fused to either the N- or C-terminus of a peptide segment.
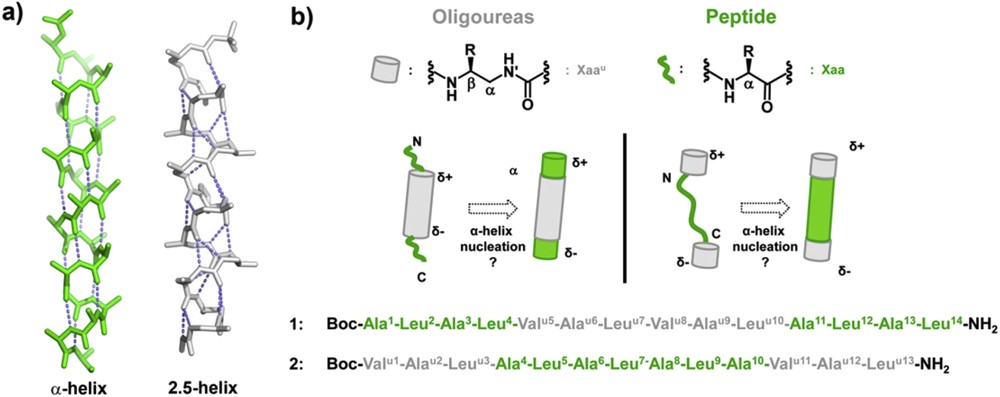
a) Comparison of peptide α-helical and oligourea 2.5-helical backbones (green and light grey, respectively); b) principle of α-helix nucleation by oligourea segments in triblock oligomers and primary sequences of triblock chimeras 1 and 2 studied in this work. The standard three letter code is used for α-amino acid residues (green), and a superscript “u” is added for the corresponding urea units (light grey).
To gain additional insight into the α-helix stabilization properties of oligoureas, we have now investigated the folding properties of peptide-oligourea-peptide and oligourea-peptide-oligourea chimeras 1 and 2 containing 14 and 13 residues, respectively (Fig. 1b). Sequence 1 which contains a 6-mer oligourea motif flanked by two tetrapeptide units is aimed at testing the ability of a central oligourea chain to induce helix formation concurrently in the two peptide regions. Oligomer 2 was designed to evaluate whether two triurea units fused at the N- and C-termini of a peptide segment can work synergistically to promote α-helicity. In both sequences, the peptide segment is composed of alternating Ala and Leu residues, both known for their helix forming tendency [39,40].
2 Results and discussion
Both oligomers 1 and 2 have been synthesized by a solid-phase methodology starting from a Sieber amide resin [41] as shown in Scheme 1. Oligourea segments were assembled by coupling succinimidyl (2-azidoethyl)carbamate derivatives 3 (with side chains of Val, Ala and Leu) using microwave assistance followed by the reduction of azido groups under Staudinger conditions as previously described [42]. The peptide segments were built using standard Fmoc chemistry. Only the last Ala residue in 1 and the last urea unit in 2 were coupled as N-Boc protected monomers. Cleavage from the resin was performed by treatment with 1% TFA in CH2Cl2 thus allowing the N-terminal Boc protecting group to be preserved. The two oligomers were purified to homogeneity by C18 RP-HPLC and their identity was confirmed by mass spectrometry and NMR in CD3OH. The frequencies of all 1H, 13C and 15N atoms were assigned by using homonuclear COSY, TOCSY and NOESY experiments and heteronuclear experiments (Tables 1 and 2). The assignment of spin systems and conformational investigation of the two chimeras were facilitated by the dispersion and by the distinct ranges of chemical shifts of amide and urea NH resonances, amide NHs appearing systematically downfield to urea NHs (in the range of 7.2–9.1 ppm in 1 and 7.2–8.9 ppm in 2).

(a) Fmoc-Xaa-OH or Boc-Ala-OH (4 equiv), HBTU (4 equiv), HOBt (4 equiv), DIPEA (8 equiv), DMF, MW (50 W, 50 °C), 10 min; (b) 20% piperidine in DMF, MW (50 W, 50 °C), 8 min; (c) 3a–c or 4 (3 equiv), DIPEA (6 equiv), DMF, MW (50 W, 50 °C), 10 min; (d) PMe3, 1,4-dioxane/H2O (7:3, v/v), MW (50 W, 50 °C), 30 min; (e) 1% TFA in CH2Cl2.
1H, 13C and 15N chemical shifts for compound 1 in CD3OH (700 MHz) at 4 mM.
| Residue | HN | HN′ | Hα | Hβ | Hγ | Hδ | Hδ1 | Hδ2 | Hɛ1 | Hɛ2 |
| N | N′ | Cα | Cβ | Cγ | Cδ | Cδ1 | Cδ2 | Cɛ1 | Cɛ2 | |
| Ala (1) | 7.35 | 3.91 | 1.37 | |||||||
| 125.2 | 55.9 | 19.7 | ||||||||
| Leu (2) | 8.52 | 4.07 | 1.59/1.70 | 1.76 | 0.92 | 0.99 | ||||
| 116.9 | 58.2 | 42.6 | 28.1 | 24.3 | 25.0 | |||||
| Ala (3) | 7.86 | 4.29 | 1.49 | |||||||
| 118.8 | 55.0 | 19.5 | ||||||||
| Leu (4) | 7.49 | 4.19 | 1.65/1.78 | 1.77 | 0.92 | 0.98 | ||||
| 117.5 | 58.0 | 43.5 | 28.1 | 24.3 | 25.0 | |||||
| Val u (5) | 7.28 | 5.78 | 2.80/3.61 | 3.97 | 1.73 | 0.91 | 0.94 | |||
| 119.4 | nd | 44.7 | 57.4 | 34.1 | 23.1 | 22.1 | ||||
| Ala u (6) | 5.65 | 6.06 | 2.50/3.57 | 3.87 | 1.05 | |||||
| 126.2 | nd | 50.0 | 48.9 | 20.6 | ||||||
| Leu u (7) | 5.98 | 6.48 | 2.52/3.65 | 3.94 | 1.21/1.27 | 1.75 | 0.93 | 0.93 | ||
| 123.9 | 109.7 | 49.0 | 51.4 | 44.9 | 28.1 | 24.3 | 24.3 | |||
| Val u (8) | 6.40 | 6.66 | 2.42/3.77 | 3.52 | 1.62 | 0.90 | 0.94 | |||
| 120.9 | 110.8 | 46.7 | 59.0 | 34.3 | 20.8 | 22.1 | ||||
| Ala u (9) | 6.06 | 6.48 | 2.33/3.57 | 4.13 | 1.03 | |||||
| 127.5 | 109.7 | 50.4 | 48.8 | 20.6 | ||||||
| Leu u (10) | 5.92 | 6.65 | 2.31/3.57 | 3.98 | 1.10/1.22 | 1.75 | 0.85 | 0.92 | ||
| 121.7 | 110.8 | 47.4 | 50.0 | 44.3 | 28.1 | 24.1 | 26.2 | |||
| Ala (11) | 7.33 | 3.91 | 1.37 | |||||||
| 124.3 | 55.9 | 19.7 | ||||||||
| Leu (12) | 9.06 | 4.25 | 1.55/2.05 | 1.88 | 0.91 | 0.96 | ||||
| 115.0 | 57.2 | 41.5 | 28.3 | 23.1 | 25.7 | |||||
| Ala (13) | 8.5 | 4.3 | 1.5 | |||||||
| 120.6 | 53.9 | 19.5 | ||||||||
| Leu (14) | 7.57 | 4.27 | 1.65/1.79 | 1.75 | 0.88 | 0.94 | ||||
| 117.6 | 55.4 | 43.5 | 28.1 | 16.6 | 25.7 |
1H, 13C and 15N chemical shifts for compound 2 in CD3OH (700 MHz) at 4 mM.
| Residue | HN | HN′ | Hα | Hβ | Hγ | Hδ | Hδ1 | Hδ2 | Hɛ1 | Hɛ2 |
| N | N′ | Cα | Cβ | Cγ | Cδ | Cδ1 | Cδ2 | Cɛ1 | Cɛ2 | |
| Val u (1) | 6.61 | 6.00 | 3.57/2.47 | 3.38 | 1.56 | 0.84 | 0.88 | |||
| 123.8 | 107.4 | 46.2 | 60.1 | 34.2 | 25.7 | 22.3 | ||||
| Ala u (2) | 5.84 | 5.70 | 3.52/2.24 | 4.04 | 0.98 | |||||
| 125.7 | nd | 50.5 | 48.8 | 20.0 | ||||||
| Leu u (3) | 5.90 | 6.63 | 3.55/2.26 | 3.91 | 1.09/1.09 | 1.66 | 0.80 | 0.83 | ||
| 121.8 | 110.4 | 49.7 | 50.2 | 28.1 | 24.4 | 25.7 | ||||
| Ala (4) | 7.19 | 3.84 | 1.31 | |||||||
| 124.0 | 57.2 | 20.2 | ||||||||
| Leu (5) | 8.90 | 4.18 | 1.88/1.61 | 1.72 | 0.84 | 0.91 | ||||
| 116.0 | 57.9 | 42.0 | 28.3 | 24.0 | 25.2 | |||||
| Ala (6) | 7.99 | 4.03 | 1.48 | |||||||
| 121.6 | 56.1 | 18.8 | ||||||||
| Leu (7) | 7.77 | 3.97 | 1.67/1.58 | 1.65 | 0.81 | 0.87 | ||||
| 118.4 | 58.8 | 42.5 | 28.1 | 21.0 | 22.3 | |||||
| Ala (8) | 8.01 | 3.92 | 1.45 | |||||||
| 120.7 | 55.9 | 18.4 | ||||||||
| Leu (9) | 7.66 | 4.08 | 1.80/1.49 | 1.79 | 0.81 | 0.86 | ||||
| 117.5 | 58.2 | 43.2 | 27.8 | 21.6 | 24.0 | |||||
| Ala (10) | 8.05 | 4.03 | 1.43 | |||||||
| 121.4 | 56.1 | 20.0 | ||||||||
| Val u (11) | 7.47 | 5.81 | 3.50/2.75 | 3.85 | 1.62 | 0.81 | 0.81 | |||
| 118.9 | nd | 44.4 | 55.7 | 34.4 | 21.0 | 21.0 | ||||
| Ala u (12) | 5.4 | 5.8 | 3.42/2.44 | 3.99 | 0.96 | |||||
| nd | nd | 50.1 | 47.8 | 20.8 | ||||||
| Leu u (13) | 6.00 | 6.40 | 3.50/2.68 | 3.85 | 1.13/1.23 | 1.63 | 0.85 | 0.85 | ||
| 123.8 | nd | 47.7 | 51.5 | 46.0 | 28.1 | 25.70 | 25.70 |
Oligomer 1 consists of a 6 mer oligourea chain prolonged at both ends by a Ala-Leu-Ala-Leu tetrapeptide sequence. Inspection of the oligourea NH/CH fingerprint region in the 1H NMR spectra reveals features typical of a helically folded oligourea chain, i.e., the large vicinal coupling constants between NH and CH(R) protons (3J > 9 Hz) and a high degree of anisochronicity (Δδ) of main chain methylene protons (0.81 < Δδ < 1.35 ppm) [43–45]. Furthermore, the presence of unambiguous non-sequential nuclear Overhauser enhancements (NOEs) along the oligourea backbone such as βN(i,i + 2) and βN′(i,i + 2) are fully consistent with helix formation in the oligourea segment [43]. The observation in the two peptide regions of an uninterrupted NN(i, i + 1) pattern of NOEs indicates that the peptide backbone also adopts a helical conformation (Fig. 2). This is supported by the presence of non-sequential medium range (i,i + 2), (i,i + 3) NOEs in the peptide regions and at the junction between the peptide and oligourea segments (e.g., βN(9,12), βN(9,13), and α2N(9,13) connectivities, Fig. 2b). However, because medium range NOEs between peptide and oligourea protons are non canonical, it is difficult to discriminate between a possible α-helical conformation and a 310-helical conformation for the short peptide segments. The values of the vicinal 3J (NH, αCH) coupling constants in the peptide segments are slightly above those reported for stable α- and 310 helical structures (<6 Hz) [46] thus suggesting helix fraying. Nevertheless, this NMR study tends to suggest that a helical structure develops without a break between the three segments, thus confirming the compatibility of both helix geometries and related H-bonding networks.
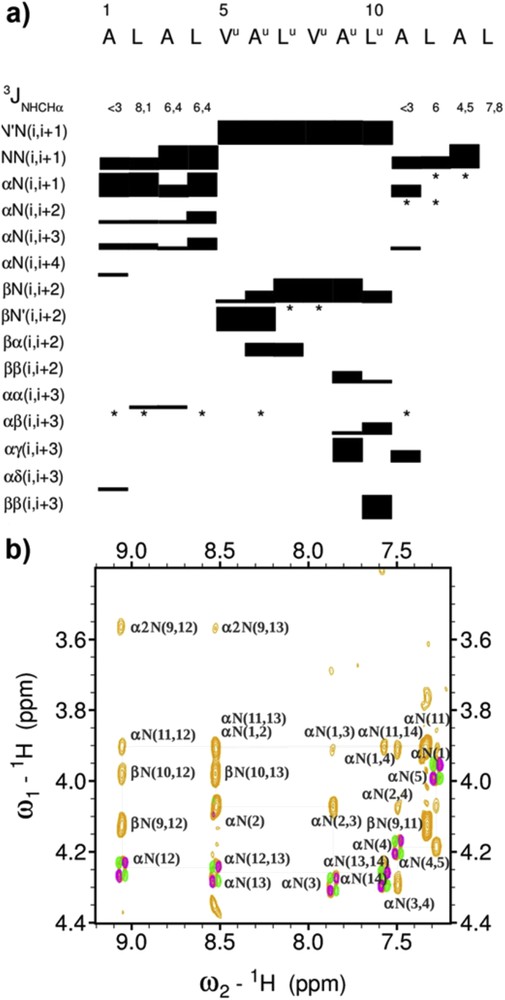
(a) Overview of sequential and medium-range 1H–1H NOEs observed in the peptide and oligourea domains of 1 and spin-spin-coupling constants 3J(NH, αCH) measured in the peptide chain. The thickness of the bars is proportional to the intensity of the observed NOEs. *indicates signal overlaps precluding unambiguous NOE assignment; (b) some key medium range NOE connectivities (light orange) between oligourea and peptide segments in 1.
The 1H NMR spectrum of triblock chimera 2 shares a number of features with that of chimera 1, including large >9.5 Hz 3J(NH, βCH) and anisochronicity of main chain methylene protons along the oligourea backbone, which altogether are indicative of a helical conformation [43]. Remarkably, five out of the seven vicinal 3J (NH, αCH) coupling constants in the peptide segment are below 6 Hz (3–5.8 Hz), supporting the view that the peptide region is also helical in solution. The non-sequential medium range βN(i,i + 2) and βN′(i,i + 2) in the two oligourea segments as well as NN(i, i + 1), αN(i,i + 3), αN(i,i + 2) and αN(i,i + 4) NOEs along the peptide chain observed in the NOESY spectrum of 2 are also consistent with the formation of a seamless helical conformation with no apparent interruption at the two peptide/oligourea junctions (Fig. 3).

(a) Overview of sequential and medium-range 1H–1H NOEs observed in the peptide and oligourea domains of 2 and spin-spin-coupling constants 3J(NH, αCH) measured in the peptide chain. The thickness of the bars is proportional to the intensity of the observed NOEs. *indicates signal overlaps precluding unambiguous NOE assignment; (b) some key medium range NOE connectivities (light orange) between oligourea and peptide segments in 2.
Circular dichroism also proved useful to characterize the conformational preference of chimera 2. The spectrum recorded at 50 μM in MeOH (see Supporting information) was found to combine the hallmarks of both helically folded oligourea and α-peptide backbones. The spectrum is largely dominated by the contribution of the urea chromophore with a strong positive maximum at ca 203 nm characteristic of the oligourea 2.5-helical structure. A negative maximum at ca 222 nm is also observed that is not present in the spectra of the cognate homooligourea 5 and chimera 1 (see Supporting information). This signal can thus be tentatively ascribed to the peptide α-helical conformation and the corresponding molar ellipticity at 222 nm is likely to reflect the degree of α-helicity in chimera 2. However, due to the presence of two distinct chromophores and helix types in the main chain, the spectra of chimeras should be analyzed with caution and our interpretation at that stage remains largely tentative. Nevertheless, the ellipticity value at 222 nm for 2 in MeOH surpasses that measured for the parent heptapeptide 6 in both MeOH or TFE, supporting a stabilizing effect of the two oligourea domains in 2.
Additional insight into the conformation of triblock chimeras was obtained by high resolution crystallographic structural studies. Single crystals of chimera 2 suitable for X-ray diffraction were grown from a DMF solution and the structure was solved in the P212121 space group (Table 3). The structure shows a continuous and regular helix that spans the entire sequence with no apparent distortion and with all intramolecular complementary H-bonding sites being satisfied. (Fig. 4). Closer inspection of the structure revealed useful details about the propagation of the H-bonding network and the way the oligourea and peptide helices communicate. The geometry of the helical peptide segment in the crystal structure of 2 is particularly notable. The Φ and Ψ dihedral angles of amino acid in the structure of 2 (mean Φ and Ψ values for the seven residues : −69°, −41°) match almost perfectly those of a canonical α-helix (−63°, −42°) [47]. The two terminal oligourea segments adopt a canonical 2.5-helical conformation [37,44]. A series of specific, non canonical bifurcated H-bonds are observed at the junction between the oligourea and peptide parts at both ends of the peptide sequence. In particular, urea carbonyls of Alau2 and Leuu3 are both engaged in H-bonds with i+3 and i+4 amide protons (i.e., NHs of L5, A6 and L7). In a similar manner, amide carbonyls of Leu7 and Ala8 form a continuous network of four H-bonds with NHs of Leuu11 and Alau12. In contrast, amide carbonyls of Leu9 and Ala10 form two centered i,i+3 H-bonds with N′H and NH of Alau12 and Leuu13, closing 15- and 14-membered pseudocycles, respectively. Overall, the medium range NOEs between main chain NH and CH protons of the junction region (see for example αN(2,5), βN(2,5), αN(7,11), and αN(8,11) connectivities in Fig. 3b) are in good agreement with the short distances measured in the crystal structure, thus further supporting the conclusion that the molecule is largely helical in solution.
X-ray crystallographic parameters for chimera 2.
| CCDC number | CCDC 1041244 |
| Formula | C69H132N20O15 |
| Crystal System | Orthorhombic |
| Space Group | P212121 |
| Z | 4 |
| Unit cell parameters | |
| a, Å | 46.115(4) |
| b, Å | 11.5920(9) |
| c, Å | 16.7751(13) |
| α, ° | 90 |
| β, ° | 90 |
| γ, ° | 90 |
| Temperature, K | 130(2) |
| Volume, Å3 | 8967.4(12) |
| FW, g mol−1 | 1157.59 |
| ρ, g cm−3 | 1.098 |
| λ, Å | 1.54187 |
| Mu, E | 0.638 |
| θ min/θ max | 2.803/60.948 |
| Radiation source | Rotating anode |
| Reflections measured | 21785 |
| Reflections unique ( [Fo > 2σFo)]) | 11484 |
| Parameters/restraints | 938/58 |
| GOF | 1.091 |
| R1 (I > 2σ(I)) | 0.1067 |
| wR2 (all data) | 0.2547 |
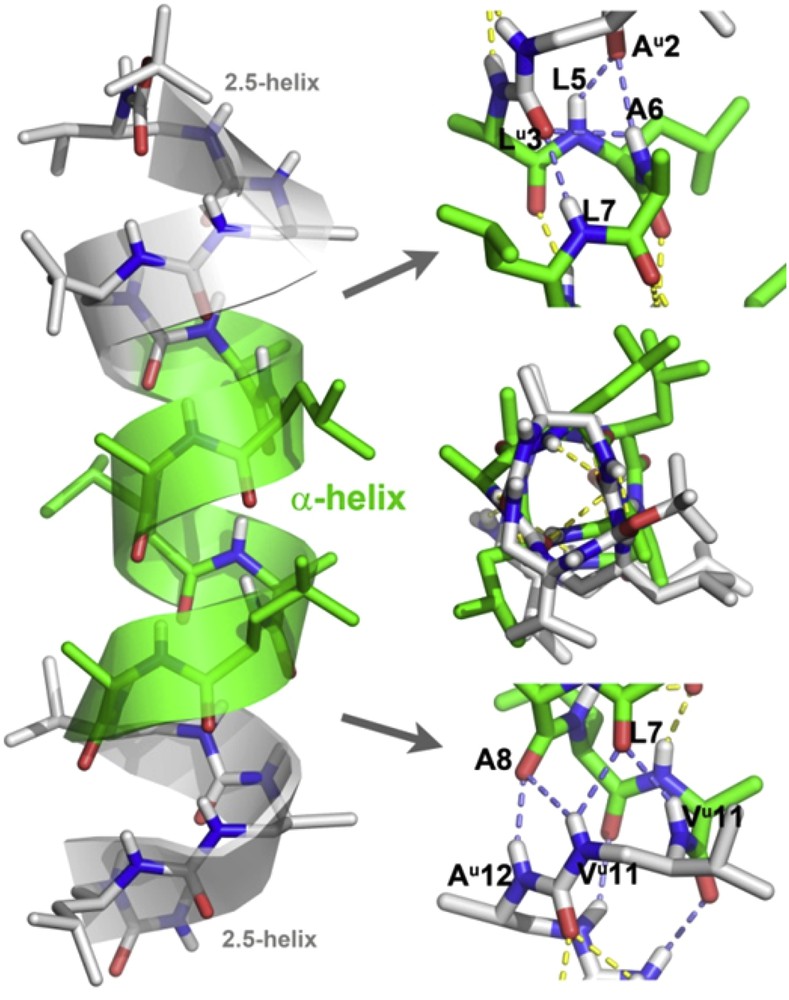
X-ray crystal structure representations (side view and top view) of chimera 2 with details of the H-bonding network at the two peptide/oligourea junctions. Intramolecular H-bonds between peptide and oligourea segments are shown in slate blue. C atoms in the oligourea and peptide segments are shown in light grey and light green, respectively.
3 Conclusion
Herein, we have studied helix propagation and α-helix nucleation in peptide-oligourea-peptide and oligourea-peptide-oligourea triblock oligomers. Our results obtained in a polar organic solvent (methanol) and in the crystal state confirm the ability of oligourea segments as short as triureas to serve as nucleation templates when located either at the N- or C-terminus [31,32] or at both ends of a short peptide sequence. The crystal structure of the 13-mer chimera 2 reveals the formation of a regular helix that spans the entire sequence. In this structure, the peptide part in the middle of the sequence adopts a canonical α-helical conformation which is locked at both ends by two short 2.5 oligourea helices. This observation suggests a synchronized effect of both accessory foldamers in nucleating the α-helical structure and a possible strategy to stabilize α-helical peptides targeting protein surfaces. The oligourea backbone which is strongly biased towards helix formation provides a pre-organized cap for the initial four peptide NHs and final four carbonyl groups. It is noteworthy that most synthetic α-helical templates [8,14,15] reported in the literature are designed to engage H-bonds in the initial turn of the helix but relatively few molecules have been shown to effectively cap the carbonyl groups in the final turns of the helix [48]. Similar α-helix capping effects also take place in proteins [49]. To some extent, the complementary H-bonded network observed in 2 can be compared to that of native capping motifs found at the N- and C-termini of α-helices in proteins such as the N-terminal capping box [50,51], or the C-terminal Schellman motif [52,53]. Overall these findings and the high resolution structural data reported here may suggest a possible strategy based on oligourea foldamers to stabilize helical peptide epitopes for protein ligand design. Our progress in this direction will be reported in due course.
4 Experimental
4.1 General
Abbreviations : DIPEA (diisopropylethylamine), DMF (dimethylformamide), HBTU (N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate), HOBT (1-Hydroxybenzotriazole), and TFA (trifluoroacetic acid). Analytical RP-HPLC analyses were performed on a Dionex U3000SD using a Macherey–Nagel Nucleodur column (4.6 × 100 mm, 3 μm) at a flow rate of 1 ml/min with UV detection at 200 nm. The mobile phase was composed of 0.1% (v/v) TFA-H2O (Solvent A) and 0.1% (v/v) TFA-MeOH (Solvent B). Semi-preparative purification of chimeras was performed on a Dionex U3000SD using a Macherey–Nagel Nucleodur column (10 × 250 mm, 5 μm) at a flow rate of 4 ml/min with UV detection at 200 nm. ESI-MS analyses were carried out on a ThermoElectron LCQ Advantage spectrometer equipped with an ion trap mass analyzer and coupled with a ThermoElectron Surveyor HPLC system. Succinimidyl (2-azido-1-X-ethyl)carbamate monomers with side chains of Ala (3a), Val (3b) and Leu (3c) and succinimidyl {(2S)-2-{[(tert-butoxy)carbonyl]amino}}-3-methylbutyl} carbamate 4 were prepared using previously described procedures [42,54].
4.2 Synthesis
4.2.1 General procedure for oligomer synthesis on a solid support (GP1)
Solid-phase synthesis of chimeras 1 and 2 was conducted with microwave irradiation using the Discover Bio® System from CEM. All steps were performed under microwave irradiation at atmospheric pressure. The temperature was maintained by modulation of power and controlled with a fiber optic sensor. The starting Sieber amide resin [41] (≈100 mg, loading 0.62 mmol/g) was swelled in DMF (2 mL) for 30 min prior to the synthesis. The Fmoc-group was then removed with 20% piperidine in DMF (2 mL), under microwave irradiation (50 W, 50 °C, 8 min). Amide bond and urea bond formation were monitored by the Kaiser test [55] and the chloranil test, respectively.
4.2.2 General procedure for coupling α-amino acids (GP2)
N-Fmoc-α-amino acids or Boc-Ala-OH (used as N-terminal residue in the synthesis of 1) (0.248 mmol, 4 equiv), HBTU (0.094 g, 0.248 mmol), HOBt (0.038 g, 0.248 mmol) and DIPEA (0.086 mL, 0.496 mmol) were dissolved in DMF and after 5 min the mixture was added into a reaction vessel (CEM). The vessel was then placed inside the microwave reactor and irradiated for 10 min (50 W, 50 °C). The resin was then filtered and washed with DMF (4 × 2 mL).
4.2.3 General procedure for the Fmoc deprotection (GP3)
The N-Fmoc protecting group was removed with 20% piperidine in DMF (2 mL), under microwave irradiation (50 W, 50 °C, 8 min).
4.2.4 General procedure for coupling succinimidyl carbamates (GP4)
Activated monomer 3 or 4 (used as the N-terminal residue in the synthesis of 2) (0.186 mmol, 3 equiv) was dissolved in DMF (2 mL) and was added to the reaction vessel, followed by DIPEA (0.065 mL, 0.372 mmol, 6 equiv). The reaction was performed under microwave irradiation for 20 min (50 W, 50 °C). The resin was then filtered and washed with DMF (4 × 2 mL).
4.2.5 General procedure for the reduction of the azide moiety on a solid support (GP5)
The reduction of the azido group was performed in a mixture of 1,4-dioxane/H2O (7:3 v/v). The resin was first washed with this mixture of solvents and the Staudinger reaction was then performed under microwave conditions (50 W, 50 °C, 30 min) by treating a suspension of the resin in 1,4-dioxane/H2O (2 mL) with a 1 M PMe3 solution in THF (0.62 mL, 10 equiv). After completion, the resin was filtered off and washed with 1,4-dioxane/H2O (7:3 v/v, 1 × 2 mL) and DMF (4 × 2 mL).
4.2.6 General procedure for the cleavage from the resin (GP6)
After completion of the last coupling (Boc protected monomer), the resin was transferred into a syringe with a frit, washed with DMF (5 × 2 mL), CH2Cl2 (5 × 2 mL), and Et2O (5 × 2 mL) and dried in a desiccator. Before the cleavage step, the resin was swelled in CH2Cl2 (2 mL) for 2 h. The cleavage was performed under mild acidic conditions, with 1% TFA in CH2Cl2 (1 mL) for 2 min. This step was repeated 10 times. After every 2 min, the resin was filtered directly into the solution of 10% pyridine in MeOH (2 mL) to neutralize the TFA. The mixture was concentrated to about 5% of the volume and cooled in an ice/water bath. H2O was added to precipitate the product as a creamy solid. The precipitation was filtered and washed few times with H2O, and dried in the desiccator.
Boc-Ala-Leu-Ala-Leu-Valu-Alau-Leuu-Valu-Alau-Leuu-Ala-Leu-Ala-Leu-NH2 (1). Chimera 1 was synthesized according to the general procedures GP1–GP6 starting from Sieber amide resin (100 mg, 0.062 mmol). The final product 1 was purified by semi preparative C18 RP-HPLC to give 8 mg (5% overall yield); RP-HPLC (H2O (0.1% TFA), MeOH (0.1% TFA); gradient 50–100%, 5 min; 100%, 5 min) tR = 7.79 min; 1H NMR (CD3OH): Table 1; 13C NMR (CD3OH): Table 1; ESI-MS (M = 1594.1): m/z 798.0 [M+2H]2+, 1594.9 [M+H]+, 1617.1 [M+Na]+.
Boc-Valu-Alau-Leuu-Ala-Leu-Ala-Leu-Ala-Leu-Ala-Valu-Alau-Leuu-NH2 (2). Chimera 2 was synthesized according the general procedures GP1–GP6 starting from Sieber amide resin (162 mg, 0.10 mmol). The final product 2 was purified by semi preparative C18 RP-HPLC to give 28 mg (18% overall yield); RP-HPLC (H2O (0.1% TFA), MeOH (0.1% TFA); gradient 50–100%, 5 min; 100%, 5 min) tR = 7.55 min; 1H NMR (CD3OH): Table 2; 13C NMR (CD3OH): Table 2; ESI-MS (M = 1481.02): m/z 763.5 [M+2H]2+, 1504.1 [M+Na]+.
4.3 NMR conformational analysis
Experiments have been performed on a Bruker Avance III 700 MHz spectrometer equipped with a 5 mm diameter BBI Gradient probe. Water suppression was achieved with a watergate sequence. Experiments were processed with the Bruker software (Bruker BioSpin, Courtaboeuf, France) and analyzed with the Sparky program (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Chimeras 1 and 2 were dissolved in 100% CD3OH to a final concentration of 4 mM. For each peptide, a complete series of 2D homonuclear and heteronuclear spectra was acquired at 293 K. A number of scans of 16, 8, and 32 were used, respectively, for COSY-DQF, TOCSY and ROESY experiments. The mixing times were set up to 60 ms for TOCSY and 300 ms for ROESY experiments. The 2D homonuclear spectra were acquired with 215 increments in F1 dimension and 2048 points in F2 dimension. The heteronuclear 1H15N and 1H13C spectra were acquired, respectively, with 38 and 128 increments in F1 dimension and with a number of scans of 1024 and 128. The 1H, 15N and 13C spectral width was set up, respectively, to 14, 22 and 120 ppm.
4.4 X-ray diffraction studies
Crystallographic data were collected at the IECB X-ray facility on a high flux microfocus Rigaku FRX rotating anode at the copper kα wavelength equipped with a Dectris Pilatus 200 K hybrid detector and Varimax HF optics at 130 K. The crystal was mounted on a cryo-loop after quick soaking on Paratone—N oil from Hampton research and flash-frozen. The data were processed using the CrystalClear suite version 2.1b25. The crystal structure was solved using SHELXD and refined with SHELXL 2013 version [56]. Full-matrix least-squares refinement was performed on F2 for all unique reflections, minimizing w(Fo2-Fc2)2, with anisotropic displacement parameters for non-hydrogen atoms. Hydrogen atoms were positioned at idealized positions and refined with a riding model, with Uiso constrained to the 1.2 Ueq value of the parent atom (1.5 Ueq when CH3). The positions and isotropic displacement parameters of the remaining hydrogen atoms were refined freely. SIMU and DELU commands were used to restrain some side chains as rigid groups and restrain their displacement parameters. The BYPASS/SQUEEZE [57] procedure was used to take into account the electron density in the potential solvent area.
CCDC-1041244 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request.cif.
Acknowledgements
This work was supported by the CNRS, Univ. Bordeaux, the Conseil Regional d’Aquitaine (Project #20091102003), ANR (Project #ANR-12-BS07-0019) and UREkA. CIFRE support from UREkA and ANRT to L. M. is gratefully acknowledged. We thank Jonathan Clayden for sharing results with us and for fruitful discussions.