

1 Introduction
The propensity of foldamers to fold and the properties of their folded conformations directly express features encoded in the structure of their building blocks [1]. As such, foldamer research critically depends on the development of new desirable monomers and their easy incorporation into sequences. Thus, synthetic foldamers do not face the same limitations as biopolymers which are based on small ensembles of units (e.g. twenty amino acids for proteins and four nucleobases for nucleic acids). In this manuscript, we present the synthesis of quinoline amino acids bearing oligoethylene glycol side-chains of various lengths, their incorporation into oligomers via solid-phase synthesis (SPS), and the determination of solubility properties endowed by these side-chains.
Aromatic amide foldamers are a growing class of oligomers that combine relatively easy synthetic access and stable and predictable conformations [2]. The folding of aromatic oligoamide foldamers arises from backbone rigidity due to local electrostatic repulsions and hydrogen bonds between amide function and adjacent units. The size and orientation of substituents on each monomer determine the strand curvature, leading to linear [3], zig-zag [4], or bent shapes [5], which eventually give rise to macrocycles [6], hydrogen-bonded duplexes [7], single [8] and multiple [9] helices, and aromatic β-sheets [10]. In sheets and helices, aromatic stacking, which may include electrostatic and solvophobic components, brings about additional stabilization [11]. Thus aromatic oligoamide folding depends primarily on main chain features, regardless of side-chains which, with a few exceptions [9(a)] and unlike in peptides and nucleotides, have little effect on the folded conformations. Monomer development for aromatic amide foldamers thus allows the consideration of main chain and side-chain attributes in an orthogonal manner. Over the years, a great variety of benzenic and heterocyclic building blocks have been introduced that can be combined at will [2,12].
For their part, side-chains determine the solubility. This is not as trivial a question as it may appear as aromatic oligoamides are inherently very poorly soluble molecules. Nevertheless, appropriate substitution with groups diverging from the folded structures allows solubility in all kinds of media. Some side-chains like isobutoxy groups provide high solubility in organic media and also favor crystallogenesis [8,9,10,13]. Interest in controlling the solubility of aromatic oligoamides in a wide range of solvents stems from the fact that folding has been shown to occur in solvents as diverse as chloroform, toluene, dimethyl sulfoxide, methanol or water [11(b),14]. While folded conformations do not depend much on the solvent, their stability may be greatly influenced as illustrated by considerable variations of the rate of helix handedness inversion in helical foldamers [11(b)]. These variations may have practical applications. For example, helix handedness induction by a chiral residue may be effective in a solvent in which handedness inversion is fast. Once handedness induction is complete, the same molecule may be transferred into another solvent in which the conformation is kinetically inert [15].
Side-chains also determine the ability of the folded structures to interact with other molecules, in particular protein surfaces [14(b),16] and nucleic acids [17]. This essential property has motivated the development of aromatic amino acids bearing a wide range of side-chains [14,16], and of microwave-assisted SPS methods to conveniently assemble them into varied sequences [18]. This allows the rapid synthesis of oligomers where any position in the sequence can be substituted for another, without the laborious preparation and purification of intermediates required by a solution-phase strategy.
In this manuscript, we focus on oligoamide foldamers derived from 8-amino-2-quinolinecarboxylic acid having side-chains in position 4 (Fig. 1) [8,11,14,15,17,18(a)]. These oligomers adopt very robust helical conformations that have been used to construct organic soluble, isobutoxy-substituted structures, of the size of small proteins [13]. The installation of an aminopropyl (i.e. ornithine-like) side-chain endows oligomers with good water solubility, with cell penetration properties [14(a),19], and with high affinity for G-quadruplex DNA [17]. Upon combining various proteinogenic side-chains, interactions with protein surfaces have been evidenced [14(b)]. In this context, we have been interested in introducing oligoethylene glycol functionalized monomers. Such side-chains would ensure the solubility of the same molecules in a wide range of solvents allowing, for example, the locking of helix handedness in water after inducing it in an organic solvent [15]; they may also potentially reduce toxicity [20]; and avoid non-specific interactions between foldamer surfaces and biomolecules [21], discouraging any interference with interactions involving designed recognition spots.
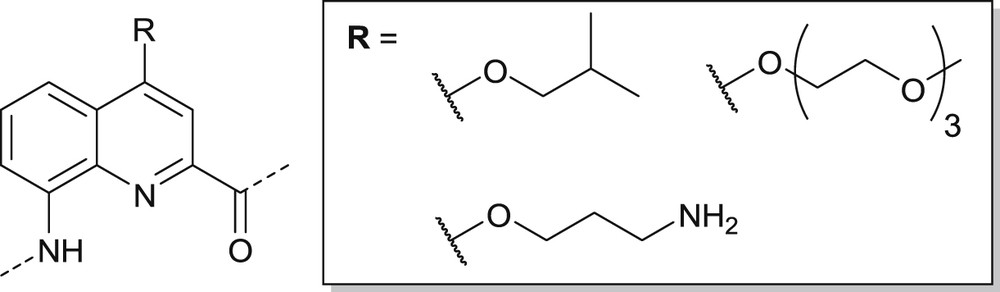
Isobutoxy, 3-aminopropoxy and triethylene glycol monomethyl ether side-chains at position 4 of 8-amino-2-quinolinecarboxamide monomers.
In a previous study [11(b)], we successfully incorporated a triethylene glycol side-chain on a monomer and prepared oligomers up to the octamer in solution (Fig. 1). These oligomers were shown to be soluble in solvents ranging from diethyl ether to methanol/water mixtures. However, it was noted that the aryl–alkyl ether linkage connecting the side-chain to the quinoline position 4 was somewhat acid-labile, most likely via a beta-elimination process, and that it would not be compatible with the acidic conditions required to cleave oligomers from the Wang resin after SPS [18]. Here we present a strategy for the synthesis of Fmoc protected quinoline monomers possessing oligoethylene glycol (OEG) side-chains ranging from mono- to tetra-ethylene glycol attached at the quinoline position 4 via an acid-resistant thioether linkage. These monomers were subsequently assembled in the SPS of four octameric sequences each consisting exclusively of one monomer type. We demonstrate that these side-chains are compatible with helix handedness separation by HPLC on a chiral stationary phase, and also investigate the effect of OEG side-chain length on the solubility profile of each octameric sequence.
2 Results and discussion
2.1 Synthesis
Our strategy was to design a generic synthesis for Fmoc-acid quinoline monomers ready for SPS which would allow the attachment of potentially any length of OEG side-chains. Thus, side-chains were introduced by the formation of the thioether linkage via aromatic nucleophilic substitution of a 4-bromoquinoline intermediate [14(b)] by the corresponding thiol (Scheme 1). Only the shortest side-chain precursor (2-methoxyethanethiol) is commercially available. Other precursors were prepared in three steps from OEG monomethyl ethers which are commercially available for a wide range of oligo- and poly-ethylene glycols. Following previously reported methods [22], these precursors were tosylated, subsequently substituted by thiourea and hydrolyzed to yield the desired thiol products in 24–64% yields over the three steps.

Synthesis of Fmoc quinoline building blocks with OEG side-chains.
Aromatic nucleophilic substitution of methyl 8-nitro-4-bromo-2-quinolinecarboxylate [14(b)] afforded 8-nitro 2-ester monomer intermediates in good yields (54–78%). These could be obtained with excellent purity due to their high crystallinity, even for the tri- and tetra-ethylene glycol chains. From there, the desired monomers (1–4) could be obtained through saponification of the methyl ester, reduction of the nitro group by catalytic hydrogenation and Fmoc protection of the amine.
Of these three steps, only the reduction step required some optimization; H2/Pd–C was found to be somewhat sluggish, presumably due to gradual poisoning of the catalyst by traces of thiol intermediates. This problem was easily overcome by using the more rapid NH4HCO2/Pd–C system. Fmoc monomers were therefore afforded in 30–50% overall yields on 1 g scale syntheses from the bromoquinoline intermediate. Interestingly all the Fmoc monomers were found to be amenable to crystallization. It was possible to use this as an extra purification step for achieving monomers of high purity. Fig. 2 shows the crystal structure of the tetraethylene glycol-functionalized monomer 4.

Crystal structure of monomer 4. Hydrogen atoms have been omitted for clarity.
SPS of quinoline carboxamide oligomers is commonly carried out on Wang resin and affords a free C-terminal carboxylic acid function after resin cleavage [18]. However, in order to afford oligomers which would be amenable to purification by both normal and reverse-phase HPLC, recycling gel permeation chromatography (GPC), or even flash chromatography, we decided to avoid the carboxylic acid function and to follow a strategy which would provide a primary amide at the C-terminus. This was anticipated to significantly reduce oligomer polarity without causing problems with solubility. Our initial attempts with SPS on Rink amide resin [23] were unsuccessful due to an unexpected resistance to the normal acidic cleavage conditions (95% TFA). We therefore utilized Sieber amide resin [24] which has a higher degree of acid lability. The first monomer could be loaded onto the resin either using HBTU/HOBt in DMF, or via the acid chloride, pre-formed by treatment of monomer acid with the Ghosez reagent (1-chloro-N,N,2-trimethyl-1-propenylamine) [25], in THF under microwave heating (Scheme 2). Fmoc deprotection was carried out at room temperature using 20% v/v piperidine in DMF and SPS continued in this fashion by coupling of the pre-formed monomer acid chlorides using the microwave-assisted methodology [18]. The sequence was terminated by coupling the nitro acid intermediate of each monomer in the same manner.
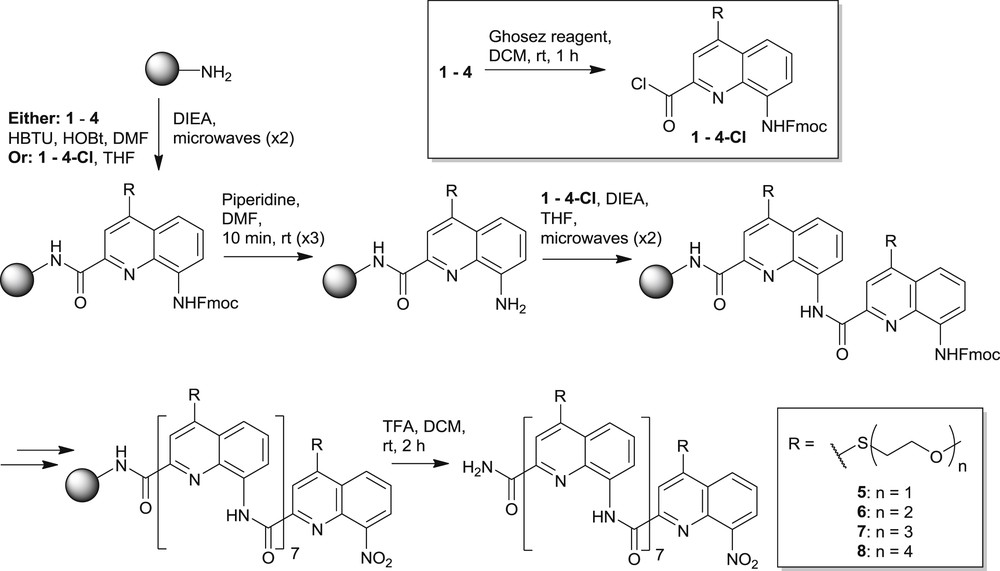
SPS of OEG functionalized octamers.
Thus, homomeric octamers 5–8 were prepared from the corresponding monomers 1–4 on a 16–32 μmol scale with crude purities ranging from 80 to 86%. Each oligomer could be purified by RP-HPLC, except for the monoethylene glycol-functionalized compound 5, due to poor solubility in water/acetonitrile mixtures. Instead, this was purified by recycling GPC in chloroform (see Experimental section). The tetraethylene glycol-functionalized octamer 8 was first purified by RP-HPLC, however it was clear by NMR and MS analysis that even after chromatographic treatment, the material also contained a nonameric sequence which could not be separated from the desired octamer due to very similar retention times on the C18 reverse-phase preparative column. Fortunately this nonameric by-product was sufficiently different in size from the desired octamer that it could be removed by recycling GPC. The insertion of an additional monomer presumably reflects slightly too harsh coupling conditions during one of the steps that may result in partial Fmoc cleavage while some activated acid is still present. To avoid this kind of side reaction and the subsequent difficult purification, it is recommended not to exceed the reported heating temperature and coupling duration.
2.2 Solubility studies
We anticipated that one critical difference between the four octamers synthesized would be their solubility profile. Previous experience with a triethylene glycol-functionalized octamer similar to 7 had demonstrated that this side-chain afforded solubility in a wide range of solvents at concentrations sufficient to allow the measurement of circular dichroism [11(b)]. However, while methanol/water mixtures containing up to 50% v/v water were tolerated, this octamer was not soluble in pure water. By increasing the side-chain length by one ethylene glycol unit, we hoped to access this property with compound 8. In addition, we aimed to investigate the solubility behavior of octamers 5 and 6, which during their purification had shown themselves to be more compatible with low polarity organic solvents.
A quantitative assessment of the solubility of 5–8 in eight typical solvents was carried out via UV absorbance measurement. A saturated solution of the studied oligomer was produced by adding excess solid material to a particular solvent, stirring and allowing a mixing time of 15 min at 20 °C. The mixture was then centrifuged at 6000 rpm for 5 min, or until a clear supernatant had been achieved as assessed by light microscopy. An accurately measured volume of the supernatant was then removed and evaporated to dryness. The residue was dissolved in DMSO, a solvent in which all foldamers were well soluble, and diluted to a concentration appropriate for UV absorbance measurement. This was subsequently carried out at 375 nm, corresponding to a local maximum of the quinoline oligoamide foldamer absorbance. The extinction coefficient at this wavelength had previously been calculated by measuring the absorbance of serial dilutions of an NMR sample of 8, for which the concentration had been assessed by comparison with an internal calibrant (see Supporting information). From this, the original concentration of foldamer in each solvent could be extrapolated, serving as an accurate measure of solubility. As shown in Table 1, there are significant differences in the solubility profile imparted by the adjustment of side-chain length. As we had anticipated, the tetraethylene glycol side-chain permitted a reasonable (mM) solubility of 8 in pure water. In contrast, the other octamers showed negligible aqueous solubility. The octamer 8 also possessed excellent solubility in most of the other solvents apart from n-hexane and isopropanol, similarly to the remaining compounds.
Estimated solubility (mM) of foldamers 5–8 in each solvent.
| Solvent | Estimated solubility (mM) | |||
| 5 | 6 | 7 | 8 | |
| Hexane | <0.01 | <0.01 | <0.01 | <0.01 |
| Toluene | 0.20 | 0.28 | 55 | 69 |
| Chloroform | 158 | 39 | 86 | 59 |
| THF | 13 | 70 | 64 | 85 |
| DMF | 21 | 73 | 105 | 88 |
| Isopropanol | 0.02 | 0.03 | 0.03 | 0.27 |
| Methanol | 0.04 | 0.13 | 1.5 | 20 |
| Water | <0.01 | <0.01 | <0.01 | 2.5 |
The behavior of the triethylene glycol-functionalized octamer 7 matched well with our previous experiment, which demonstrated similar properties to 8, with the difference that solubility in protic solvents was diminished by at least one order of magnitude. Decreasing the side-chain length further to diethylene glycol (6) decreased the solubility of the octamer to sub-millimolar levels in both toluene and methanol, with limited effects on other solvents. Finally, as anticipated, the monoethylene glycol-functionalized octamer 5 showed a marked reduction in solubility in all polar solvents, although this was also accompanied by a near doubling of solubility in chloroform compared to 7, the next most soluble oligomer in this solvent. These results show that the solubility properties of the quinoline-based foldamers can be effectively tailored by altering the length of OEG-based side-chains, allowing access to highly soluble oligomers compatible with both water and a range of organic solvents.
2.3 Chiral HPLC
During studies of the tetraethylene glycol-functionalized octamer 8, it became clear to us that its highly soluble nature could be taken as an advantage, with respect to the separation of helix handedness forms. We have previously demonstrated that the handedness inversion kinetics of the quinoline oligoamide foldamers are highly solvent dependent, with an octameric sequence similar to that of 7 demonstrating half lives varying from minutes in aprotic solvents to days in protic solvents [11(b)]. In water, kinetics are essentially frozen: aqueous solutions of separated P and M helices of octamers have been left at room temperature for months and demonstrated no evidence of handedness inversion [15]. Separation of P and M helices of water-soluble oligomers was previously carried out using a phenylalanine unit installed at the C-terminus to permit RP-HPLC separation of the two helix forms as diastereoisomers. In contrast, we envisaged that the good solubility of 8 in both organic and aqueous solvents would permit the use of chiral HPLC to separate the two helix handedness forms as enantiomers. If left in the solid state, or if dissolved in water handedness would remain ‘locked’. This strategy proved to be valid as we successfully used a standard Chiralpak-IA chiral analytical HPLC column to separate the P and M helix forms of compound 8 at low temperature (Fig. 3a, b) in organic solvents. In-line circular dichroism demonstrated elution of the M-helix as the first peak, followed by the P-helix; these could be isolated with ee values of >99% and 93% respectively (Fig. 3c, d). Conveniently, handedness inversion kinetics in the solvent mixture and temperature used for chiral HPLC (MeOH/CHCl3, 75:25, −5 °C) appeared to be extremely slow: the isolated M helix was subsequently incubated for up to eight hours under these conditions, with no visible racemization (see Supporting information). We could also use the same column to separate one-handed forms of the monoethylene glycol-functionalized octamer 5 at room temperature, and this also demonstrated the same order of elution as compound 8 (see Supporting information).

Chiral HPLC separation of P and M helices of compound 8 at −5 °C in MeOH/CHCl3 (75:25). Expansion of: (a) UV detector trace (254 nm); (b) CD detector trace (385 nm). Overlaid expansions of isolated P-8 (gray) and M-8 (black); (c) UV detector traces (254 nm); (d) CD detector traces (385 nm).
In summary, the tetraethylene glycol side-chains allow facile separation of P and M helices by using chiral HPLC in organic solvents, yet also permit solubility under aqueous conditions where helix handedness inversion kinetics are essentially frozen. For oligomers bearing these side-chains, this could conceivably be a convenient method to isolate usable quantities of separate handedness forms without requiring the installation of chiral groups for handedness induction, or separation of diastereoisomers.
3 Conclusions
Four Fmoc-acid quinoline monomers have been synthesized with OEG side-chains installed at position four through an acid-stable thioether linkage. The general methodology for their preparation is compatible with the production of monomers possessing longer OEG units; indeed probably the main limiting factor is the commercial availability of the OEG monomethyl ether starting material. From these monomers four octameric sequences, each consisting exclusively of one monomer type, were synthesized by a microwave-assisted SPS methodology. Their solubility profiles were investigated and it was shown that elongation of the side-chain unit to tetraethylene glycol permitted excellent solubility of the corresponding octameric sequence (8) in the majority of organic solvents studied. More importantly, this compound was also found to possess modest solubility in pure water, which has not been demonstrated before in quinoline oligoamide foldamers without the presence of multiple charged side-chains. The P and M helix handedness forms of octamer 8 were separated using a standard chiral HPLC stationary phase, providing for the first time a method to isolate water-soluble helices as their separate enantiomers, without requiring chiral groups for handedness induction, or separation of the respective diastereoisomers.
Future work will investigate the use of OEG functionalized monomer units in sequences designed to interact with biomolecules. While providing foldamers with the necessary aqueous solubility, we foresee that monomers such as 8 can also have a potential role in eliminating undesired non-specific interactions of the foldamer backbone with a biological target.
4 Experimental
4.1 General methods
Sieber amide resin was purchased from Novabiochem. Ghosez reagent (1-chloro-N,N,2-trimethyl-1-propenylamine) was purchased from Sigma–Aldrich. N,N-diisopropylethylamine was distilled over calcium hydride. Analytical grade organic solvents were used for solid phase synthesis. Anhydrous THF and CH2Cl2 for solution and solid phase synthesis were dispensed from an MBRAUN SPS-800 solvent purification system. HPLC grade acetonitrile and MilliQ water were used for RP-HPLC analyses and purification. Reactions requiring anhydrous conditions were performed under nitrogen. SPS was carried out manually using a CEM Discover microwave and vacuum station, and in proprietary reactor vessels. 1H NMR spectra were measured at 300 MHz and 13C NMR spectra were measured at 75 MHz. Chemical shifts are reported in ppm and are calibrated against residual solvent signals of CDCl3 (δ 7.26, 77.2), or DMSO-d6 (δ 2.50, 39.4). All coupling constants are reported in hertz (Hz). Signals were abbreviated as s, singlet; br s, broad singlet; d, doublet; t, triplet; q, quartet; m, multiplet, dd, doublet of doublets. Silica gel chromatography was performed using a Merck Kieselgel Si 60. Thin layer chromatography was performed using Merck Kieselgel Si 60 F254 plates. Recycling gel permeation chromatography was performed at 37 °C on a JAI LC-9130G NEXT using a JAIGEL 2.5H or a JAIGEL 3H column (20 × 600 mm) with chloroform (HPLC grade, ethanol stabilized) as the eluent running at 3.5 mL min−1. Chiral analytical HPLC was performed using a Daicel Chiralpak IA column with either isocratic ethyl acetate/chloroform (50:50) or methanol/chloroform (75:25) as the eluent running at 0.5 mL min-1 at room temperature (compound 5) or 1.0 mL min−1 at −5 °C (compound 8). RP-HPLC analyses were performed at 1.5 mL min−1 using a Macherey-Nagel Nucleodur C18 or C8 Gravity column (4.6 × 100 mm, 3 μm). The mobile phase was composed of 0.1% (v/v) TFA-H2O (Solvent A) and 0.1% TFA-CH3CN (Solvent B) running the following gradients: 50–100% B over 15 min (System A), 40–90% B over 20 min (System B), or 50–100% B over 20 min (System C). Monitoring by UV detection was carried out at 214 nm, 254 nm and 300 nm using a diode array detector. Semi-preparative purifications of oligomers were performed at 4 mL min−1 on a semipreparative HPLC using a Macherey-Nagel Nucleodur C18 HTEC column (21 mm × 125 mm, 5 μm). The mobile phase was the same as that for the analytical system, running the following gradients: 50–100% B over 20 min (System D), 40–90% B over 15 min (System E), or 10–100% B over 25 min (System F). Monitoring by UV detection was carried out at 254 nm and 300 nm using a diode array detector. High resolution electrospray ionization time of flight (ESI-TOF) mass spectra were measured in the positive ion mode on a Waters/Micromass Q-Tof Ultima. X-ray diffraction experiments were carried out on a high flux RIGAKU FRX rotating anode at the Cu Kα wavelength.
CCDC-1056802 for monomer 4 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
4.2 Monomer synthesis
2-Methoxyethanethiol was commercially available while the other 3 thiols [2-(2-methoxyethoxy)ethanethiol, 2-(2-(2-methoxyethoxy)ethoxy)ethanethiol, 2,5,8,11-tetraoxatridecane-13-thiol] were synthesized according to literature procedures [22]. Methyl 4-bromo-8-nitroquinoline-2-carboxylate was prepared as previously reported [14(b)]. Fmoc-monomers 1–4 were produced using the same general methodology. This is exemplified below by the synthesis of compound 1 and its corresponding intermediates:
4.2.1 General procedure for nucleophilic aromatic substitution
4.2.1.1 Methyl 4-(2-methoxyethylthio)-8-nitroquinoline-2-carboxylate
Methyl 4-bromo-8-nitroquinoline-2-carboxylate (2.5 g, 8.0 mmol) and Cs2CO3 (3.9 g, 12.0 mmol, 1.5 equiv) were suspended in 75 mL anhydrous DMF. 2-Methoxyethanethiol (815 mg, 8.8 mmol, 1.1 equiv) was added and the resulting slurry stirred at room temperature for 48 h under N2. The reaction mixture was then diluted with brine, extracted with EtOAc and the combined organic phases were dried over Na2SO4, filtered, and evaporated. Recrystallization in DCM/MeOH afforded the title compound as an orange solid (1.8 g, 71%). 1H NMR (CDCl3): δ 8.39 (1H, dd, J = 8.6, 1.3), 8.12 (1H, s), 8.08 (1H, dd, J = 7.5, 1.2), 7.69 (1H, dd, J = 8.6, 7.4), 4.04 (3H, s), 3.81 (2H, t, J = 6.0), 3.44 (2H, t, J = 5.9), 3.44 (3H, s). 13C NMR (CDCl3): δ 128.17, 127.67, 126.92, 124.79, 117.02, 69.91, 59.26, 53.59, 31.76. HRMS (ES+): m/z calcd for C14H15N2O5S [M+H]+ 323.0696 found 323.0702.
4.2.1.2 Methyl 4-(2-(2-methoxyethoxy)ethylthio)-8-nitroquinoline-2-carboxylate
Synthesized using the general methodology mentioned above. Purification by silica gel chromatography (cyclohexane/EtOAc (6:4) to cyclohexane/EtOAc (4:6)) followed by precipitation in DCM/MeOH afforded the title compound as a yellow solid (2.3 g, 78%). 1H NMR (CDCl3): δ 8.38 (1H, dd, J = 8.5, 1.3), 8.12 (1H, s), 8.07 (1H, dd, J = 7.4, 1.3), 7.69 (1H, dd, J = 8.5, 7.5), 4.03 (3H, s), 3.90 (2H, t, J = 6.2), 3.71–3.66 (2H, m), 3.58–3.54 (2H, m), 3.46 (2H, t, J = 6.3), and 3.38 (3H, s). 13C NMR (CDCl3): δ 165.60, 150.74, 149.34, 148.91, 138.73, 128.19, 127.69, 126.88, 124.77, 117.10, 72.06, 70.85, 68.85, 59.30, 53.56, 31.69. HRMS (ES+): m/z calcd for C16H19N2O6S [M+H]+ 367.0958 found 367.0968.
4.2.1.3 Methyl 4-(2-(2-(2-methoxyethoxy)ethoxy)ethylthio)-8-nitroquinoline-2-carboxylate
Synthesized using the general methodology mentioned above. Purification by silica gel chromatography (cyclohexane/EtOAc (8:2) to cyclohexane/EtOAc (1:1)) afforded the title compound as a yellow solid (2.3 g, 71%). 1H NMR (CDCl3): δ 8.36 (1H, dd, J = 8.6, 1.3), 8.11 (1H, s), 8.06 (1H, dd, J = 7.5, 1.3), 7.68 (1H, dd, J = 8.4, 7.5), 4.02 (3H, s), 3.89 (2H, t, J = 6.2), 3.72–3.60 (6H, m), 3.54–3.49 (2H, m), 3.44 (2H, t, J = 6.2), 3.35 (3H, s). 13C NMR (CDCl3): δ 165.64, 150.86, 149.38, 148.94, 138.75, 128.21, 127.71, 126.95, 124.81, 117.14, 72.15, 70.98, 70.85, 68.89, 59.27, 53.60, 31.82. HRMS (ES+): m/z calcd for C18H23N2O7S [M+H]+ 411.1226 found 411.1226.
4.2.1.4 Methyl 4-(2,5,8,11-tetraoxatridecan-13-ylthio)-8-nitroquinoline-2-carboxylate
Synthesized using the general methodology mentioned above. Purification by silica gel chromatography (cyclohexane/EtOAc (8:2) to cyclohexane/EtOAc (1:1)) followed by crystallization from Et2O at 4 °C afforded the title compound as a white solid (2.0 g, 54%). 1H NMR (CDCl3): δ 8.39 (1H, dd, J = 8.6, 1.3), 8.13 (1H, s), 8.07 (1H, dd, J = 7.5, 1.3), 7.69 (1H, dd, J = 8.5, 7.5), 4.04 (3H, s), 3.90 (2H, t, J = 6.2), 3.73–3.61 (10H, m), 3.56–3.51 (2H, m), 3.45 (2H, t, J = 6.2), 3.37 (3H, s). 13C NMR (CDCl3): δ 165.73, 150.91, 149.46, 149.02, 128.30, 127.79, 126.99, 124.86, 117.20, 72.22, 71.04, 70.94, 70.90, 70.88,70.82, 68.92, 59.32, 53.67, and 31.88. HRMS (ES+): m/z calcd for C10H26N2O8S [M+H]+ 455.1483 found 455.1493
4.2.2 General procedure for saponification of methyl ester
4.2.2.1 4-(2-Methoxyethylthio)-8-nitroquinoline-2-carboxylic acid
4-(2-methoxyethylthio)-8-nitroquinoline-2-carboxylate (1.8 g, 5.6 mmol) was dissolved in 280 mL THF/H2O (4:1). LiOH·H2O (470 mg, 11.2 mmol, 2 equiv) was added and the reaction was stirred for 45 min at room temperature. 5% w/v citric acid solution was then added to reach pH 4, THF was evaporated under reduced pressure and the resulting aqueous mixture was extracted with DCM. The combined organic phases were washed with H2O and brine, dried over Na2SO4, filtered and evaporated to yield the title compound as a yellow solid (1.7 g, 97%). 1H NMR (CDCl3): δ 8.47 (1H, dd, J = 8.6, 1.2), 8.21 (1H, dd, J = 7.6, 1.1), 8.21 (1H, s), 7.77 (1H, dd, J = 8.3, 7.6), 3.83 (2H, t, J = 5.9), 3.49 (2H, t, J = 5.9), 3.44 (3H, s). 13C NMR (CDCl3): δ 163.53, 153.81, 148.03, 146.48, 136.91, 128.43, 128.25, 127.44, 126.00, 114.76, 69.70, 59.27, 31.95. HRMS (ES+): m/z calcd for C13H13N2O5S [M+H]+ 309.0540 found 309.0551.
4.2.2.2 4-(2-(2-Methoxyethoxy)ethylthio)-8-nitroquinoline-2-carboxylic acid
Synthesized using the general methodology mentioned above to afford the title compound as a yellow solid (1.8 g, 96%). 1H NMR (CDCl3): δ 8.44 (1H, dd, J = 8.6, 1.2), 8.20 (1H, s), 8.19 (1H, dd, J = 7.0, 1.3), 7.75 (1H, dd, J = 8.6, 7.6), 3.92 (2H, t, J = 5.9), 3.71–3.66 (2H, m), 3.59–3.54 (2H, m), 3.49 (2H, t, J = 6.1), 3.37 (3H, s). 13C NMR (CDCl3): δ 163.51, 153.83, 148.02, 146.50, 136.90, 128.45, 128.28, 127.39, 125.96, 114.92, 72.03, 70.85, 68.71, 59.26. HRMS (ES+): m/z calcd for C15H17N2O6S [M+H]+ 353.0802 found 353.0812.
4.2.2.3 4-(2-(2-(2-Methoxyethoxy)ethoxy)ethylthio)-8-nitroquinoline-2-carboxylic acid
Synthesized using the general methodology mentioned above to afford the title compound as a yellow solid (1.9 g, 98%). 1H NMR (CDCl3): δ 8.46 (1H, dd, J = 8.6, 1.2), 8.24 (1H, s), 8.20 (1H, dd, J = 7.6, 1.2), 7.76 (1H, dd, J = 8.5, 7.7), 3.93 (2H, t, J = 6.0), 3.74–3.62 (6H, m), 3.55–3.51 (2H, m), 3.49 (2H, t, J = 6.0), 3.36 (3H, s). 13C NMR (CDCl3): δ 163.50, 153.95, 148.10, 146.53, 136.96, 128.53, 128.31, 127.40, 125.98, 114.98, 72.11, 71.00, 70.81, 68.80, 59.22, 32.02. HRMS (ES+): m/z calcd for C17H21N2O7S [M+H]+ 397.1069 found 397.1073.
4.2.2.4 4-(2,5,8,11-Tetraoxatridecan-13-ylthio)-8-nitroquinoline-2-carboxylic acid
Synthesized using the general methodology mentioned above to afford the title compound as a yellow solid (1.6 g, 93%). 1H NMR (CDCl3): δ 8.43 (1H, d, J = 8.5), 8.20 (1H, s), 8.19 (1H, d, J = 7.3), 7.76 (1H, t, J = 8.0), 3.94 (2H, t, J = 6.0), 3.75–3.60 (10H, m), 3.58–3.52 (2H, m), 3.49 (2H, t, J = 6.0), 3.37 (3H, s). 13C NMR (CDCl3): δ 163.72, 153.53, 147.99, 146.76, 136.93, 128.28, 128.15, 127.26, 125.80, 115.05, 71.99, 70.85, 70.71, 70.66, 70.55, 68.64, 59.09, 53.58, 31.89. HRMS (ES+): m/z calcd for C19H25N2O8S [M+H]+ 441.1326 found 441.1331.
4.2.3 General procedure for reduction of nitro group
4.2.3.1 8-Amino-4-(2-methoxyethylthio)quinoline-2-carboxylic acid
4-(2-methoxyethylthio)-8-nitroquinoline-2-carboxylic acid (1.6 g, 5.2 mmol) was dissolved in 375 mL EtOAc/EtOH (4:1), 10% Pd/C (0.16 g, 10% by mass) and NH4VO3 (124 mg, 1.1 mmol, 0.2 equiv) were added and the mixture was heated to reflux (bath temperature 95 °C). A solution of NH4HCO2 (15.7 g, 248 mmol, 48 equiv) in 12 mL H2O was then added drop-wise to the reaction mixture over 10 min and the mixture stirred for 4 h under reflux. The reaction mixture was cooled to room temperature, EtOAc added and the resulting solution filtered on Celite. The organic phase was washed with water and brine, dried over Na2SO4, filtered, and solvents were evaporated under reduced pressure to yield the title compound as an orange-red oil, which was used directly in the next step without further purification. 1H NMR (DMSO-d6): δ 12.83 (1H, s), 7.90 (1H, s), 7.42 (1H, t, J = 8.0), 7.12 (1H, dd, J = 8.2, 1.0), 6.89 (1H, dd, J = 7.8, 1.0), 6.63 (2H, br s), 3.70 (2H, t, J = 6.0), 3.43 (2H, t, J = 6.0), 3.31 (3H, s).
4.2.3.2 8-Amino-4-(2-(2-methoxyethoxy)ethylthio)quinoline-2-carboxylic acid
1H NMR (DMSO-d6): δ 12.83 (1H, s), 7.92 (1H, s), 7.42 (1H, t, J = 8.0), 7.12 (1H, dd, J = 8.3, 1.1), 6.89 (1H, dd, J = 7.7, 1.1), 6.63 (2H, br s), 3.77 (2H, t, J = 6.1), 3.61–3.56 (2H, m), 3.48–3.39 (4H, m), 3.23 (3H, s).
4.2.3.3 8-Amino-4-(2-(2-(2-methoxyethoxy)ethoxy)ethylthio)quinoline-2-carboxylic acid
1H NMR (DMSO-d6): δ 7.91 (1H, s), 7.37 (1H, t, J = 7.9), 7.11 (1H, d, J = 8.0), 6.87 (1H, d, J = 7.5), 6.45 (2H, br s), 3.77 (2H, t, J = 6.1), 3.62–3.46 (6H, m), 3.44–3.35 (4H, m), 3.21 (3H, s).
4.2.3.4 4-(2,5,8,11-Tetraoxatridecan-13-ylthio)-8-aminoquinoline-2-carboxylic acid
1H NMR (CD3OD): δ 8.13 (1H, s), 7.43–7.30 (2H, m), 6.98 (1H, dd, J = 7.1, 1.3), 3.88 (2H, t, J = 6.4), 3.72–3.56 (10H, m), 3.54–3.48 (2H, m), 3.43 (2H, t, J = 6.4).
4.2.4 General procedure for Fmoc installation
4.2.4.1 8-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-4-((2-methoxyethyl)thio)quinoline-2-carboxylic acid (1)
8-Amino-4-(2-methoxyethylthio)quinoline-2-carboxylic acid (1.2 g, 4.1 mmol) was dissolved in 22 mL 1,4-dioxane, 73 mL of a 10% w/v aqueous solution of NaHCO3 was added, and the mixture was stirred at 0 °C (ice-bath) under N2. 1.2 g of Fmoc-Cl (1.2 g, 4.5 mmol, 1.1 equiv) was dissolved in 63 mL 1.4-dioxane, and added dropwise via a dropping funnel under N2 over approximately 1 h. The mixture was then stirred at 0 °C for a further 1 h and then at rt for 20 h. Approximately 100 mL H2O was then added, and the resulting mixture acidified to pH 6 by portion-wise addition of 1 M HCl. The mixture was then extracted with DCM and the combined organic phases washed with brine, dried over Na2SO4 filtered and evaporated under reduced pressure. Purification by silica gel chromatography (100% DCM to DCM/MeOH 9:1), followed by crystallization in DCM afforded the title compound as a white solid (1.1 g, 43%). 1H NMR (DMSO-d6): δ 13.59 (1H, s), 10.44 (1H, s), 8.06 (1H, s), 7.93 (2H, d, J = 7.4), 7.80–7.72 (3H, m), 7.67 (1H, t, J = 8.3), 7.47–7.33 (4H, m), 4.62 (2H, d, J = 6.7), 4.45 (1H, t, J = 6.8), 3.72 (2H, t, J = 6.0), 3.51 (2H, t, J = 6.0), 3.31 (3H, s). 13C NMR (DMSO-d6): δ 165.40, 153.52, 149.98, 144.30, 143.78, 140.89, 136.42, 135.62, 129.33, 127.85, 127.28, 126.65, 125.23, 120.29, 116.50, 116.05, 114.75, 69.41, 66.54, 58.13, 46.68, 30.38. HRMS (ES+): m/z calcd for C28H25N2O5S [M+H]+ 501.1479 found 501.1478.
4.2.4.2 8-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-4-(2-(2-methoxyethoxy)ethylthio)quinoline-2-carboxylic acid (2)
Synthesized using the general methodology mentioned above to afford the title compound as a white solid (0.96 g, 68%). 1H NMR (DMSO-d6): δ 7.99 (1H, s), 7.92 (2H, d, J = 7.2), 7.74 (2H, d, J = 7.3), 7.66 (1H, d, J = 8.1), 7.53 (1H, t, J = 7.8), 7.47–7.32 (4H, m), 4.64 (2H, d, J = 6.1), 4.43 (1H, t, J = 6.2), 3.76 (2H, t, J = 6.2), 3.60–3.55 (2H, m), 3.47–3.42 (2H, m), 3.40 (2H, t, J = 6.1), 3.23 (3H, s). 13C NMR (DMSO-d6): δ 167.67, 152.85, 150.73, 147.84, 143.59, 140.70, 135.79, 135.34, 128.87, 127.63, 127.12, 125.70, 124.96, 121.32, 119.97, 116.27, 115.89, 115.05, 69.24, 66.33, 57.85, 54.89, 46.49, 30.22. HRMS (ES+): m/z calcd for C30H29N2O6S [M+H]+ 545.1741 found 545.1750.
4.2.4.3 8-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-4-(2-(2-(2-methoxyethoxy)ethoxy)ethylthio)quinoline-2-carboxylic acid (3)
Synthesized using the general methodology mentioned above. Purification by silica gel chromatography (100% DCM to DCM/MeOH 92:8) followed by precipitation in DCM afforded the title compound as a white solid (1.2 g, 67%). 1H NMR (DMSO-d6): δ 13.60 (1H, s), 10.47 (1H, s), 8.38 (1H, br s), 8.08 (1H, s), 7.93 (2H, d, J = 7.2), 7.81–7.72 (3H, m), 7.67 (1H, t, J = 8.1), 7.47–7.31 (4H, m), 4.62 (2H, d, J = 6.8), 4.45 (1H, t, J = 6.6), 3.80 (2H, t, J = 6.1), 3.62–3.45 (8H, m), 3.40–3.35 (2H, m), 3.31 (2H, br s), 3.20 (2H, s). 13C NMR (DMSO-d6): δ 165.35, 153.41, 149.86, 144.36, 143.67, 140.79, 136.31, 135.56, 129.20, 127.74, 127.18, 126.58, 125.12, 120.19, 116.35, 115.97, 114.83, 71.22, 69.81, 69.74, 69.59, 68.05, 66.44, 64.89, 57.99, 46.57, 30.47, 15.13. HRMS (ES+): m/z calcd for C32H33N2O7S [M+H]+ 589.2003 found 589.2017.
4.2.4.4 8-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-4-(2,5,8,11-tetraoxatridecan-13-ylthio)-8-aminoquinoline-2-carboxylic acid (4)
Synthesized using the general methodology mentioned above. Purification by silica gel chromatography (100% DCM to DCM/MeOH 9:1) afforded the title compound as a white solid (1.1 g, 73%). 1H NMR (DMSO-d6): δ 10.01 (1H, s), 8.31 (1H, s), 8.25 (1H, s), 8.05 (1H, s), 7.89 (2H, d, J = 7.4), 7.74–7.64 (3H, m), 7.58 (1H, t, J = 7.8), 7.40 (2H, t, J = 7.3), 7.31 (2H, t, J = 7.2), 4.55 (2H, d, J = 6.4), 4.36 (1H, t, J = 6.6), 3.74 (2H, t, J = 5.8), 3.58–3.54 (14H, m), 3.20 (3H, s). 13C NMR (DMSO-d6): δ 153.06, 148.55, 143.61, 140.72, 135.73, 128.87, 128.25, 127.68, 127.15, 126.06, 125.02, 121.33, 120.16, 119.97, 115.97, 115.70, 115.53, 79.16, 71.21, 69.77, 69.73, 69.70, 69.51, 67.98, 66.38, 57.98, 46.48, 30.40. HRMS (ES+): m/z calcd for C34H37N2O8S [M+H]+ 633.2265 found 633.2259.
4.3 General procedure for SPS of octamers
Octamers 5–8 were produced using the same general methodology, exemplified here by the synthesis of compound 5:
4.3.1 Monomer activation
4.3.1.1 Compound 1-Cl
Compound 1 (49 mg, 0.098 mmol, 6 equiv relative to resin loading) was suspended in anhydrous CH2Cl2 (2 mL) under N2. Ghosez reagent was added (26 μL, 0.195 mmol, 2 equiv) and the reaction was stirred for 1 h at rt under N2. Solvents were evaporated under reduced pressure on a vacuum line (approximately 2 h).
4.3.1.2 4-(2-Methoxyethylthio)-8-nitroquinoline-2-carbonyl chloride
4-(2-Methoxyethylthio)-8-nitroquinoline-2-carboxylic acid (30.2 mg, 0.098 mmol, 6 equiv) was activated in the same manner as described above.
4.3.2 SPS
4.3.2.1 Compound 5
Synthesized on a 16.3 μmol scale (25 mg Sieber amide resin with manufacturer's loading: 0.65 mmol g−1). The resin was initially swollen for 1 h in DMF. Fmoc deprotection: the resin was stirred in a 20% v/v solution of piperidine in DMF at rt for 10 min, reagents were removed under vacuum, the resin was washed with DMF briefly, and treatment was repeated twice. The resin was then washed thoroughly with DMF. Coupling of monomer acid chlorides (also the loading method for compounds 5–7): The resin was washed thoroughly with anhydrous THF, then suspended in 1.3 mL anhydrous THF, to which anhydrous DIEA (34 μL, 0.196 mmol, 6 equiv) was added. Compound 1-Cl (assuming complete conversion to acid chloride, 0.098 mmol, 6 equiv) was dissolved in 2.6 mL anhydrous THF under N2, and 1.3 mL of this solution (i.e. 3 equiv of acid chloride) was added to the resin which was then treated with microwaves (50 W, 50 °C, 5 min). The resin was washed briefly with anhydrous THF and the process was repeated with the remaining acid chloride solution. The resin was then thoroughly washed with anhydrous THF, then DMF. Coupling of the N-terminal nitro-functionalized quinoline monomer 4-(2-methoxyethylthio)-8-nitroquinoline-2-carbonyl chloride was carried out in the same manner. Resin cleavage: the resin was washed thoroughly with DMF, DCM and DCM/MeOH (1:1), dried and desiccated. It was then suspended in 2 mL of TFA/DCM (1:1 v/v) and stirred at rt for 1 h. The resin was removed by filtration and solvents were evaporated under reduced pressure to yield the crude product. This was purified by recycling GPC (due to the limited solubility under RP-HPLC conditions) and followed by an additional crystallization in DCM/MeOH to afford the title compound as a yellow solid (8 mg, 20%). 1H NMR (CDCl3): δ 11.43 (1H, s), 11.13 (1H, s), 11.03 (1H, s), 10.99 (3H, s), 10.91 (1H, s), 8.23 (1H, dd, J = 7.0, 2.6), 8.17–8.11 (3H, m), 8.03 (1H, dd, J = 7.8, 1.1), 8.00 (1H, dd, J = 8.4, 1.2), 7.95 (1H, dd, J = 8.5, 1.1), 7.82–7.74 (3H, m), 7.67–7.57 (3H, m), 7.56–7.47 (3H, m), 7.47–7.28 (9H, m), 7.22 (1H, d, J = 7.8), 7.11 (1H, d, J = 8.1), 7.07 (1H, s), 7.01 (1H, s), 6.96 (1H, s), 6.91 (1H, s), 6.88 (1H, s), 6.57 (1H, s), 6.40 (1H, d, J = 4.0), 4.18 (1H, d, J = 4.0), 4.08–3.96 (4H, m), 3.93–3.73 (10H, m), 3.68 (3H, s), 3.66 (3H, s), 3.61 (3H, s), 3.56 (3H,s), 3.55 (3H, s), 3.52 (3H, s), 3.49 (3H, s), and 3.47–3.07 (21H, m). 13C NMR (CDCl3): δ 164.74, 163.72, 163.12, 160.61, 160.51, 160.03, 159.96, 159.33, 58.89, 158.82, 154.92, 152.28, 151.34, 150.86, 150.78, 150.63, 150.52, 150.14, 149.89, 149.73, 149.63, 149.36, 147.17, 146.31, 146.20, 146.12, 145.97, 145.56, 145.47, 144.67, 144.51, 137.37, 136.28, 136.10, 136.02, 135.97, 135.64, 135.48, 134.73, 134.20, 133.53, 133.41, 133.32, 133.00, 132.89, 129.15, 128.44, 128.34, 128.22, 127.66, 127.63, 127.56, 127.45, 127.41, 127.35, 127.23, 127.12, 127.02, 126.98, 126.91, 126.86, 126.68, 125.88, 125.82, 125.15, 125.12, 118.86, 118.70, 118.40, 118.22, 117.94, 117.24, 117.09, 116.73, 116.03, 115.63, 114.47, 113.99, 113.23, 112.83, 112.41, 112.25, 112.13, 111.95, 70.71, 70.44, 70.11, 59.47, 59.45, 59.42, 59.40, 59.34, 59.31, 59.24, 31.70, 31.48, 31.38, 31.27, 31.23. HRMS (ES+): m/z calcd for C104H98N17O18S8 [M+H]+ 2128.5042 found 2128.5109.
4.3.2.2 Compound 6
Synthesized on a 32.5 μmol scale (50 mg Sieber amide resin with manufacturer's loading: 0.65 mmol g−1) using the general methodology mentioned above. The crude product was purified by preparative RP-HPLC (System D) to afford the title compound as a yellow solid (22 mg, 28%). RP-HPLC (System A): Rt = 18.17 min 1H NMR (CDCl3): δ 11.42 (1H, s), 11.13 (1H, s), 11.03 (1H, s), 11.00 (1H, s), 10.99 (1H, s), 10.97 (1H, s), 10.90 (1H, s), 8.27–8.20 (1H, m), 8.19–8.11 (3H, m), 8.06–8.92 (3H, m), 7.82–7.74 (3H, m), 7.68–7.56 (3H, m), 7.55–7.47 (3H, m), 7.46–7.28 (8H, m), 7.20 (1H, d, J = 7.2), 7.15–7.05 (3H, m), 7.04 (1H, s), 6.96 (1H, s), 6.93 (1H, s), 6.88 (1H, s), 6.56 (1H, s) 6.38 (1H, br s), 4.59 (1H, br s) 4.20–3.97 (14H, m), 3.90–3.60 (m, 50H), 3.57 (3H, s), 3.56 (3H, s), 3.55 (3H, s), 3.49 (3H, s), 3.46 (3H, s), 3.44 (3H, s), 3.43 (3H, s), 3.40 (3H, s). HRMS (ES+): m/z calcd for C120H130N17O26S8 [M+H]+ 2480.7139 found 2480.7204.
4.3.2.3 Compound 7
Synthesized on a 32.5 μmol scale (50 mg Sieber amide resin with manufacturer's loading: 0.65 mmol g−1) using the general methodology mentioned above. The crude product was purified by preparative RP-HPLC (System E) to afford the title compound as a yellow solid (14 mg, 15%). RP-HPLC (System B): Rt = 8.98 min 1H NMR (CDCl3): δ 11.42 (1H, s), 11.12 (1H, s), 11.03 (1H, s), 10.98 (3H, s), 10.89 (1H, s), 8.26–8.19 (1H, m), 8.18–8.10 (3H, m), 8.07–7.92 (3H, m), 7.82–7.73 (3H, m), 7.69–7.56 (3H, m), 7.55–7.47 (3H, m), 7.47–7.29 (8H, m), 7.22–7.18 (1H, m), 7.12 (1H, s), 7.10–7.05 (1H, m), 7.04 (1H, s), 6.96 (1H, s), 6.93 (1H, s), 6.88 (1H, s), 6.56 (1H, s), 6.35 (1H, s), 4.67 (1H, s), 4.19–4.05 (4H, m), 4.00–3.52 (92H, m), 3.48 (6H, s), 3.45 (3H, s), 3.42 (3H, s), 3.41 (3H, s), 3.39 (3H, s), 3.37 (3H, s), 3.31 (3H, s). HRMS (ES+): m/z calcd for C136H162N17O34S8 [M+H]+ 2832.9236 found 2832.9309.
4.3.2.4 Compound 8
Synthesized on a 16.3 μmol scale (25 mg Sieber amide resin with manufacturer's loading: 0.65 mmol g−1) using the general methodology mentioned above, with the exception that loading of the first monomer was carried out using HBTU (18.5 mg, 0.049 mmol, 3 equiv), and HOBt (7.5 mg, 0.049 mmol, 3 equiv) in the presence of DIEA (17 μL, 0.098 mmol, 6 equiv) with 1 mL DMF as the solvent and treated with microwaves (25 W, 70 °C, 10 min). Loading was repeated once. The crude product was purified by preparative RP-HPLC (System F) and then by recycling GPC to afford the title compound as a yellow solid (7 mg, 14%). RP-HPLC (System C): Rt = 11.33 min 1H NMR (CDCl3): δ 11.45 (1H, s), 11.14 (1H, s), 11.06 (1H, s), 11.01 (2H, s), 11.00 (1H, s), 10.91 (1H, s), 8.27 (1H, m), 8.20 (3H, m), 8.11–7.98 (3H, m), 7.82 (3H, m), 7.71–7.35 (15H, m), 7.23 (1H, m), 7.19 (1H, s), 7.12 (1H, m), 7.08 (1H, s), 7.00 (1H, s), 6.97 (1H, s), 6.92 (1H, s), 6.60 (1H, s), 6.43 (1H, d, J = 3.0), 4.65 (1H, m), 4.16 (4H, m), 3.98–3.55 (108H, m), 3.47–3.40 (32H, m), 3.30–3.26 (6H, m), 3.17 (2H, m). HRMS (ES+): m/z calcd for C152H193N17O42S8 [M+H]+ 3187.1410 found 3187.1431.
Acknowledgments
The authors would like to thank Dr P. Prabhakaran and Dr. B. Kauffmann for the crystallization and diffraction measurements of monomer 4, respectively. We would also like to thank the Servier Research Institute of Medicinal Chemistry for their assistance in developing monomer synthesis protocols. This work was supported by the European Union's Seventh Framework Programme through Marie Curie actions (FP7-PEOPLE-2010-ITN-264645 DYNAMOL, pre-doctoral fellowship to C.T.) and through the European Research Council (Grant Agreement Number ERC-2012-AdG-320892, postdoctoral fellowship to S. D.).